Agricultural Sciences
Vol.5 No.6(2014), Article ID:46222,10 pages DOI:10.4236/as.2014.56055
Development of a Multiplex Ligation Dependent Probe Amplification (MLPA) Module for Simultaneous Detection of Five Genetically Modified Rapeseed Events
Patrick Guertler*, Ottmar Goerlich, Ulrich Busch
Bavarian Health and Food Safety Authority, Oberschleissheim, Germany
Email: *patrick.guertler@lgl.bayern.de
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 27 February 2014; revised 25 April 2014; accepted 14 May 2014
ABSTRACT
For the official surveillance of genetically modified crops, efficient and simple detection methods need to be on hand to implement the strict requirements regarding approval, labelling and traceability determined by the European Union. Therefore, a multiplex ligation dependent probe amplification (MLPA) module was developed for simultaneous detection of five genetically modified rapeseed events (MS8, RF3, GT73, Falcon GS40/90 and T45). Probes were designed and concentrations were adapted in order to obtain high sensitivity and specificity. This MLPA module was validated using certified reference materials and its applicability was tested analyzing routine honey, mustard and rapeseed samples. The limit of detection was determined by analyzing a dilution series (n = 16 for each concentration) of respective transgenic DNA. After optimization, the MLPA revealed limits of detection between 10 to 50 copies of the transgene DNA/assay. The method proved to be sensitive and highly reproducible. When analyzing routine samples, results obtained applying the MLPA module were similar compared to real-time PCR.
Keywords:MLPA, Genetically Modified Organism, Rapeseed, Capillary Electrophoresis, Detection

1. Introduction
The legislation of the European Union (EU) dictates stringent requirements for approval of genetically modified (gm) crops, for their use as seed, feed or food, as well as for labelling and traceability (as outlined in EU directive 2001/18/EC, EU council directive 90/220/EEC and EU regulations 1829/2003 and 1830/2003). Products that contain authorized gm material above a content of 0.9% need to be labelled. Unapproved gm crops or products containing unauthorized gm crops may not be placed on the EU market (zero tolerance policy). As a consequence, products entering the EU market must be analyzed for genetic modification, which is a major challenge for the surveillance laboratories due to the increasing amount of gm crops grown worldwide.
Rapeseed (Brassica napus) is becoming a very important food crop in Europe. While no gm rapeseed is being grown or cultivated in Europe, over 9.3 million hectares of land was utilized in 2012 for growing gm rapeseed in the US, Canada, Australia and Chile [1] . Biotech rapeseed has mainly been altered to induce herbicide tolerance (e.g. events ACS-BNØØ5-8 (MS8), ACS-BNØØ3-6 (RF3), ACS-BNØØ8-2 (T45), ACS-BNØ1Ø-4 (Falcon GS 40/90) and MON-ØØØ73-7 (GT73)) by introducing bacterial genes bar (phosphinotricinacetyltransferase), pat (phosphinotricinacetyltransferase) or epsps (5-enolpyruvylshikimate-3-phosphate synthase). MS8, RF3, T45 and GT73 are authorized as food and feed in the EU and, as a consequence, products containing these gm events need to be labelled above a content of 0.9%. Cultivation of these events is prohibited in the EU and seeds containing any of these gm rapeseed events may not be placed on the EU market.
The most common technique for gm crop surveillance is the quantitative real-time polymerase chain reaction (qPCR), which allows a sensitive and quantitative analysis of the target [2] [3] . By combining several qPCR reactions in one assay (multiplex qPCR), a simultaneous detection of two or more targets can be achieved [4] -[6] . However, the use of several pairs of primers leads to different amplification efficiencies for each target. Multiplex qPCR assays are prone to PCR inhibition and mispriming, and are laborious to validate. Furthermore, multiplex qPCR is not particularly flexible as single primer sets which cannot be removed or added easily without influencing the whole assay. Other DNA-based detection methods for gm crops were developed but have not yet been applied for official surveillance of seed, feed and food [7] . A very promising DNA detection method is the multiplex ligation depended probe amplification (MLPA) [8] , which has already been applied in the area of medical diagnostics [9] or for the determination of allergens in food products [10] [11] . Moreano et al. [12] and Ehlert et al. [13] described the applicability of MLPA for the detection of gm events MONØØ81Ø-6 (MON810 corn) and MONØ4Ø32-6 (GTS 40-3-2 soybean), as well as for the detection of commonly used regulatory elements in gm crops like the P-35S promoter.
For the official food and feed surveillance, MLPA is an interesting analytical tool to cope with the increasing amount of gm crops worldwide by detecting several gm events in one reaction. Hence, the scope of this work was to develop an MLPA assay for the simultaneous detection of major gm rapeseed events and to evaluate its use for the official seed surveillance.
2. Material and Methods
2.1. Sample Material
For assay development and validation, the following certified reference materials were used: MS8 (0306-F3), RF3 (0306-G3) and T45 (0208-A4). These reference materials are distributed by the American Oil Chemists’ Society (Urbana, IL, USA). Leaf tissue (GT73) and seed (Falcon GS 40/90) were provided by the Bavarian Health and Food Safety Authority (LGL). The LGL also provided 25 routine samples (honey, rapeseed and white mustard), which were used to demonstrate the applicability of the developed detection method.
2.2. DNA Extraction
Genomic DNA was extracted using an automated DNA extraction protocol for rapeseed samples as previously described by Guertler et al. [14] . As a slight modification, 20 µl RNase A (10 mg∙ml−1) and 20 µl thioglycerol (Promega GmbH, Mannheim, Germany) were added to the CTAB buffer/sample/proteinase K mix prior incubation at 65˚C for 90 minutes. Honey samples were extracted as described by Guertler et al. [15] . The Quant-iTPicoGreen technology was applied according to the manufacturer’s instructions (Life Technologies, Darmstadt, Germany) to quantify isolated DNA.
2.3. Verification of Target Sequences and MLPA Probe Design
Prior to MLPA probe design, event-specific sequences obtained from the NCBI (http://www.ncbi.nlm.nih.gov) or GMDD database [16] were verified by PCR and subsequent sequencing (data not shown). Sequencing primers (Metabion, Martinsried, Germany) were designed for amplification of the genomic plant DNA/transgene junction. This sequence information is required for the development of an event-specific detection assay.
Amplicons were separated by gel electrophoresis and extracted from the gel using the Wizard SV Gel and PCR clean-up System (Promega, Mannheim, Germany) according to the manufacturer’s instructions. Sequencing reactions were performed by GATC Biotech AG (Konstanz, Germany). These verified DNA sequences were compared to already published sequences and used for MLPA probe design according to the suggestions by MRC Holland (Amsterdam, Netherlands; http://www.mrc-holland.com). Positions of amplicons in the respective genome of different rapeseed events are illustrated in Figure 1. Designed probes were checked for secondary structures using Mfold [17] and for specificity by using NCBI Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
2.4. MLPA Workflow
MLPA reactions were performed in a Primus 96 plus thermocycler (Eurofins MWG Operon, Ebersberg, Germany) using the SALSA MLPA EK20 reagent kit (MRC Holland, Amsterdam, Netherlands) according to the manufacturer’s instructions. All components, except DNA mix, probe mix, water and TE buffer are included in the SALSA MLPA EK20 reagent kit.
DNA samples (20 ng∙µl−1 or adjusted to certain DNA copy numbers) contained DNA from one of the following gm rapeseed events: GT73, MS8, RF3, Falcon GS 40/90 and T45. A mixture containing all rapeseed DNA samples in equal concentration (20 ng∙µl−1) was also applied. A non-gm rapeseed sample was used as a nontemplate control (NTC) and for sample dilution. The optimal probe concentration was determined by adding probes in different concentrations to the probe mix. Optimal probe concentrations are outlined in Table 1. The probe mix consisted of all probes premixed in a 1.5 mL tube and 0.01 M Tris/HCl buffer (pH = 9.0), which was added to a final volume of 600 µl. In addition to the event-specific probes, the probe mix also contained a set of probes for the detection of a reference gene (cruA) as a positive control for the presence of rapeseed DNA.
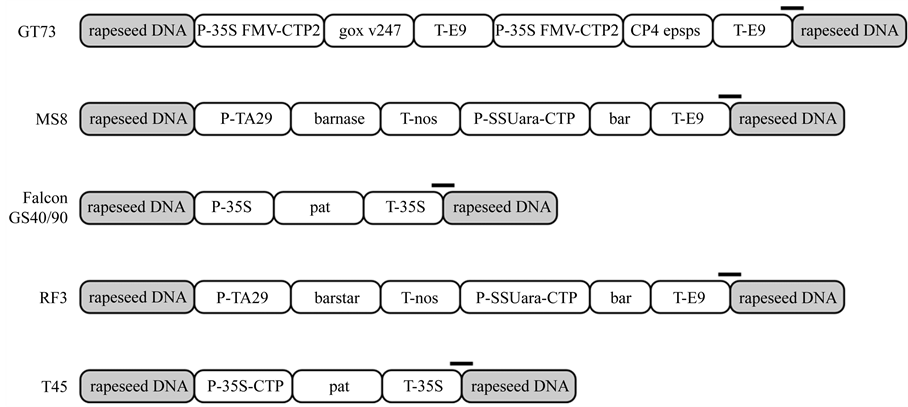
Figure 1. Schematic schema of the genetic structure of different genetically modified rapeseed events. Position of the amplicon as amplified in the MLPA is indicated by black bars. P-35S FMV-CTP2 = 35S promoter from Figwort mosaic virus and chloroplast transit peptide (CTP) from Arabidopsis thaliana; CP4 epsps = gene encoding for 5-enolpyruvoylshikimate-3-phosphate synthetase isolated from Agrobacterium tumefaciens strain CP4; T-E9 = non-translated region of the ribulose-1,5-bisphosphate carboxylase small subunit from Pisumsativum; P-TA29 = pollen specific TA29 promoter from Nicotianatabacum; barnase = gene encoding for barnaseribonuclease from Bacillus amyloliquefaciens; T-nos = terminator from Agrobacterium tumefaciensnopalin synthase gene; P-SSUara-CTP = promoter of the small subunit of the ribulose-1,5-bisphosphate carboxylase and chloroplast transit peptide (CTP) from Arabidopsis thaliana; bar = gene coding for phosphinothricin N-acetyltransferase from Streptomyces hygroscopicus; P-35S = 35S promoter from Cauliflower mosaic virus; pat = gene coding for phosphinothricin N-acetyltransferase from Streptomyces viridochromogenes; T-35S = 35S terminator from Cauliflower mosaic virus; barstar = gene coding for barnaseribonuclease inhibitor from Bacillus amyloliquefaciens.

Table 1. Sequences of probes used in the MLPA assay. Common primer binding sequences are L = GGGTTCCCTAAGGGTTGGA and R = TCTAGATTGGATCTTGCTGGCAC. Forward primers are 5’-FAM-labelled. LPO = left probe oligo, RPO = right probe oligo. Upper bold = hybridization sequence, lower italic = stuffer sequence. The RPO is 5’-phosphorylated (Pho). c = final concentration in the probe mix.
Five µl DNA solutions were incubated at 95˚C for 5 minutes for initial denaturation. Afterwards, 1.5 µl MLPA buffer and 1.5 µl probe mix were added and incubated at 60˚C for 16 hours. As the long duration of the hybridization step is a negative aspect of this method, we also compared results after different hybridization times (16 hours and 3 hours).
The ligation was performed by adding 3 µl buffer A, 3 µl buffer B, 25 µl H2O and 1 µl Ligase-65 before incubating the mixture for 15 minutes at 55˚C and then for 5 minutes at 98˚C. For the subsequent PCR reaction, 7.5 µl H2O, 2 µl PCR Primer Mix and 0.5 µl Salsa Polymerase were added. The PCR reaction was performed under the following conditions: 40 cycles of denaturation at 95˚C for 30 seconds, annealing at 60˚C for 30 seconds and extension at 72˚C for 1 minute, followed by 1 cycle of extension at 72˚C for 20 minutes. Finally, samples were cooled down to 4˚C and used for fragment length analysis or stored at −20˚C. The whole workflow is depicted in Figure 2.
2.5. Fragment Length Analysis
Fragment length analysis was performed using a 3130 Genetic Analyzer (Applied Biosystems, Carlsbad, USA) and a POP-7 performance optimized polymer (Life technologies, Darmstadt, Germany). Samples were diluted 1:40 using HPLC grade water and 1 µl was added to 15 µl HiDi-formamide containing 0.15 µl GeneScan 500 LIZ 3130 size standard (Life technologies, Darmstadt, Germany).
Fragment length analysis was performed using the following settings: oven temperature: 60˚C; prerun voltage: 15 kV; prerun time: 180 s; injection voltage: 1.2 kV; injection time: 16 s; run voltage: 15 kV and run time: 1200 s.
2.6. Assay Validation
Combined hybridization sequences of two adjacent binding probes were virtually tested for specificity by using NCBI Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Specific probe pairs were then tested practically for specificity by applying DNA isolated from different gm events and non-gm materials. Afterwards, these probe pairs were compiled to the MLPA rapeseed probe mix. This probe mix was applied to DNA from single gm rapeseed events and to a mixture consisting of all targeted gm rapeseed events. Optimization of the probe mix was achieved by varying the probe concentrations.
Limit of detection (LOD), dynamic range and repeatability were determined by analyzing a dilution series of DNA from each targeted gm rapeseed event. DNA was diluted from 10,000 copies to 1 copy of transgenic DNA/assay and analyzed 4-times using 4 replicates in each run. The LOD was defined as the lowest analyte concentration for which all 16 replicates gave positive signals.
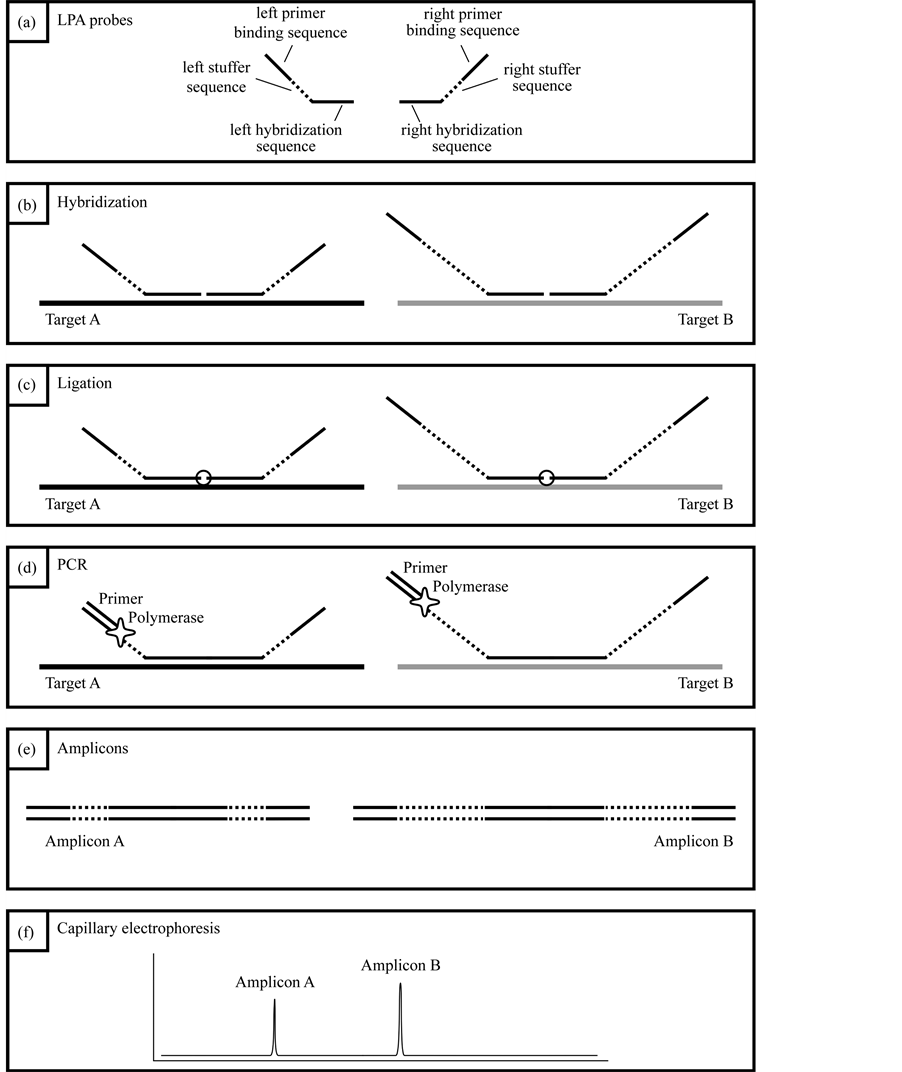
Figure 2. (a) Each LPA probe consists of a hybridization sequence, a stuffer sequence which defines the length of the amplicon, and a universal primer binding site; (b) both LPA probes bind adjacent to each other to the target sequence; (c) after hybridization, both LPA probes are ligated; (d) in a subsequent PCR reaction, the ligated probes are amplified using FAM-labelled universal primers; (e) amplicons of different probes differ in length; (f) amplicons of different probes can be discriminated due to their unique length by using capillary electrophoresis.
2.7. Routine Samples
DNA from honey, mustard and rapeseed samples (n = 25), isolated and analyzed as part of the routine food and seed surveillance of the Bavarian Health and Food Safety Authority, was used to compare results obtained by qPCR [19] -[21] with results obtained by MLPA. 5 µl DNA was used in the MLPA assay as described before and a hybridization time of 16 hours was applied. After MLPA, samples were diluted 1:40 before capillary electrophoresis.
3. Results
3.1. DNA Extraction
Automated DNA extraction resulted in DNA concentrations varying from 28 ng∙µl−1 to 74 ng∙µl−1. DNA purity was not checked as previous isolations from rapeseed samples using the Maxwell 16 extraction robot resulted in high quality DNA [14] . We assume only slight differences between DNA isolations as the procedure is automated and highly reproducible.
3.2. Validation of the MLPA Rapeseed Module
Probe concentrations used in this MLPA assay were reduced (T45, cruA) or increased (GT73, MS8, RF3, Falcon GS 40/90) compared to the instructions set by MRC Holland (see Table 1), which was needed to achieve a similar amplification signal for all targets.
By analyzing a DNA-Mix containing all rapeseed events, a specific peak was obtained for each event (Figure 3(a)). When analyzing DNA from only one event, peaks specific for the reference gene cruA and the respective gm rapeseed event were obtained (Figure 3(b)-3(f)).
Validation experiments revealed mean fragment sizes of 95.5 bp ± 0.1 for the reference gene cruA, 98.7 bp ± 0.1 for GT73, 102.6 bp ± 0.07 for MS8, 107.3 bp ± 0.07 for Falcon GS 40/90, 110.7 bp ± 0.09 for RF3 and 115.7 bp ± 0.13 for T45. No specific peak was observed in the NTC control. The obtained fragment sizes are in line with the expected fragment sizes.
The LOD was determined by analyzing serial dilutions of DNA from the respective rapeseed events. LOD values of 10 copies for T45, 25 copies for events MS8 and Falcon GS 40/90, and 50 copies for events GT73 and RF3 were obtained. Reduction of hybridization time to 3 hours resulted in slightly better or similar results compared to a hybridization time of 16 hours (Figure 4).
3.3. Routine Samples
Rapeseed, white mustard and honey samples, previously analyzed by qPCR as part of the official gm crop surveillance, were reanalyzed applying the developed MLPA rapeseed module. This assay revealed similar results as obtained by qPCR, which are summarized in Table 2. Rapeseed samples, which were negative after qPCR analysis, also showed no positive signal when applying the MLPA rapeseed module. White mustard samples were either tested negative or contained MS8 and RF3. Honey samples were tested positive for rapeseed events GT73, MS8 and RF3 by using MLPA, however, one honey samples only contained GT73. All results obtained using MLPA go in line with results of the official gm crop surveillance. Lower Cq values in qPCR also corresponded to larger peak areas and higher peak heights after capillary electrophoresis.
4. Discussion
The MLPA is an easy-to-learn technique, as it is very similar to conventional PCR regarding the hands-on steps and it can be implemented in every lab with a capillary sequencer. Compared to qPCR assays, the probes used in MLPA are inexpensive as merely the 5’-end of the right probe needs a modification (phosphate). Only minute amounts of probes are used when setting up a probe mix. Therefore, these probes can be used for a large number of samples once synthesized and stored properly (−20˚C).
By combining an automated DNA extraction method with MLPA, a large number of samples can be screened for the presence of major gm rapeseed events at the same time [14] . The applicability of this method was also improved by reducing the hybridization time from 16 hours, as set by MRC Holland, to 3 hours. Therefore, the whole workflow (automated DNA extraction in combination with a MLPA assay) can be carried out in a single day.
The developed MLPA rapeseed module is highly flexible and can easily be modified based on the actual scope of GM surveillance. Probes for the detection of other rapeseed events can be added when needed; how-
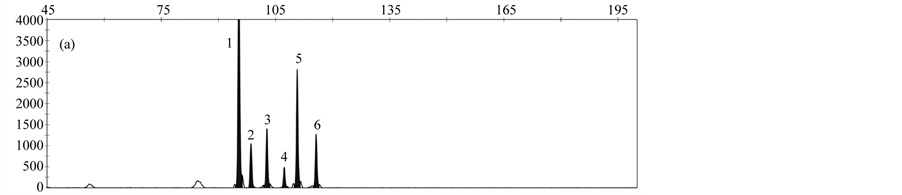
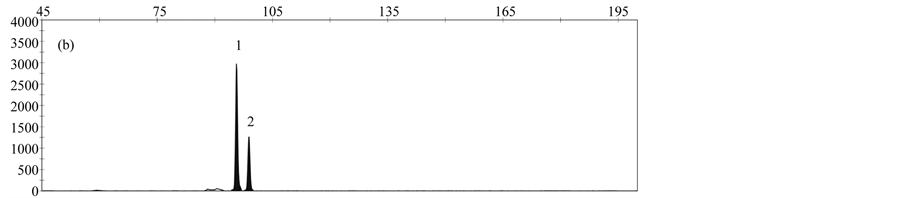
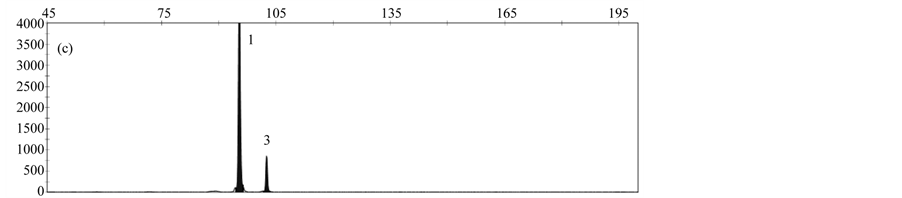
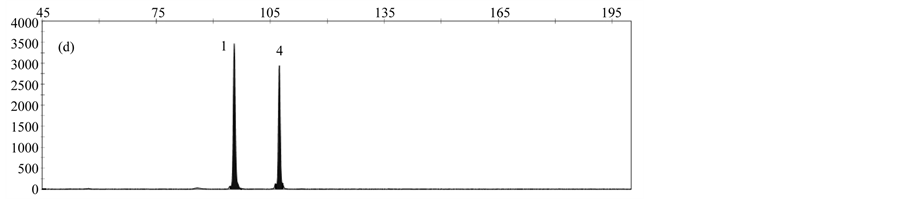
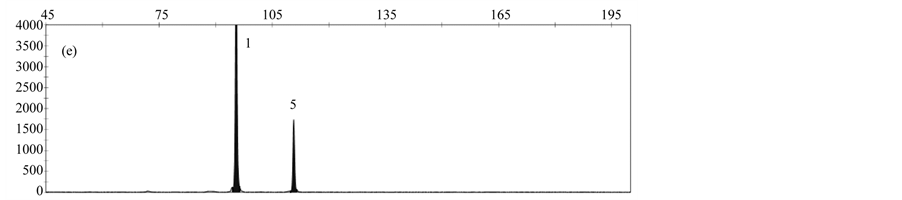
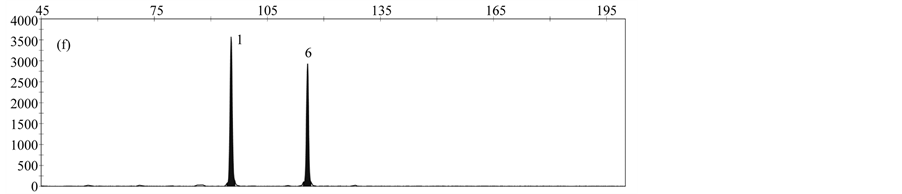
Figure 3. Results obtained by applying the MLPA rapeseed module with (a) DNA mix containing reference material from all gm rapeseed events: 1-cruA (reference gene), 2-GT73, 3-MS8, 4-Falcon GS 40/90, 5-RF3, 6-T45; (b) DNA mix containing GT73 only: 1-cruA, 2-GT73; (c) DNA mix containing MS8 only 1-cruA, 2-MS8; (d) DNA mix containing Falcon GS 40/90 only 1-cruA, 2-Falcon GS 40/90; (e) DNA mix containing RF3 only: 1-cruA, 2-RF3; (f) DNA mix containing T45 only 1-cruA, 2-RF3; (f) 1-cruA, 2-T45.
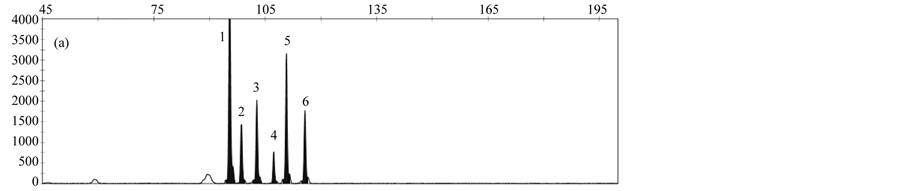
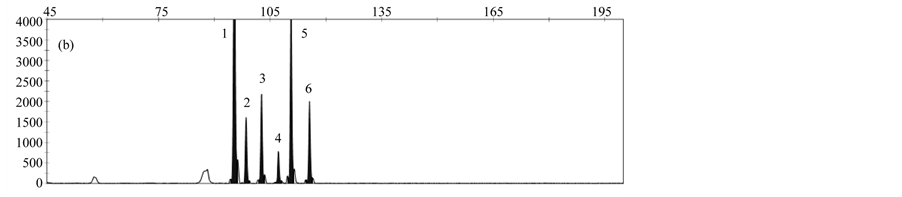
Figure 4. Comparison of different hybridization times; (a) 16 hours; (b) 3 hours; Peaks represent the following gm rapeseed events: 1-cruA (reference gene), 2- GT73, 3-MS8, 4-Falcon GS 40/90, 5-RF3, 6-T45.
Table 2. Different kinds of routine samples, which were originally analyzed by qPCR, underwent MLPA analysis and the two detection methods were compared. Data are presented as Cq ± standard deviation and MLPA peak area (Cq = cycle of quantification, A = peak area, n.d. = not detected).
ever, the maximum number of probe pairs in a probe mix is limited. In its synthetic probe design instructions, MRC Holland sets out that the optimal length for combined MLPA probe pairs is between 100 and 140 bp. This would lead to a maximum of 11 probe pairs that can be combined in a probe mix, provided that the amplified fragments differ in a fragment size of 4 bp. A difference in amplicon size of at least 4 bp is needed to clearly differentiate specific peaks after capillary electrophoresis.
The gm rapeseed events, targeted by the developed MLPA module, were chosen according to their importance worldwide [22] and the availability of sequence information and reference material. As soon as certified reference material and sequence information for other rapeseed events are available, the MLPA module can be extended. By using the actual MLPA rapeseed module, amplicons with a fragment size between 96 and 116 bp are obtained. As a consequence, according to the probe design instructions by MRC Holland, an additional set of 6 probe pairs could be added to this module.
So far, there is no multiplex qPCR assay available that can detect up to 5 gm rapeseed events in one reaction. And most of multiplex detection assays have been performed using conventional PCR [22] [23] . In multiplex qPCR, you need to add different sets of primers and probes in order to detect different targets in one reaction. This results in different PCR efficiencies and potential interaction of the oligonucleotides. In an MLPA assay, you use one set of primers for all targets. Therefore, the PCR efficiency is equal or very similar and interactions between the primers can be excluded. The obtained detection limits of our developed MLPA assay are in the range of a rapeseed qPCR [24] [25] . One should keep in mind that we used genomic DNA as starting material, whereas plasmids are often used as calibrators in qPCR [26] . By doing so, the possibility of contamination is reduced and potential matrix effects are also taken into account. MLPA and qPCR were directly compared analyzing different routine samples: honey, rapeseed and white mustard. Even when using a very complex matrix like honey as a starting material, the developed MLPA system revealed reliable results, similar to those obtained after qPCR [15] . The sensitivity of this MLPA rapeseed module is high enough to detect even traces of gm rapeseed in white mustard and honey samples.
In summary, the developed MLPA assay is reliable, fast and easy to perform and offers a high degree of multiplexing. For routine gm crop surveillance, this module can be applied as a useful screening tool for the simultaneous detection of five commonly used gm rapeseed events. However, the developed MLPA assay is a qualitative assay. Once the samples are screened for the presence of gm rapeseed, a quantitative method needs to be performed, if quantification is required.
Acknowledgements
The Bavarian State Ministry of the Environment and Public Health is gratefully acknowledged for funding this project. We thank Dr. Norbert Hess for providing integration sequences for Falcon GS 40/90.
References
- James, C. (2012) Global Status of Commercialized Biotech/GM Crops: 2012. ISAAA Brief, No. 44, ISAAA, Ithaca, New York.
- Engel, K.-H., et al. (2006) Quantification of DNA from Genetically Modified Organisms in Composite and Processed Foods. Trends in Food Science & Technology, 17, 490-497. http://dx.doi.org/10.1016/j.tifs.2006.04.008
- Waiblinger, H.-U., et al. (2010) A Practical Approach to Screen for Authorised and Unauthorised Genetically Modified Plants. Analytical and Bioanalytical Chemistry, 396, 2065-2072. http://dx.doi.org/10.1007/s00216-009-3173-2
- Elenis, D.S., et al. (2008) Advances in Molecular Techniques for the Detection and Quantification of Genetically Modified Organisms. Analytical and Bioanalytical Chemistry, 392, 347-354. http://dx.doi.org/10.1007/s00216-008-1868-4
- Huber, I., et al. (2013) Development and Validation of Duplex, Triplex, and Pentaplex Real-Time PCR Screening Assays for the Detection of Genetically Modified Organisms in Food and Feed. Journal of Agricultural and Food Chemistry, 61, 10293-10301. http://dx.doi.org/10.1021/jf402448y
- Waiblinger, H.-U., et al. (2008) Validation and Collaborative Study of a P35S and T-Nos Duplex Real-Time PCR Screening Method to Detect Genetically Modified Organisms in Food Products. European Food Research and Technology, 226, 1221-1228. http://dx.doi.org/10.1007/s00217-007-0748-z
- Morisset, D., et al. (2008) Alternative DNA Amplification Methods to PCR and Their Application in GMO Detection: A Review. European Food Research and Technology, 227, 1287-1297.
- Schouten, J., et al. (2002) Relative Quantification of 40 Nucleic Acid Sequences by Multiplex Ligation-Dependent Probe Amplification. Nucleic Acids Research, 30, e57. http://dx.doi.org/10.1093/nar/gnf056
- Lunkka-Hytonen, M., et al. (2008) G.P.9.04 Development of the Multiplex Ligation-Dependent Probe Amplification (MLPA) Method for Identifying Large Scale Mutations in the Nebulin Gene. Neuromuscular Disorders, 18, 787. http://dx.doi.org/10.1016/j.nmd.2008.06.216
- Luber, F., et al. (2012) Apricot DNA as an Indicator for Persipan: Detection and Quantitation in Marzipan Using Ligation-Dependent Probe Amplification. Journal of Agricultural and Food Chemistry, 60, 5853-5858. http://dx.doi.org/10.1021/jf301202s
- Mustorp, S.L., Drømtorp, S.M. and Holck, A.L. (2011) Multiplex, Quantitative, Ligation-Dependent Probe Amplification for Determination of Allergens in Food. Journal of Agricultural and Food Chemistry, 59, 5231-5239. http://dx.doi.org/10.1021/jf200545j
- Moreano, F., et al. (2006) Ligation-Dependent Probe Amplification for the Simultaneous Event-Specific Detection and Relative Quantification of DNA from Two Genetically Modified Organisms. European Food Research and Technology, 222, 479-485.
- Ehlert, A., et al. (2008) Development of a Modular System for Detection of Genetically Modified Organisms in Food Based on Ligation-Dependent Probe Amplification. European Food Research and Technology, 227, 805-812. http://dx.doi.org/10.1007/s00217-007-0790-x
- Guertler, P., et al. (2013) Development of a CTAB Buffer-Based Automated gDNA Extraction Method for the Surveillance of GMO in Seed. European Food Research and Technology, 236, 599-606. http://dx.doi.org/10.1007/s00217-013-1916-y
- Guertler, P., et al. (2014) Automated DNA Extraction from Pollen in Honey. Food Chemistry, 149, 302-306. http://dx.doi.org/10.1016/j.foodchem.2013.10.129
- Dong, W., et al. (2008) GMDD: A Database of GMO Detection Methods. BMC Bioinformatics, 9, 260. http://dx.doi.org/10.1186/1471-2105-9-260
- Zuker, M. (2003) Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Research, 31, 3406-3415. http://dx.doi.org/10.1093/nar/gkg595
- Hess, N., Ulrich, A. and Hoffmann, T. (2002) Insertionsspezifische Nachweisverfahren für transgene Pflanzenlinien unter Anwendung der inversen PCR. Bundesgesundheitsblatt-Gesundheitsforschung-Gesundheitsschutz, 45, 626-633. http://dx.doi.org/10.1007/s00103-002-0444-x
- Mazzara, M., et al. (2007) Quantitative PCR Method for Detection of Oilseed Rape Event MS8. European Union Reference Laboratory for GM Food and Feed.
- Mazzara, M., et al. (2007) Quantitative PCR Method for Detection of Oilseed Rape Event GT73. European Union Reference Laboratory for GM Food and Feed.
- Savini, C., et al. (2007) Quantitative PCR Method for Detection of Oilseed Rape Event RF3. European Union Reference Laboratory for GM Food and Feed.
- Demeke, T. and Ratnayaka, I. (2008) Multiplex Qualitative PCR Assay for Identification of Genetically Modified Canola Events and Real-Time Event-Specific PCR Assay for Quantification of the GT73 Canola Event. Food Control, 19, 893-897. http://dx.doi.org/10.1016/j.foodcont.2007.08.020
- James, D., et al. (2003) Reliable Detection and Identification of Genetically Modified Maize, Soybean, and Canola by Multiplex PCR Analysis. Journal of Agricultural and Food Chemistry, 51, 5829-5834. http://dx.doi.org/10.1021/jf0341159
- Yang, L., et al. (2008) Qualitative and Quantitative Event-Specific PCR Detection Methods for Oxy-235 Canola Based on the 3’ Integration Flanking Sequence. Journal of Agricultural and Food Chemistry, 56, 1804-1809. http://dx.doi.org/10.1021/jf073465i
- Yang, L., et al. (2006) Event-Specific Qualitative and Quantitative Polymerase Chain Reaction Analysis for Genetically Modified Canola T45. Journal of Agricultural and Food Chemistry, 54, 9735-9740. http://dx.doi.org/10.1021/jf061918y
NOTES

*Corresponding author.