Case Reports in Clinical Medicine
Vol.2 No.2(2013), Article ID:31119,3 pages DOI:10.4236/crcm.2013.22033
Efficacy of deferasirox for the treatment of iron overload in a child affected by Juvenile Hemochromatosis
![]()
1Department of Pediatrics, University of Milano-Bicocca, Ospedale S. Gerardo, Monza, Italy; *Corresponding Author: n.masera@hsgerardo.org
2Consortium for Human Molecular Genetics, Monza, Italy
3Department of Health Sciences, University of Milano-Bicocca, Ospedale S. Gerardo, Monza, Italy
Copyright © 2013 Nicoletta Masera et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 3 March 2013; revised 11 April 2013; accepted 20 April 2013
Keywords: Juvenile Hemochromatosis; Deferasirox; Iron Chelation; Child; Iron Overload
ABSTRACT
We report the case of a 7 years old girl with Juvenile Hemochromatosis, due to homozygous mutation of HJV, which had increased serum iron indices and liver iron overload in the absence of any clinical sign of disease. Oral iron chelation with low dose deferasirox showed good efficacy and no side effects. The oral iron chelator deferasirox could be a valid option for removing excess iron in early Juvenile Hemochromatosis.
1. INTRODUCTION
Juvenile hemochromatosis (JH), or hemochromatosis type 2, is a rare form of hemochromatosis (HH), characterized by early and marked iron deposition in tissues and severe clinical course [1,2]. Clinical complications include hypogonadotropic hypogonadism, liver cirrhosis, diabetes mellitus, cardiomyopathy and arthropathy occurring typically in the second to third decades of life [1,2]. JH is determined by homozygous mutations in two genes, HAMP and HJV, which account for about 10% and 90% of cases, respectively. HAMP encodes hepcidin, the master regulator of iron homeostasis, and HJV encodes hemojuvelin, a membrane protein which acts as co-receptor for bone morphogenetic protein signaling which positively regulates hepcidin expression in liver cells [1,2]. Suppression of hepcidin synthesis induced by both HAMP and HJV mutations causes increased intestinal iron absorption and iron release from macrophages leading to very high transferrin saturation and progressive tissue iron overload [3]. The complications of iron overload can be avoided by early diagnosis and appropriate removal of excess iron. The mainstay of treatment in HJV is phlebotomy, but oral chelation could be a new therapeutic approach [4].
2. CASE DESCRIPTION
A 7 years old girl of Albanese origin, living in the neighbourhood of Milan, was referred to our Pediatric Haemathology Outpatient Service in Monza, in October 2008 for occasional detection of elevated serum iron parameters (transferrin saturation: 101%, serum ferritin: 194 μg/L). The girl was growing well, had a normal blood count, normal liver enzymes and no clinical signs of disease. One month after, the analyses confirmed high iron indices (serum iron 254 µg/dL, transferrin saturation 79%, ferritin: 181 μg/L) and HH was suspected. She was negative for the p.Cys282Tyr and p.His63Asp variants of HFE (Smart Cycler, Cepheid, Sunnyvale, CA, USA), and sequence analyses (ABI PRISM 3130 Avant Automatic Sequencer, PE Applied Biosystem, Foster City, CA, USA) of HFE, Transferrin receptor 2 (responsible for HH type 3) and HAMP were negative, too. Sequencing of HJV showed the presence of p.Gly320Val mutation in the homozygous state. Family study confirmed the presence of the mutation in the heterozygous state in her parents and in the sister, who had normal iron indices.
The child performed non invasive quantitation of iron in liver and heart showing increased liver iron concentration without cardiac iron overload (Table 1). Considering the risk of iron-related damage inherent to JH, a therapeutic option was considered. Phlebotomy therapy was excluded for two main reasons: 1) even in skilled hands,
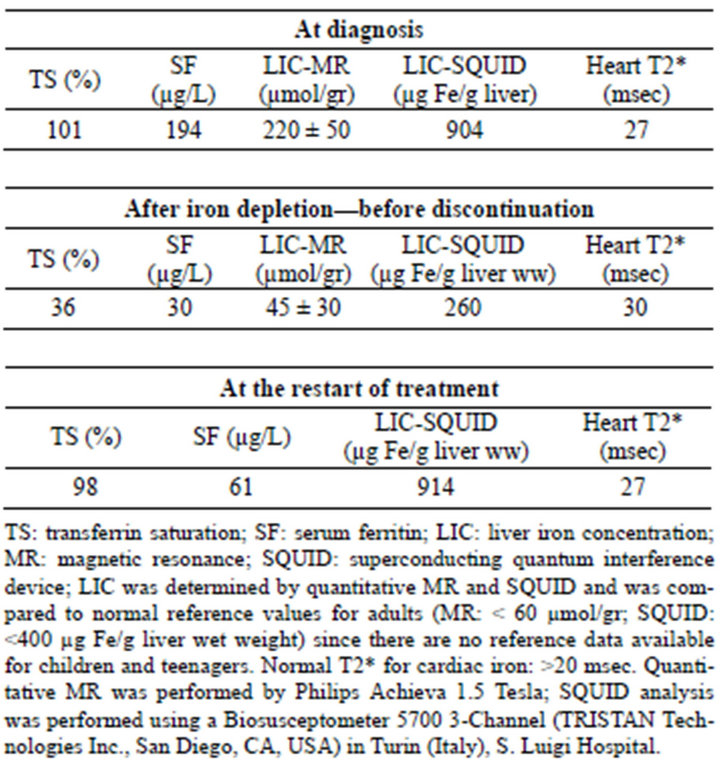
Table 1. Serum and tissue (liver and heart) iron indices in different phases of iron chelation.
placement of peripheral intravenous (IV) catheters in children is a difficult task, and when multiple IV attempts are required, the child’s stress is increased [5]; 2) repeated hospital admittances would be needed further increasing family distress (transit from home to hospital, loss of school days). Both factors might reduce child and parental compliance to care. The recent report on safety and efficiency of deferasirox in HH type 1 [6] led us to propose deferasirox as an off-label treatment, at the dose of 10 mg/kg/day. Informed written consent was obtained by both parents.
The therapy was well tolerated and effective. A progressive reduction of serum iron, transferrin saturation and ferritin concentration was observed. One year after starting chelation, serum indices were normalized; both SQUID (Superconducting quantum interference device) and magnetic resonance revealed a normal value of liver iron concentration (LIC) (Table 1). Thus, iron chelation was discontinued.
In the following months, iron indices were periodically investigated. Fifteen months after treatment discontinuation, transferrin saturation and serum ferritin were increased and LIC detected by SQUID was again increased to the original level (Table 1). Based on these data, we reintroduced deferasirox at the dosage of 4 mg/kg/day. After a further 12 months period of follow-up, no side effects were noticed and serum iron indices progressively reduced.
3. DISCUSSION
In the present report there are two main findings. Firstthe demonstration of the efficacy and safety of deferasirox in the treatment of iron overload in JH children; second, the importance of early diagnosis of JH, preventing the development of severe complications and allowing intermittent and low-dosage therapy to be performed.
Phlebotomy therapy remains the best therapeutic option in HH, but not all patients are candidates for phlebotomy due to underlying anemia, heart disease, or poor venous access, and compliance with regular phlebotomy may be an issue. Although deferoxamine has been used successfully in a limited number of HH patients, due to the absent oral bioavailability and short plasma half-life, it has to be given by slow subcutaneous or intravenous infusion that limits patients’ compliance to treatment [4]. Oral chelation therapy is a possible alternative in selected cases of HH. A phase 1/2, dose-escalation trial of deferasirox was recently performed in uncomplicated patients with HH and results were encouraging [6]. A starting dose of 10 mg/kg/day was the most appropriate in this population in term of safety and efficacy. In transfusional iron overload, deferasirox has been extensively used at higher dosages (usually 20 to 40 mg/kg/day) according to the amount of transfusional iron loading even in children [7]. At the best of our knowledge there are only two reports on the use of deferasirox in the treatment of iron overload in JH in humans [8,9] and none in children. In a murine model of JH characterized by massive iron overload, deferasirox effectively reduced liver and heart iron at a dosage of 30 - 100 mg/kg/day [10]. Differently from the markedly iron overloaded murine model, our young patient had moderate hepatic iron overload, no cardiac iron and no biochemical or clinical signs or iron-related complications. Accordingly, Kaikov et al. [11] reported three children affected by JH (aged 7, 6 and 4 years) that showed markedly increased LIC without clinical complications. Overall, these data indicate that an early diagnosis of JH is mandatory to avoid iron-related organ damage. Unfortunately, this does not commonly occur in this rare and severe disease. In fact, we previously showed that diagnosis of JH frequently occurs later (mean ± SD: 23 ± 5 years) when 96.1% of the patients already had hypogonadotrophic hypogonadism, 34.6% cardiac complication, 42.1% cirrhosis, 57.7% glucose intolerance or diabetes, and 26.9% arthropathy [1]. These findings indicate that particular attention should be given to children and young adolescents with unexpected high transferrin saturation value, which could suggest the presence of JH or HH type 3 at an early stage [12]. Although the rarity of these forms of HH limits the possibility of statistically valuable studies, the present report indicates that the oral iron chelator deferasirox could be a valid option for removing excess iron in early JH, thus avoiding the development of clinical complications and changing the natural history of the disease.
REFERENCES
- De Gobbi, M., Roetto, A., Piperno, A., et al. (2002) Natural history of juvenile haemochromatosis. British Journal of Haematology, 117, 973-979. doi:10.1046/j.1365-2141.2002.03509.x
- Pietrangelo, A. (2004) Non-HFE hemochromatosis. Hepatology, 39, 21-29. doi:10.1002/hep.20007
- Piperno, A., Mariani, R., Trombini, P. and Girelli, D. (2009) Hepcidin modulation in human diseases: From research to clinic. World Journal of Gastroenterology, 15, 538-551. doi:10.3748/wjg.15.538
- Clark, P., Britton, L.J. and Powell, L.W. (2010) The diagnosis and management of hereditary haemochromatosis. Clinical Biochemist Reviews, 31, 3-8.
- Larsen, P., Eldridge, D., Brinkley, J., et al. (2010) Pediatric peripheral intravenous access: Does nursing experience and competence really make a difference? Journal of Infusion Nursing, 33, 226-235. doi:10.1097/NAN.0b013e3181e3a0a8
- Phatak, P., Brissot, P., Wurster, M., et al. (2010) A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology, 52, 1671-1779. doi:10.1002/hep.23879
- Cappellini, M.D., Porter, J., El-Beshlawy A., et al. (2010) Tailoring iron chelation by iron intake and serum ferritin: The prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica, 95, 557-566. doi:10.3324/haematol.2009.014696
- Santos, P.C., Cancado, R.D., Pereira, A.C., et al. (2010) HJV hemochromatosis, iron overload, and hypogonadism in a Brazilian man: Treatment with phlebotomy and deferasirox. Acta haematologica, 124, 204-205. doi:10.1159/000321493
- Maeda, T., Nakamaki, T., Saito, B., et al. (2011) Hemojuvelin hemochromatosis receiving iron chelation therapy with deferasirox: Improvement of liver disease activity, cardiac and hematological function. European Journal of Haematology, 87, 467-469. doi:10.1111/j.1600-0609.2011.01693.x
- Nick, H., Allegrini, P.R., Fozard, L., et al. (2009) Deferasirox reduces iron overload in a murine model of juvenile hemochromatosis. Experimental Biology and Medicine (Maywood), 234, 492-503. doi:10.3181/0811-RM-337
- Kaikov, Y., Wadsworth, L.D., Hassall, E., Dim-mick, J.E. and Rogers, P.C. (1992) Primary hemochro-matosis in children: Report of three newly diagnosed cases and review of the pediatric literature. Pediatrics, 90, 37-42.
- Piperno, A. (2013) Molecular diagnosis of he-mochromatosis. Expert Opinion on Medical Diagnostics, 7, 161- 177. doi:10.1517/17530059.2013.763794