American Journal of Plant Sciences
Vol.4 No.6(2013), Article ID:32899,15 pages DOI:10.4236/ajps.2013.46149
Proteomics: A Successful Approach to Understand the Molecular Mechanism of Plant-Pathogen Interaction
![]()
Department of Biotechnology, Visva-Bharati University, Santiniketan, India.
Email: #jolly.basak@visva-bharati.ac.in
Copyright © 2013 Tushar Dilipchand Lodha et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 14th, 2013; revised April 20th, 2013; accepted May 15th, 2013
Keywords: Proteome; Plant-Pathogen Interaction; Systemic Acquired Resistance; Mass Spectrometry; Pathogenicity Related Proteins
ABSTRACT
In recent years, proteomics has played a key role in identifying changes in protein levels in plant hosts upon infection by pathogenic organisms and in characterizing cellular and extracellular virulence and pathogenicity factors produced by pathogens. Proteomics offers a constantly evolving set of novel techniques to study all aspects of protein structure and function. Proteomics aims to find out the identity and amount of each and every protein present in a cell and actual function mediating specific cellular processes. Structural proteomics elucidates the development and application of experimental approaches to define the primary, secondary and tertiary structures of proteins, while functional proteomics refers to the development and application of global (proteome wide or system-wide) experimental approaches to assess protein function. A detail understanding of plant defense response using successful combination of proteomic techniques and other high throughput techniques of cell biology, biochemistry as well as genomics is needed for practical application to secure and stabilize yield of many crop plants. This review starts with a brief introduction to geland non gel-based proteomic techniques followed by the basics of plant-pathogen interaction, the use of proteomics in recent pasts to decipher the mysteries of plant-pathogen interaction, and ends with the future prospects of this technology.
1. Introduction
Plant-pathogen interaction is a multifaceted process [1, 2]. At the beginning of the interaction, plants develop two pathways to recognize and resist pathogen attack. One pathway involves the formation of danger-associated molecular patterns (DAMPs) and pathogen associated molecular patterns (PAMPs) while other one results in effector-triggered immunity (ETI) and PAMPs-triggered immunity (PTI) after recognition by specific pathogen effector molecules [1-3]. Accordingly, downstream signaling cascades are activated, producing antimicrobial compounds that kill the pathogen and thereby maintain homeostasis [1-4]. Numerous proteins and signaling pathways are engaged in this precisely controlled multifaceted process [5]. This intricacy of plant-pathogen interactions makes it very difficult to determine which anatomical features, metabolites, and signaling pathways are triggered: conventional biochemical and genetic experimental methods are insufficient tools for the task [3]. Currently, proteomics provides a comprehensive insight to understand the intricacy of plant-pathogen interactions. Proteome is defined as the total protein components of the cell that are specified by the genome at specified condition and study of the structure and function of all these proteins present in a cell, organ or organism at a particular time is known as the proteomics [6]. Proteomics aims to find out the identity and amount of each and every protein present in a cell and actual function mediating specific cellular processes [7]. Structural proteomics elucidates the development and application of experimental approaches to define the primary, secondary and tertiary structures of proteins, while functional proteomics refers to the development and application of global (proteome wide or system-wide) experimental approaches to assess protein function.
Why we need proteomics at all in the successful post-genomic era?
1) In multicellular organism, although the DNA in each type of cell is same, different sets of cells express different sets of genes. Protein component of cell varies from cell to cell even under different stress conditions. Therefore, by studying the proteome of individual cell, we can identify and analyze the proteins actually present therein.
2) Function of many genes identified by genome sequence remains a mystery [8].
3) Genome sequence tells us about the sequence of proteins, but there are many post translational modifications that are taking place in eukaryotic cells. Genomics fails to explain these modifications. Post translational modifications, the biological relevance of such modifications and transcript advances can only be interpreted through proteomics [9-12].
Modern techniques of proteomics allow us to study the protein content of cells which are differentially synthesized under diverse physiological and stress related conditions.
2. Different Techniques Used in Proteomics
2.1. Two Dimensional Polyacrylamide Gel Electrophoresis (2D-PAGE)
2D-PAGE exploits the two independent properties (Isoelectric point pI and molecular size) of proteins to separate particular protein from the mixture of proteins [13,14]. Isoelectric focusing separates the proteins according to the charge present on them in first direction. For second direction, sodium dodecyl sulfate (SDS) PAGE is used to separate proteins according to their size [15]. 2D-PAGE provides a good platform to analyze the mixture of proteins. Under favorable circumstances, upto 5000 protein spots can be identified on a large format of 2D-gel. The separated protein spots are visualized by staining the gel with different staining techniques; Coomassie blue staining, silver staining or SYPRO staining [7,9]. SYPRO is a collection of molecular probe for protein detection. It stains for luminescent detection of proteins separated by PAGE. Digital images of 2D-PAGE are captured by sensitive scanners [9] and analyzed using softwares such as Melanie, PDQuest, Phoretix, Progenesis, Z3 or Z4000 [16]. The spots of interest are excised either manually or robotically from the gel [7]. After digestion with sequence specific proteases, the identity of proteins are analysed by mass spectrometry [17,18]. Analysis of 2D-PAGE gels simultaneously provides several pieces of informations such as molecular weight, isoelectric point, quantity as well as post translational modifications (PTMs) [15,19]. Various PTMs can be profiled via 2D-PAGE. Some specific applications are a) phosphor-protein expression profiling, based on the shift of pIs of phosphorylated protein to a more acidic region on the gel; b) acetylated-protein expression profiling based on the principle that acetylation of proteins at their amino termini or on lysine side-chains will cause them to shift to a more acidic region on the gel and (c) glycosylated-protein expression profiling based on the fact that attachment of relatively simple glycans (O-glycosylation) or more complex glycans (N-glycosylation) can have varying effects on protein molecular weight, pI, and protein functions.
2.2. Fluorescent Two-Dimensional “Difference Gel Electrophoresis” (2D-DIGE)
DIGE proteomics uses 2D gel electrophoresis to analyze differential protein regulation between control and target samples. In this technique, the two samples to be compared are each treated with one of two different but structurally similar fluorescent dyes (cy2, cy3 and cy5 etc.). Each dye reacts with amino groups, so that each protein is fluorescently labeled by the dye binding to lysine residues and the N-terminal amino group. The two protein mixtures to be compared are then mixed together and run on a single 2D gel. Thus every protein in one sample superimposes with its differentially labeled identical counterpart in other sample. Scanning of the gel at two different wavelengths that excite two dye molecules reveal whether any individual spot is associated with only one dye molecule rather than two [20]. The resulting images are then analysed by softwares which are specifically designed for 2D-DIGE analysis [21]. Advantages of DIGE is that it exhibits higher sensitivity as well as linearity, accuracy of quantification and enhanced reproducibility by directly comparing hundreds of protein samples under similar electrophoresis conditions [22, 23]. It allows rapid identification of protein changes between two samples on the same 2D gel without influence of gel to gel variation. Using DIGE technique, it is possible to compare hundred of proteins in a single experiment under quantitative and reproducible conditions. Since DIGE technique allows running an internal strand as part of each separation, more subtle abundance changes can be demonstrated with statistical confidence using the pooled internal standard methodology, thereby detecting true difference in protein expressions. Biomarker development involves reproducible, quantitative, highly sensitive and high-throughput experimental techniques, and 2D-DIGE meets these criteria, so it has also a great prospect in cancer proteomics toward personalized medicine.
Gel-based techniques, although very widely used, have several drawbacks including insensitivity to low copy proteins, poor reproducibility and inability to characterize the entire protein content. Because of the complexity of any given proteome and separation limits of 2D PAGE techniques, only fraction of the proteome can be analyzed [17]. An alternative approach is the gel-free proteomics technology, which is more suitable for the analysis of proteins with low abundance in complex samples. [7,17,24]. Separation of proteins using gel-free techniques revolutionized the study of proteome in the cell [24,25]. Several gel-free proteomic techniques are discussed below.
2.2.1. ICAT (Isotop-Coded Affinity Tags)
It is a gel-free method for quantitative proteomics that relies on chemical labeling reagents to identify and quantify most of the proteins present in two cell populations [7,10]. The isotope used in ICAT is C13. Two populations are tagged with two different ICAT reagents (one with light and another with heavy), combined and enzymatically cleaved to generate peptide fragments, some of which are tagged. The tagged peptides are isolated by avidin affinity chromatography and subsequent analysis is done by microcapillary liquid chromatography-electron spray tandem mass spectrometry [7,10]. Combinations of the results are generated by MS and subsequent analysis of ICAT-labeled peptides determines the relative quantities of the components of protein mixtures in a single automated operation [10]. Gygi et al., demonstrated that they could follow the differential expression of more than 1400 different proteins in yeast [10]. This technique gives a quantitative as well as qualitative measure of changes in protein levels induced by different stress conditions. ICAT has been extensively used to find proteins associated with centrosomes (protein structures) that accumulate in abnormal ways in cancer cells and tumors. These proteins have the possibility to be used as diagnostic biomarkers and therapeutic targets (26).
2.2.2. ITRAQ (Isobarric Tagged for Relative and Absolute Quantitation)
ITRAQ is a non-gel based technique used to quantify protein from different sources in a single experiment [27-30]. Similar to ICAT, this technique also uses isotope coded covalent tags. The difference lies in the labeling of proteins. This method is based on covalent labeling of N-terminus and side chain amine of peptides from protein digestion with isotopic tag of varying mass. The peptides from the mixture are separated by nano chromatography. The separated peptides are then analyzed by tandem mass spectrometry (MS/MS). A database search is then performed using the fragmentation data to identify the labeled peptides and hence the corresponding proteins. The fragmentations of attached tag generate a low molecular mass reporter ion that can be used to relatively quantify the peptides and the proteins from which they originate using softwares (eg, i-tracker). The advantage of iTRAQ is its ability to multiplex several samples, quantification, simplified analysis and increased analytical precision and accuracy. It has the potential to comparing upto four different samples in one MS-based experiment.
2.2.3. MudPIT (Multidimensional Protein Identification Technology)
MudPIT is used especially for the analysis of complex protein mixtures [31]. In this technique, protein samples are subjected to sequence-specific enzymatic digestion with trypsin and endoproteinase lysc, and the resultant peptide mixtures are separated by strong cation exchange ( SCX ) and reverse phase ( RP ) high performance liquid chromatography (HPLC ) [31,32]. Peptides from the RP column enter the mass spectrometer and MS data is used to search the protein databases [32]. The advantage of this technique is that it is fast and sensitive with good reproducibility and generates an exhaustive list of proteins present in a particular protein sample [18,32]. As MudPIT combines both SCX and RP-HPLC separation, it is successfully used in a broad range of proteomics experiments, including large-scale cataloging of proteins in cells and organisms, profiling of membrane and organelle proteins, identification of protein complexes, determination of PTMs and quantitative analysis of protein expression[33-35].
3. Mass Spectrometry
Application of mass spectrometry to analyze amino acids and peptides was first reported in 1958 by Carl - Ove Anderson [36]. Mass spectrometry (MS) is an analytic technique which plays central role in the field of proteomics [37,38]. In MS, test samples are converted into gaseous ions that are separated in mass spectrometer according to their mass to charge ratio (m/z) and detected. In case of protein samples, proteins or peptides are fragmented by using the trypsin. The fragmented proteins are then separated using the liquid chromatography. The samples are then ionized and converted into the gaseous phase. Two soft ionization techniques, namely, Matrix-assisted laser desorption/ionization (MALDI) and Electron spray ionization (ESI) are used for the ionization. The invention of the time of flight mass spectrometry (TOF-MS) and relatively nondestructive methods to convert proteins into volatile ions made the revolution in the mass spectrometry [36]. Gaseous ions are separated in the mass analyzer. Peptides having a specific mass are fragmented using collision-induced dissociation and sent through a second mass spectrometry which generates a set of fragment peaks from which the amino acid sequence of the peptide are inferred. Protein identification algorithms are used to compare the results obtained with the known standards. Such algorithms fall into two categories; database search algorithms and de novo search algorithms. Some of the examples of the algorithms are listed in the Table 1. Mass spectrometry has a wide range of application in biotechnological development, pharmaceutical discovery, clinical testing as well as environmental analysis. Some of the common applications for MS are listed in the Table 2.

Table 1. List of the protein identification algorithms and data analysis softwares.
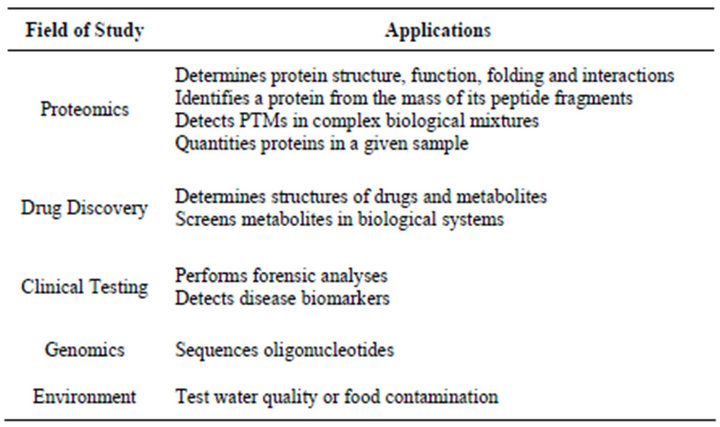
Table 2. Applications and fields that uses mass spectrometry.
4. Protein Microarray
Protein microarray is used for profiling complex samples in large quantities at a time and provides platform for both the classical and functional proteome analysis [56-60]. The first antibody microarray used for protein-protein interaction and post translational modification analysis in mammalian cells was reported in 2004 by Ivanov et al. [61]. Antibody array hold potential promise for the high-throughput profiling of a smaller number of proteins. In protein microarray antibodies or other affinity reagents such as short peptides, aptamers, polysaccharides, allergens or synthetic small molecules are spotted onto a surface such as glass, plastic or silicon chip; a complex mixture of cell lysate is passed over the surface to allow the antigen present to bind to their cognate antibodies. The screening of bound antigen is done by using fluorescently tagged or radioactively labeled proteins or by using secondary antibodies against each antigen of interest. Three types of protein microarray are currently used to study activity of protein. Analytical microarray is used to study differential expression profile, functional microarray is used for the study of protein-protein, protein-DNA, protein-RNA interaction and reverse phase protein microarray is used for determination of presence of altered protein that may be the result of disease.
5. Plant-Pathogen Interaction
To gain an in depth understanding of plant-pathogen interactions, numerous studies have already been carried out from the plant as well as from the pathogen perspectives, which reveals that plant-pathogen interaction results from precise communication between the plant and the invading pathogen [1,3,60,62]. Plants serve as host for vast numbers of pathogens such as bacteria, fungi, nematodes, oomycetes, viruses, and insects [63]. In compatible interactions, plants are incapable of mounting effective anti-infectious defense responses, allowing the pathogens to complete their life cycle [3,63]. During incompatible interactions, plants trigger a series of complex defense responses against pathogenic interactions to forestall pathogen growth [3,60]. In general, two types of defenses are induced in plants during interaction with pathogens; local and systemic defense responses [3,60, 64]. Local response begins with recognition, when plant sense pathogens or pathogen isolates via pathogen associated molecular pattern (PAMPs) located on the cell surface (e.g. pattern recognition receptors) of plant [5]. This type of recognition is mediated by the plant resistance proteins and results in much more reliable, high impact defense responses, leading to rapid ion fluxes across the plasma membrane, cell wall reinforcement, MAP kinase activation, active production of reactive oxygen species (ROS) and culminating in programmed cell death (PCD) known as the hypersensitive response [5,60,64,65]. A systematic or long-term response not only restrict pathogen infection locally, but also induce signals that enhance defense responses to pathogens in distal systemic tissues, a phenomenon known as systemic acquired resistance (SAR). SAR plays crucial role in developing resistance to disease as well as to recover from the disease damage. Activation of SAR require endogenous salicylic acid (SA) and elevates the level of ethylene, jasmonic acid (JA), nitric oxide (NO), and pathogenesis related (PR) proteins, which in-turn activates many downstream processes [3,66-70]. The signaling pathways triggered by different stressors (e.g., pathogen attack and herbivores/wounding) have been widely documented [71]. Disentangling such complex defense systems require the investigation of plant reactions to different stressors in comparable conditions.
6. Use of Proteomics to Study Plant-Pathogen Interactions
Proteomics of plant-pathogen interactions started almost two decades ago when electrophoretic and protein identification techniques were not as high-tech as of today. Differential protein profiling of seedlings of Pinus lambertiana (sugar-pine), that were susceptible or resistant to the white pine blister rust fungus Cronartium ribicola, was carried out by Ekramoddoullah and Hunt in 1993 using 2D PAGE [72]. Foliage proteins were extracted and separated by 2D gel electrophoresis from three and nine days old pine seedlings after inoculating them with the C. ribicola. Two acidic proteins were detected in relatively large amounts, of which the bigger one (36.7 kDa) was significantly suppressed in susceptible seedlings at day three while the smaller one (28.1 kDa) was significantly enhanced in resistant seedlings at day nine. The differential expression of these two proteins accounts for the nature of the outcome of the infection process in the susceptible and resistant seedlings. Additionally, an increase of few proteins involved in protein biosynthesis and a decrease of others involved in the same process was observed in resistant seedlings whereas proteins involved in this vital cellular process were decreased only in susceptible seedlings [72].
Medicago truncatula is considered as the model plant for proteomic studies due to its small genome size (454- 526 Mbp). Numerous papers are published throughout the globe on proteomic study of M. trancatula [73-81]. Mathesius et al. [82] used a proteomic approach to detect the M trancatula response to the N-acyl homoserine lactone (AHL) signals from pathogenic Pseudomonas aeruginosa. Compared to the AHL from nitrogen fixing bacteria Sinorhizobium meliloti, they established that one third of the responsive elements are different. Using proteomics it could be confirmed that plant can distinguish between concentrations and time of exposure of AHLs from closely similar structures. Mathesius et al. has reported that in response to AHL, plant produces AHL “signal mimic” substances which manipulate the bacterial behavior controlled by quorum sensing.
Erwinia chrysanthemi is the causative agent of soft rot disease in ornamental plants and vegetables [83]. The proteome analysis of the secretome (extracellular proteins) of uninduced E. chrysanthemi using 2D-PAGE and MS showed that there are 55 spots on the 2D-PAGE gel from which 25 were uniquely present. Cellulase (Cel5), protease (PrtA, B, C), flagelin (Fli C) and some intracellular proteins were the major proteins found in uniduced condition. On the other hand, 14 unique proteins were successfully identified from the secretome of culture induced by a heat soluble extract prepared from Chrysanthemum leaves. Majority of these proteins are pectin lyases, pectin acetylsterase, pectin methylsterase and polygalacturonase. From the analysis of mutants, authors showed that the type-I system of secretome is playing key role in secretion of proteases and type-II system is responsible for the secretion of the esterase and of the Avr-like protein AvrL. AvrL is homologous to the avirulence protein reported in Xanthomonas campestries which is the causative agent of black rot disease in crucifers [84]. Watt et al., [85] did the proteome analysis of the X. campestries pv campestries using 2D PAGE and MALDI-TOF-MS. They found 97 distinct protein spots on coomassie brilliant blue stained gels, which were excised and tryptic digested fragments were analysed by MALDI-TOF-MS and 68 different proteins were identified. By Signal P software, authors concluded that only 53% of proteins have putative secretion signals which are secreted in sec-dependent pathway. Results showed that many of these extracellular proteins are involved in degradative activities and important factors allowing for the infection of susceptible plant hosts [85].
Zhang et al., [86] analyzed the accumulation of PR proteins induced by compatible and incompatible interactions of pepper mild mottle virus (PMMoV) in Capsicum chinense L3 plants using proteomics. Spanish strain (PMMoV-S) results in incompatible interaction while the Italian strain (PMMoV-I) results in compatible interaction, although both viruses are nearly identical at their nucleotide sequence level (98%). PMMoV-S induces a HR in C. chinense plants with the formation of necrotic local lesions and restriction of the virus at the primary infection sites [86]. 2D-PAGE and image analysis with PDQuest software detected differential accumulation of C. chinense PR proteins, of which isoforms belonging to the PR-1, β-1, 3-glucanases (PR-2), chitinases (PR-3), osmotin-like protein (PR-5), peroxidases (PR-9), germin-like protein (PR-16), and PRp27 (PR-17) were identified using MALDI-TOF and MS/MS and subsequent database search [68]. In HR condition, an earlier and higher accumulation of three of these isoforms, namely a basic PR-1, PR2 and PR5 were specifically induced during PMMoV-S-activation of C. chinense L3 plants. However, the accumulation rates of PR proteins did not correlated with maximal accumulation levels of viral RNA, indicating that PR protein expression reflects the physiological status of the plant [86].
Fagus sylvatica is a broad leaf species in Central and Eastern Europe and is highly susceptible to Phytophthora cambivora, P cinnamoni, P. citricola, P kernoviae, P gonapodyides, P. ramorum and P. cactorum. Out of these, P. citricola is particularly aggressive and the morphological and physiological consequences of F. sylvatica root infection with this pathogen have been addressed in several studies. The proteome analysis of the plant pathogen interaction between F. sylvatica and P. citrocola was studied by Valcu et al. in 2009. A total of 188 spots were identified in 2D-PAGE, out of which 142 were from leaf and 46 were from root. The synthesis of the proteins was changed following root infection with P. citricola or wounding. Of them, 129 proteins spots were regulated by pathogen attack, 37 spots specifically responded to wounding and 22 were common to both stresses. Protein spots were analysed by tandem MS and LC-MS/MS and was found that some proteins were up-regulated in infection and down regulated in wounding. The differentially expressed proteins were mostly translation elongation factors, protein kinases and mRNA binding translocases responsible for regulating general metabolism as well as synthesis of defense related proteins. Changes in protein abundance following root infection suggested a rather weak and inefficient defense against the pathogen [87]. Adjustments of host energy and primary metabolism consisted mainly in decreased carbon fixation accompanied by a reallocation of nutrients. Defense reactions against the pathogen were mobilized late and consisted of rather unspecific mechanisms involving phenylpropanoid and flavonoid synthesis pathways and protection from oxidative stress. The observed patterns of protein regulation suggested either a lack of recognition or a suppression of host’s defense reaction by the invading pathogen, mimicking a sink organ and consequently manipulating host’s metabolism to support its own growth [87].
Opium poppy (Papaver somniferum) is the source of pharmacologically important compounds. Many of these compounds are the result of defence response of poppy plants. Zulak et al., [88] used cell culture of opium poppy to study proteome of plant in induced condition by fungal-derived-elicitors. Proteome analysis was performed for the 50h elicitor-treated opium poppy cell cultures by 2D-PAGE coupled with LC-MS. Out of 340 spots on gel, 219 proteins were identified [88]. Within these 219 protein spots, 129 were identified in the pre-existing plant proteome databases, 63 were identified by comparing with predicted translation products in opium poppy EST databases, and the remaining were common in both databases [88,89]. Of the several categories, 38.4% of identified proteins were involved in metabolic processes representing the core category of proteins including several glycolytic and secondary metabolic enzymes, nearly entire set of TCA cycle enzymes, and one alkaloid biosynthetic protein [88]. The other categories of proteins were heat shock proteins, protein degradation factors, proteins involved in secondary metabolic and pathogenesis-related proteins providing a comprehensive proteomics view on the coordination of plant defense responses [88]. Secondary metabolic enzymes included a catechol Omethyltransferase, phenylalanine ammonia lyase, an Nmethyltransferase, three proteins similar to dirigent (DIR) proteins and 6OMT which converts (S)-norcoclaurine to (S)-coclaurine in benzylisoquinoline alkaloid biosynthesis. Six alkaloid biosynthetic enzymes were identified using available polyclonal antibodies which were absent in non induced culture [88]. The identification of proteins reported in this study is an aid to characterize the induction of benzylisoquinoline alkaloid metabolism and other defense responses in elicitor-treated opium poppy cell cultures, a so called non-model but important medicinal plant.
Phaseolus vulgaris is a staple edible dry bean, having rust specific resistance to Uromyces appendiculatus. Lee et al., [89] studied the proteomics of P. vulgaris and U. appendiculatus interaction. Leaves were inoculated with liquid suspension of uredeospores of U. appendiculatus race 41 and 49. After 24 h and 72 h, the proteins were extracted and separated by differential centrifugation into three fractions; proteins from crude cell wall, organelle/membrane and soluble subcellular fraction. The fractions were analysed by LC-MS/MS. 1400 proteins were identified in each treatment and more than 3000 proteins were identified overall. The identified proteins obtained after 24hpi were categorized into defence (11%), energy (16%), metabolism (19%), primary destination and storage (14%), and protein synthesis (15%). At 72hpi the percentage was slightly less for defence (7%) and metabolism (14%) and slightly more for energy (22%) and protein destination and storage (17%). Lee et al., [89] assumed that similar type of early basal defense responses were functioning in both susceptible and resistant plants at 24h. Four proteins were identified with increased accumulation in both susceptible and resistant plants at 24 hpi including PR proteins, an inhibitor of bean rust differentiation and chitinase which is detrimental to chitin based cell walls of rust germlings. These results confirmed author’s assumptions that basal defences were triggered in susceptible and resistant plant at 24 h. Lipolytic enzymes with a GDSL motif were increased in amount which triggers downstream defence responses. In these experiment it was found that the MLO1-like membrane spanning protein was increased and previously MLO1 like membrane spanning proteins were reported as a negative regulator of defence and cell death [89,90]. In late basal defence responses glutathione-S-transferases, O-methyltransferase, Cyclases and isopentenyl pyrophosphate isomerases, flavanoids, phytoalexins and other proteins that probably mobilize cellular toxins were increased. Lee et al. found that susceptible beans have equivalent potential to generate a strong resistance response but failed when infected with U. appendiculatus race 41, which likely impairs the innate basal resistance. Another new observation from this work was the fluctuation of proteins with roles in regulating Fatty Acid signaling, which is a major consequence of rust infection and plays important role in rust resistance.
The analysis of inducible defence mechanism in insect resistant plant can be effectively studied in Arabidopsis thaliana and Plutella xylostella interaction [91]. Collins et al., [92] carried out proteomic analysis of A. thaliana genotypes exhibiting resistance or susceptibility to the insect herbivore, P. xylostella. The extracted proteins were separated using 2D-PAGE and gel comparisons were performed using PDQuest. Protein identification was carried out using MASCOT search engine. It was found that 50 spots were abundant in the resistant recombinant inbreed lines (RILs), out of which 19 were identified and 17 spots were abundant in susceptible RILs from which 10 were identified. In resistant RILs, several proteins were unregulated, some of which have antioxidant role and were previously identified [93,94]. It was found that the P. xylostella larvae were lower in plants having increased levels of reactive oxygen species (ROS). ROS has direct toxicity on the insect gut and upregulating defence genes [92]. Collins et al., concluded that significant differences exists in the proteomes between the identified resistant and susceptible RILs, and anticipated that enhanced production of ROS may be a major pre-existing mechanism of Plutella resistance in Arabidopsis Phytophthora sojae, a facultative pathogen of soybean, causes soybean root and stem rot, and results in an annual loss of 1 - 2 billion US dollars in soybean production worldwide. Zhang et al., [95] carried out proteomic study to determine the effects of P. sojae infection on soybean hypocotyls. Using 2D-PAGE and MALDI-TOF/ TOF, Zhang et al., identified 46 differentially expressed proteins. The expression levels of 26 proteins were significantly affected at various time points in the tolerant soybean line “Yudou25” (12 up-regulated and 14 downregulated). In contrast, in the sensitive soybean line “NG6255”, only 20 proteins were significantly affected (11 up-regulated and 9 down-regulated). Among these proteins, 26% were related to energy regulation, 15% to protein destination and storage, 11% to defense against disease, 11% to metabolism, 9% to protein synthesis, 4% to secondary metabolism, and 24% were of unknown function [95]. The result from this study offers an insight into the repertoire of mechanisms used by P. sojae during its infection and colonization into a host.
Non-host resistance (NHR), a rarely studied defence phenomenon, is very complex due to the fact that activation of any specific defense component may not be sufficient to render a plant resistance reaction. Proteomics studies play a key role in delineating the molecular mechanism of non-host resistance. Li et al., [96] studied the NHR of Oryza sativa cv. Nipponbare against the wheat leaf rust fungus, Puccinia triticina f. sp. tritici (Ptt). Proteins were extracted from leaves 3 dpi and analyzed by 2D-PAGE. A total of 33 protein spots were up-regulated and 9 were down-regulated by infection compared to the control, of which 30 were identified by MALDI-TOF-MS. The identified proteins were involved in defense/stress responses, energy/carbohydrate metabolism, oxidation-reduction processes, protein folding/turnover/cleavage/degradation, signal transduction and cell death regulation [96]. Proteomic analysis indicated that NHR of rice to Ptt is directly correlated with protein and energy metabolism, increased antimicrobial activities, possibly including phytoalexin accumulation and cell wall reinforcement, increased cell repair, antioxidative and detoxification reactions, and enhanced prevention of plant cell death [96]. Almost half of the up-regulated identified proteins were associated with chloroplast and mitochondrial physiology which suggests important roles for these organelles during NHR [96].
Till date, hundreds of proteomic experiments have been successfully carried out in the area of molecular plantmicrobe interaction, focusing mainly on the mechanisms controlling plant disease resistance, crosstalk among the signaling pathways involved, and the strategies used by the pathogens to suppress the defense. However, as space is limited, it is not possible to discuss all these interactions; Table 3 gives a summary of the important experiments involving plant-pathogen interaction using proteomics.
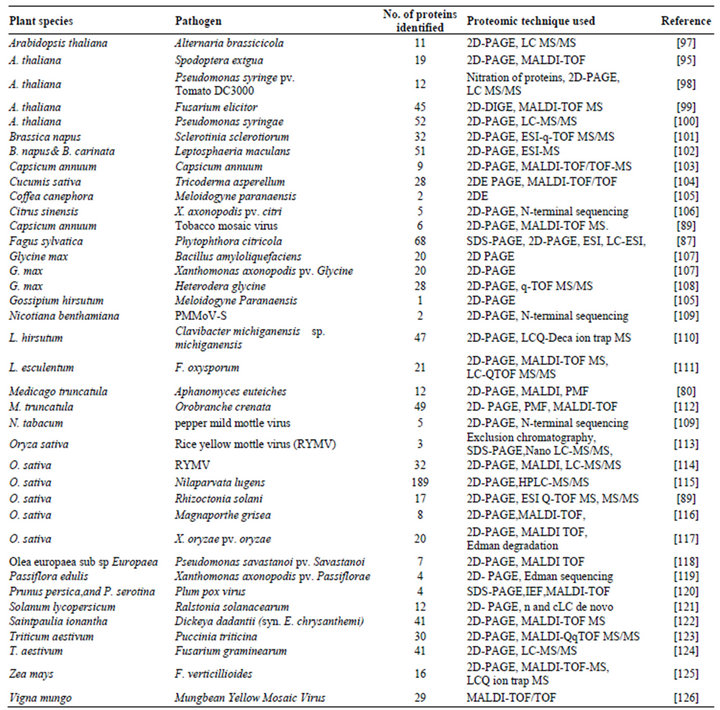
Table 3. A summary of important defense-related experiments using proteomics.
7. Future Perspectives
With the experiments of plant-pathogen interaction completed to date, we can truly consider proteomics as a mature platform for proteome analysis during plant-pathogen interaction. Proteome wide functional classification using bioinformatics approaches is becoming an important method for revealing unknown protein functions. Proteomics enables scientists to decipher the functions of proteins and get a clearer picture of the complex regulatory networks that control fundamental biological processes [127]. To meet the current challenges of food insecurity, proteins that control crop architecture and/or stress resistance in a wide range of environments need to be identified to facilitate the biological improvement of crop productivity. To identify such proteins, state-ofthe-art proteomic techniques are the best choice of scientists. However, certain disadvantages are limiting the use of proteomics. Proteins are dynamic and interacting molecules, and their changeability can make proteomic snapshots difficult. Furthermore, very closely related proteins may not guarantee a functional relationship [128]. Accordingly, there is the need for more sensitive analytical systems and effective methods for large scale data comparison in order to advance our knowledge on protein expression during plant-pathogen interactions. Nevertheless, proteomic tools are rapidly improving and novel methods and equipments are being developed. We believe that future proteomic studies, coupled with functional validation as well as bioinformatical analysis, may provide new insights into plant disease resistance and pathogenicity. Proteomics will remain to be one of the fastest growing areas in research and a symbiotic relationship between proteomics technology and systems biology approaches in the future will enable new questions to be addressed in the area of plant–pathogen interaction.
8. Acknowledgements
TDL and NT are thankful to the Department of Biotechnology, Govt. of India, for providing financial assistance. PH is thankful to University Grant Commission, Govt. of India, for providing financial assistance and JB is thankful to Visva-Bharati for necessary facilities.
REFERENCES
- P. N. Dodds and J. P. Rathjen, “Plant Immunity: Towards an Integrated View of Plant-Pathogen Interactions,” Nature Reviews Genetics, Vol. 11, No. 8, 2010, pp. 539-548. doi:10.1038/nrg2812
- P. J. De Wit, “How Plants Recognize Pathogens and Defend Themselves,” Cellular and Molecular Life Sciences, Vol. 64, No. 21, 2007, pp. 2726-2732. doi:10.1007/s00018-007-7284-7
- T. D. Lodha and J. Basak, “Plant-Pathogen Interaction: What Microarray Tells about It?” Molecular Biotechnoogy, Vol. 50, No. 1, 2012, pp. 87-97.
- J. L. Dangl and J. D. G. Jones, “Plant Pathogens and Integrated Defense Responses to Infection,” Nature, Vol. 411, No. 6839, 2001, pp. 826-833. doi:10.1038/35081161
- C. Zipfel, “Pattern-Recognition Receptors in Plant Innate Immunity,” Current Opinion in Immunology, Vol. 20, No. 1, 2008, pp. 10-16. doi:10.1016/j.coi.2007.11.003
- M. R. Wilkins, J. C. Scnchez, A. A. Gooley, R. D. Appel, I. H. Smith, D. F. Hochstrasser and K. L. Williams, “Progress with Proteome Projects: Why All Proteins Expressed by a Genome Should Be Identified and How to Do It,” Biotechnology & Genetic Engineering Reviews, Vol. 13, No. 1, 1995, pp. 19-50. doi:10.1080/02648725.1996.10647923
- N. V. K. Nat, S. Srivastava, W. Yajima and N. Sharma, “Application of Proteomics to Investigate Plant-Pathogen Interactions,” Current Proteomics, Vol. 4, No. 1, 2007, pp. 28-43. doi:10.2174/157016407781387357
- S. D. Patterson and R. H. Aebersold, “Proteomics: The First Decade and Beyond,” Nature Genetics, Vol. 33, No. 3, 2003, pp. 311-321. doi:10.1038/ng1106
- H. Duley and A. Grover, “Current Initiatives in Proteomics Research: The Plant Perspective,” Current Science, Vol. 80, No. 2, 2001, pp. 262-269.
- S. P. Gygi, B. Rist, S. A. Gerber, F. Turecek, M. H. Gelb and R. Aebersold, “Quantitative Analysis of Complex Protein Mixtures Using Isotope-Coded Affinity Tags,” Nature Biotechnology, Vol. 17, No. 10, 1999, pp. 994-999. doi:10.1038/13690
- O. K. Park, “Proteomic Study in Plants,” Journal of Biochemistry and Molecular Biology, Vol. 37, No. 1, 2004, pp. 133-138. doi:10.5483/BMBRep.2004.37.1.133
- G. Thurston, S. Regan, C. Rampitsch and T. Xing, “Proteomic and Phosphoproteomic Approaches to Understand Plant-Pathogen Interactions,” Physiological and Molecular Plant Pathology, Vol. 66, No. 1, 2005, pp. 3-11. doi:10.1016/j.pmpp.2005.03.004
- J. Klose, “Protein Mapping by Combined Isoelectric Focusing and Electrophoresis of Mouse Tissues. A Novel Approach to Testing for Induced Point Mutation in Mammals,” Human Genetics, Vol. 26, No. 3, 1975, pp. 231- 243.
- P. H. O’Farrell, “High Resolution Two Dimensional Electrophoresis of Proteins,” Journal of Biological Chemistry, Vol. 250, No. 10, 1975, pp. 4007-4021.
- A. Gorg, W. Weiss and M. J. Dunn, “Current Two-Dimensional Electrophoresis Technology for Proteomics,” Proteomics, Vol. 4, No. 12, 2004, pp. 3665-3685. doi:10.1002/pmic.200401031
- P. G. Righetti, A. Castagna, F. Antonucci, C. Piubelli, D. Cecconi, N. C. Campostrini, P. Antonioli, H. Astner, “Critical Survey of Quantitative Proteomics in Two-Dimensional Electrophoretic Approaches,” Journal of Chromatography A, Vol. 1051, No. 1, 2004, pp. 3-17. doi:10.1016/j.chroma.2004.05.106
- H. Zhu, M. Bilgin and M. Snyder, “Proteomics,” Annual Review of Biochemistry, Vol. 72, No. 1, 2003, pp. 783-812. doi:10.1146/annurev.biochem.72.121801.161511
- J. K. C. Rose, S. Bashir, J. J. Giovannoni, M. M. Jahn and R. S. Saravanan, “Tackling the Plant Proteome: Practical Approaches, Hurdles and Experimental Tools,” Plant Journal, Vol. 39, No. 5, 2004, pp. 715-733. doi:10.1111/j.1365-313X.2004.02182.x
- B. Wittmann-Liebold, H. R. Graack and T. Pohl, “TwoDimensional Gel Electrophoresis as Tool for Proteomics Studies in Combination with Protein Identification by Mass Spectrometry,” Proteomics, Vol. 6, No. 17, 2006, pp. 4688-4703. doi:10.1002/pmic.200500874
- M. Unlu, M. E. Morgan and J. S. Minden, “Difference Gel Electrophoresis: A Single Gel Method for Detecting Changes in Protein Extracts,” Electrophoresis, Vol. 18, No. 11, 1997, pp. 2071-2077. doi:10.1002/elps.1150181133
- R. Marouga, S. David and E. Hawkins, “The Development of the DIGE System: 2D Fluorescence Difference Gel Analysis Technology,” Analytical and Bioanalytical Chemistry, Vol. 382, No. 3, 2005, pp. 669-678. doi:10.1007/s00216-005-3126-3
- G. Van den Bergh and L. Arckens, “Recent Advances in 2D Electrophoresis: An Array of Possibilities,” Expert Review of Proteomics, Vol. 2, No. 2, 2005, pp. 243-252. doi:10.1586/14789450.2.2.243
- G. Zhou, H. Li and D. Camp De, “2D Differential In-Gel Electrophoresis for the Identification of Esophageal Scans Cell Cancer-Specific Protein Markers,” Molecular & Cellular Proteomics, Vol. 1, No. 2, 2005, pp. 117-124. doi:10.1074/mcp.M100015-MCP200
- J. P. Lambert, M. Ethier, J. C. Smith and D. Figeys, “Proteomics from Gel Based to Gel Free,” Analytical Chemistry, Vol. 77, No. 12, 2005, pp. 3771-3787. doi:10.1021/ac050586d
- G. Baggerman, E. Vierstraete, A. De Loof and L. Schoofs, “Gel Based Versusgel-Free Proteomics: A Review,” Combinatorial Chemistry & High Throughput Screening, Vol. 8, No. 8, 2005, pp. 669-677. doi:10.2174/138620705774962490
- M. R. Flory, A. R. Carson, E. G. Muller and R. Aebersold, “An SMC-Domain Protein in Fission Yeast Links Telomeres to the Meiotic Centrosome,” Molecular Cell, Vol. 16, No. 4, 2004, pp. 619-630. doi:10.1016/j.molcel.2004.10.027
- P. L. Ross, Y. N. Huang, J. N. Marchese, B. Williamson, K. Parker, S. Hattan, N. Khainovski and S. Pillai, “Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-Reactive Isobaric Tagging Reagents,” Molecular & Cellular Proteomics, Vol. 3, No. 12, 2004, pp. 1154-1169. doi:10.1074/mcp.M400129-MCP200
- K. Agarwal, L. H. Choe and K. H. Lee, “Shotgun Proteomics Using the iTRAQ Isobaric Tags,” Briefings in Functional Genomics and Proteomics, Vol. 5, No. 2, 2006, pp. 112-120. doi:10.1093/bfgp/ell018
- L. R. Zieske, “A Perspective on the Use of iTRAQ Reagent Technology for Protein Complex and Profiling Studies,” Journal of Experimental Botany, Vol. 57, No. 7, 2006, pp. 1501-1508. doi:10.1093/jxb/erj168
- T. C. Lund, L. B. Anderson, V. McCullar, L. Higgins, G. H. Yun, B. Grzywacz, M. R. Verneris and J. S. Miller, “iTRAQ Is a Useful Method to Screen for MembraneBound Proteins Differentially Expressed in Human Natural Killer Cell Types,” Journal of Proteome Research, Vol. 6, No. 2, 2007, pp. 644-653. doi:10.1021/pr0603912
- H. J. Issaq, K. C. Chan, G. M. Janini, T. P. Conrads and T. D. Veenstra, “Multidimensional Separation of Peptides for Effective Proteomic Analysis,” Journal of Chromatography B Analytical Technologies in the Biomedical and Life Sciences, Vol. 817, No. 1, 2005, pp. 35-47. doi:10.1016/j.jchromb.2004.07.042
- M. P. Washburn, D. Wolters and J. R. Yates, “LargeScale Analysis of the Yeast Proteome by Multidimensional Protein Identification Technology,” Nature Biotechnology, Vol. 19, No. 3, 2001, pp. 242-247. doi:10.1038/85686
- I. I. I. Yates, J. R. A. Gilchrist, K. E. Howell and J. J. Bergeron, “Proteomics of Organelles and Large Cellular Structures,” Nature Reviews, Vol. 6, No. 9, 2005, pp. 702-714. doi:10.1038/nrm1711
- A. E. Speers and C. C. Wu, “Proteomics of Integral Membrane Proteins-Theory and Application,” Chemical Reviews, Vol. 107, No. 8, 2007, pp. 3687-3714. doi:10.1021/cr068286z
- G. T. Cantin, J. D. Venable, D. Cociorva and J. R. Yates III, “Quantitative Phosphoproteomic Analysis of the Tumor Necrosis Factor Pathway,” Journal of Proteome Research, Vol. 5, No. 1, 2006, pp. 127-134. doi:10.1021/pr050270m
- C .O. Anderson, “Mass Spectrometric Studies on Amino Acid and Peptide Derivatives,” Acta Chemica Scandinavica, Vol. 12, No. 0353, 1958, p. 1353.
- W. Zhu, J. W. Smith and C. M. Huang, “Mass Spectrometry-Based Label-Free Quantitative Proteomics,” Journal of Biomedicine and Biotechnology, Vol. 2010, 2009, Article ID: 840581. doi:10.1155/2010/840518
- M. Zhu, B. Simons, N. Zhu, D. G. Oppenheimer and S. Chen, “Analysis of Abscisic Acid Responsive Proteins in Brassica Napus Guard Cells by Multiplexed Isobaric Tagging,” Journal of Proteomics, Vol. 73, No. 4, 2010, pp. 790-805. doi:10.1016/j.jprot.2009.11.002
- J. K. Eng, L. A. McCormack and J. R. Yates, “An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database,” Journal of the American Society for Mass Spectrometry, Vol. 5, No. 11, 1994, pp. 976-989. doi:10.1016/1044-0305(94)80016-2
- D. N. Perkins, D. J. C. Pappin, D. M. Creasy and J. S. Cottrell, “Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data,” Electrophoresis, Vol. 20, No. 18, 1999, pp. 3551- 3567. doi:10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2
- C. Liang, J. C. Smith and C. Hendrie, “A Comparative Study of Peptide Sequencing Software Tools for MS/ MS,” Proceedings of the 51st ASMS Conference on Mass Spectrometry and Allied Topics, Montreal, 8-13 June 2003.
- C. Jacques, A. Masselot, M. Giron, T. Dessingy and J. Magnin, “OLAV: Towards High-Throughput Tandem Mass Spectrometry Data Identification,” Proteomics, Vol. 3, No. 8, 2003, pp. 1454-1463. doi:10.1002/pmic.200300485
- Y. G. Lewis, S. P. Markey, J. A. Kowalak, L. Wagner, M. Xu, D. M. Maynard, X. Yang and W. Shi, “Open Mass Spectrometry Search Algorithm,” Journal of Proteome Research, Vol. 3, No. 5, 2004, pp. 958-964. doi:10.1021/pr0499491
- L. T. David, C. G. Fernando and M. C. Chambers, “MyriMatch: Highly Accurate Tandem Mass Spectral Peptide Identification by Multivariate Hypergeometric Analysis,” Journal of Proteome Research, Vol. 6, No. 2, 2007, pp. 654-661. doi:10.1021/pr0604054
- B. Marshall, Y. Cai and D. Goldberg, “Lookup Peaks: A Hybrid of de Novo Sequencing and Database Search for Protein Identification by Tandem Mass Spectrometry,” Analytical Chemistry, Vol. 79, No. 4, 2007, pp. 1393- 1400. doi:10.1021/ac0617013
- L. Jian, A. Erassov, P. Halina, M. Canete, N. D. Vo, C. Chung, G. Cagney and A. Ignatchenko, “Sequential Interval Motif Search: Unrestricted Database Surveys of Global MS/MS Data Sets for Detection of Putative Post-Translational Modifications,” Analytical Chemistry, Vol. 80, No. 20, 2008, pp. 7846-7854. doi:10.1021/ac8009017
- A. K. Yadav, D. Kumar and D. Dash, “MassWiz: A Novel Scoring Algorithm with Target-Decoy Based Analysis Pipeline for Tandem Mass Spectrometry,” Journal of Proteome Research, Vol. 10, No. 5, 2011, pp. 2154-2160. doi:10.1021/pr200031z
- M. S. Mikhail, M. L. Nielsen, F. Kjeldsen and R. A. Zubarev, “Proteomics-Grade de Novo Sequencing Approach,” Journal of Proteome Research, Vol. 4, No. 6, 2005, pp. 2348-2354. doi:10.1021/pr050288x
- M. Bin, K. Zhang, C. Hendrie, C. Liang, M. Li, A. Doherty-Kirby and G. Lajoie, “PEAKS: Powerful Software for Peptidede Novo Sequencing by Tandem Mass Spectrometry,” Rapid Communication in Mass Spectrometry, Vol. 17, No. 20, 2003, pp. 2337-2342. doi:10.1002/rcm.1196
- R. Winkler, “ESIprot: A Universal Tool for Charge State Determination and Molecular Weight Calculation of Proteins from Electrospray Ionization Mass Spectrometry Data,” Rapid Communication in Mass Spectrometry, Vol. 24, No. 3, 2010, pp. 285-294. doi:10.1002/rcm.4384
- M. E. Monroe, N. Tolic, N. Jaitly, J. L. Shaw, J. N. Adkins and R. D. Smith, “VIPER: An Advanced Software Package to Support High-Throughput LC-MS Peptide Identification,” Bioinformatics, Vol. 23, No. 15, 2007, pp. 2021-2023. doi:10.1093/bioinformatics/btm281
- D. B. Weatherly, J. A. Atood, T. A. Minning, C. Cavola, R. L. Tarleton and R. Orlando, “A Heuristic Method for Assigning a False-Discovery Rate for Protein Identifications from Mascot Database Search Results,” Molecular and Cellular Proteomics, Vol. 4, No. 6, 2005, pp. 762- 772. doi:10.1074/mcp.M400215-MCP200
- C. C. Paulo, J. S. G. Fischer, E. I. Chen, J. R. Yates and V. C. Barbosa, “PatternLab for Proteomics: A Tool for Differential Shotgun Proteomics,” BMC Bioinformatics, Vol. 9, 2008, p. 316. doi:10.1186/1471-2105-9-316
- M. P. Patricia, D. Walther, M. Quadroni, S. B. Catherinet, J. Burgess, C. G. Zimmermann-Ivol, J. C. Sanchez and P. A. Binz, “MSight: An Image Analysis Software for Liquid Chromatography-Mass Spectrometry,” Proteomics, Vol. 5, No. 9, 2005, pp. 2381-2384. doi:10.1002/pmic.200401244
- Z. Hans-Dieter, L. Jens., V. Khamenia, C. Schiller, A. Appel, H. Tammen, R. Crameri and H. Selle, “Datamining Methodology for LC-MALDI-MS Based Peptide Profiling,” Combinatorial Chemistry and High Throughput Screening, Vol. 8, No. 8, 2005, pp. 717-723. doi:10.2174/138620705774962481
- X. Duburcq, C. Olivier, F. Malingue, R. Desmet, A. Bouzidi, F. Zhou, C. Auriault and H. Gras-Masse, “Peptide-Protein Microarrays for the Simultaneous Detection of Pathogen Infections,” Bioconjugate Chemistry, Vol. 15, No. 2, 2004, pp. 307-316. doi:10.1021/bc034226d
- J. LaBaer and N. Ramachandran, “Protein Microarrays as Tools for Functionalproteomics,” Current Opinion in Chemical Biology, Vol. 9, No. 1, 2005, pp. 14-19. doi:10.1016/j.cbpa.2004.12.006
- G. S. Athwal, C. Lombardo, J. L. Huber, S. C. Masters, H. Fu and S. C. Huber, “Modulation of 14-3-3 Interactions with Target Proteins by Physical and Metabolic Effectors,” Plant and Cell Physiology, Vol. 41, No. 4, 2000, pp. 523-533. doi:10.1093/pcp/41.4.523
- N. Ramachandran, D. N. Larson, P. R. Stark, E. Hainsworth and J. LaBaer, “Emerging Tools for Real-Time Label-Free Detection of Interactions on Functional Protein Microarrays,” FEBS Journal, Vol. 272, No. 21, 2005, pp. 5412-5425. doi:10.1111/j.1742-4658.2005.04971.x
- [61] K. E. Hammond-Kosack and J. D. G. Jones, “Responses to Plant Pathogens,” In: B. B. Buchanan, W. Gruissem and R. L. Jones, Eds., Biochemistry and Molecular Biology of Plants, American Society of Plant Physiology, Rockville, 2000, pp. 1102-1156.
- [62] S. S. Ivanov., et al, “Antibodies Immobilized as Arrays to Profile Protein Post-Translational Modifications in Mammalian Cells,” Molecular and Cellular Proteomics, Vol. 3, No. 8, 2004, pp. 788-795. doi:10.1074/mcp.M300130-MCP200
- [63] J. A. Lucas, “Plant Pathology and Plant Pathogens,” Blackwell Science, Oxford, 1998.
- [64] P. M. Schenk, K. Kazan, L. Wilson, J. P. Anderson, T. Richmond and S. C. Somerville, “Coordinated Plant Defense Responses in Arabidopsis Revealed by Microarray Analysis,” Proceedings of National Academy of Science, Vol. 97, No. 21, 2000, pp. 11655-11660. doi:10.1073/pnas.97.21.11655
- [65] P. M. Schenk, J. H. Choo and C. L. Wong, “Microarray Analyses to Study Plant Defense and Rhizosphere Microbe Interaction,” CAB Review: Perspective in Agriculture, Veterinary, Science, Nutrition, and Natural Resources, Vol. 4, No. 045, 2009, pp. 1-14.
- [66] L. A. Mur, P. Kenton, A. J. Lloyd, H. Ougham and E. Prats, “The Hypersensitive Response; The Centenary Is upon Us but How Much Do We Know?” Journal of Experimental Botany, Vol. 59, No. 3, 2008, pp. 501-520. doi:10.1093/jxb/erm239
- [67] F. B. Andrew, W. Jinrong and D. F. Mark, “Probing Plant-Pathogen Interaction and Downstream Defense Signaling Using DNA Microarray,” Functional & Integrative Genomics, Vol. 2, No. 6, 2002, pp. 259-273. doi:10.1007/s10142-002-0080-4
- [68] M. A. Torres, D. G. Jonathan and J. L. Dangl, “Reactive Oxygen Species Signaling in Response to Pathogen,” Plant Physiology, Vol. 141, No. 2, 2006, pp. 373-378. doi:10.1104/pp.106.079467
- [69] G. E. Vallad and R. M. Goodman, “Systemic Acquired Resistance and Induced Systemic Resistance in Conventional Agriculture,” Crop Science, Vol. 44, No. 6, 2004, pp. 1920-1934. doi:10.2135/cropsci2004.1920
- [70] X. Dong, “Genetic Dissection of Systemic Acquired Resistance,” Current Opinion in Plant Biology, Vol. 4, No. 4, 2001, pp. 309-314. doi:10.1016/S1369-5266(00)00178-3
- [71] J. A. Kreps, Y. Wu, H. S. Chang, T. Zhu, X. Wang and J. F. Harper, “Transcriptome Changes for Arabidopsis in Response to Salt, Osmotic, and Cold Stress,” Plant Physiology, Vol. 130, No. 4, 2002, pp. 2129-2141. doi:10.1104/pp.008532
- [72] C. M. J. Pieterse and L. C. Van Loon, “NPR1: The Spider in the Web of Induced Resistance Signaling Pathways,” Current Opinion in Plant Biology, Vol. 7, No. 4, 2004, pp. 456-464. doi:10.1016/j.pbi.2004.05.006
- [73] A. K. M. Ekramoddoullah and R. S. Hunt, “Changes in Protein Profile of Susceptible and Resistant Sugar-Pine Foliage Infected with the Whitepine Blister Rust Fungus Cronartium ribicola,” Canadian Journal in Plant Pathology, Vol. 15, No. 4, 1993, pp. 259-264. doi:10.1080/07060669309501921
- [74] V. S. Asirvatham, B. S. Watson and L. W. Sumner, “Analytical and Biological Variances Associated with Proteomic Studies on Medicago Trancatula by Two Dimensional Polyacrylamide Gel Electrophoresis,” Proteomics, Vol. 2, No. 8, 2002, pp. 960-968. doi:10.1002/1615-9861(200208)2:8<960::AID-PROT960>3.0.CO;2-2
- [75] G. Bestel-Corre, E. Dumas-Gaudot, V. Poinsot, M. Dieu, J. F. Dierick, D. Van Tuinen, J. Remacle, V. Gianinazzipearson and S. Gianinazzi, “Proteome Analysis and Identification of Symbiosis-Related Proteins from Medicago truncatula Gaertn. By Two Dimensional Electrophoresis and Mass Spectrometry,” Electrophoresis, Vol. 23, No. 1, 2002, pp. 122-137. doi:10.1002/1522-2683(200201)23:1<122::AID-ELPS122>3.0.CO;2-4
- [76] G. Bestel-Corre, S. Gianinazzi and E. Dumass-Gaudot, “Impact of Sewage Sludge on Medicago truncatula Symbiotic Proteome,” Phytochemistry, Vol. 65, No. 11, 2004, pp. 1651-1659. doi:10.1016/j.phytochem.2004.04.037
- [77] E. Dumas-Gaudot, N. Amiour, S. Weidmann, G. BestelCorre, B. Valot, S. Lenogue, V. Gianninazzi-Pearson and S. Gianninazzi, “A Technical Trick for Studying Proteomics in Parallel to Transcriptomics in Symbiotic RootFungus Interaction,” Proteomics, Vol. 4, No. 2, 2004, pp. 451-453.doi:10.1002/pmic.200300627
- [78] K. Gallardo, C. Le Signor, J. Vandekerckhove, R. D. Thompson and J. Burstin, “Proteomics of Medicago truncaula Seed Development Establishes the Time Fram of Diverse Metabolic Processes Related to Reverse Accumulation,” Plant Physiology, Vol. 133, No. 2, 2003, pp. 664-682.
- [79] U. Mathesius, G. Keijzers, S. H. A. Natera, J. J. Weinman, M. A. Djordjeivic and B. G. Rolfe, “Establishment of a Root Proteome Reference Map for the Model Legume Medicago truncatula Using the Expressed Sequence Tag Database for Peptide Mass Fingerprinting,” Proteomics, Vol. 1, No. 11, 2001, pp. 1424-1440. doi:10.1002/1615-9861(200111)1:11<1424::AID-PROT1424>3.0.CO;2-J
- [80] B. S. Watson, Z. Lei, R. A. Dixon and L. W. Sumner, “Proteomics of Medicago sativa Cell Walls,” Phytochemistry, Vol. 65, No. 12, 2004, pp. 1709-1720. doi:10.1016/j.phytochem.2004.04.026
- [81] F. Colditz, O. Nyamsuren, K. Niehaus, H. Eubel, H. P. Braun and F. Krajinski, “Proteomic Approach: Identification of Medicago truncatula Proteins Induced in Roots after Infection with the Pathogenic Oomycete Aphanomyces euteiches,” Plant Molecular Biology, Vol. 55, No. 1, 2004, pp. 109-120. doi:10.1007/s11103-004-0499-1
- [82] M. A. Castillejo, N. Amiour, E. Dumas-Gaudot, D. Rubiales and J. V. Jorrın, “A Proteomic Approach to Studying Plant Response to Crenatebroomrape (Orobanche crenata) in Pea (Pisum sativum),” Phytochemistry, Vol. 65, No. 12, 2004, pp. 1817-1828. doi:10.1016/j.phytochem.2004.03.029
- [83] U. Mathesius, S. Mulders, M. S. Gao, M. Teplitski, G. Caetano-Anolles, B. G. Rolfe and W. D. Bauer, “Extensive and Specific Responses of a Eukaryote to Bacterial Quorum-Sensing and Signals,” Proceedings of National Academy of Science of the United States of America, Vol. 100, No. 3, 2003, pp. 1444-1449. doi:10.1073/pnas.262672599
- [84] F. Bouchart, A. Delangle, J. Lemoine, J. P. Bohin and J. M. Lacroix, “Proteomic Analysis of a Non-Virulent Mutant of the Phytopathogenic Bacterium Erwinia chrysanthemi Deficient in Osmoregulated Periplasmic Glucans: Change in Protein Expression is Not Restricted to the Envelope, but Affects General Metabolism,” Microbiology, Vol. 153, No. 3, 2007, pp. 760-767. doi:10.1099/mic.0.2006/000372-0
- [85] N. Kazemi-Pour, G. Condemine and N. HugouvieuxCotte-Pattat, “The Secretome of the Plant Pathogenic Bacterium Erwinia chrysanthemi,” Proteomics, Vol. 4, No. 10, 2004, pp. 3177-3186. doi:10.1002/pmic.200300814
- [86] S. A. Watt, A. Wilke, T. Patschkowski and K. Niehaus, “Comprehensive Analysis of the Extracellular Proteins from Xanthomonas campestris pv. campestris B100,” Proteomics, Vol. 5, No. 1, 2005, pp. 153-167. doi:10.1002/pmic.200400905
- [87] M. I. Elvira, M. M. Galdeano, P. Gilardi, I. Garcı´a-Luque, M. T. Serra, “Proteomic Analysis of PathogenesisRelated Proteins (PRs) Induced by Compatible and Incompatible Interactions of Pepper Mild Mottle Virus (PMMoV) in Capsicum chinense L3 Plants,” Journal of Experimental Botany, Vol. 59, No. 6, 2008, pp. 1253- 1265. doi:10.1093/jxb/ern032
- [88] C. M. Valcu, M. Junqueira, A. Shevchenko and K. Schlink, (2009) Comparative Proteomic Analysis of Responses to Pathogen Infection and Wounding in Fagus sylvatica,” Journal of Proteome Research, Vol. 8, No. 8, 2009, pp. 4077-4091. doi:10.1021/pr900456c
- [89] K. G. Zulak, F. K. Morgan, A. Joenel, C. S. David and J. F. Peter, “Plant Defense Responses in Opium Poppy Cell Cultures Revealed by Liquid Chromatography-Tandem Mass Spectrometry Proteomics,” Molecular & Cellular Proteomics, Vol. 8, No. 1, 2009, pp. 86-98. doi:10.1074/mcp.M800211-MCP200
- [90] J. Lee, J. Feng, K. B. Campbell, B. E. Scheffler, W. M. Garrett, S. Thibivilliers, G. Stacey, D. Q. Naiman, M. L. Tucker, M. A. Pastor-Corrales and B. Cooper , “Quantitative Proteomic Analysis of Bean Plants Infected by a Virulent and Avirulent Obligate Rust Fungus,” Molecular & Cellular Proteomics, Vol. 8, 2009, pp. 19-31. doi:10.1074/mcp.M800156-MCP200
- [91] P. Piffanelli, F. Zhou, C. Casais, J. Orme, B. Jarosch, U. Schaffrath, N. C. Collins, R. Panstruga and P. SchulzeLefert, “The Barley MLO Modulator of Defense and Cell Death Is Responsive to Biotic and Abiotic Stress Stimuli,” Plant Physiology, Vol. 129, No. 3, 2002, pp. 1076- 1085. doi:10.1104/pp.010954
- [92] D. Kliebenstein, D. Pedersen, B. Barker and T. MitchellOlds, “Comparative Analysis of Quantitative Trait Loci Controlling Glucosinolates, Myrosinase and Insect Resistance in Arabidopsis thaliana,” Genetics, Vol. 161, No. 1, 2002, pp. 325-332.
- [93] R. M. Collins, M. Afzal, D. A. Ward, M. C. Prescott, S. M. Sait, H. H. Rees and A. D. Tomsett, “Differential Proteomic Analysis of Arabidopsis thaliana Genotypes Exhibiting Resistance or Susceptibility to the Insect Herbivore, Plutella xylostella,” PLoS One, Vol. 5, No. 4, 2010, e10103. doi:10.1371/journal.pone.0010103
- [94] G. P. Biewenga, G. R. M. M. Haenen and A. Bast, “The Pharmacology of the Antioxidant Lipoic Acid,” General Pharmacology: The Vascular System, Vol. 29, No. 3, 1997, pp. 315-331.doi:10.1016/S0306-3623(96)00474-0
- [95] A. Koshkin, G. M. Knudsen and P. R. O. de Montellano, “Intermolecular Interactions in the AhpC/AhpD Antioxidant Defence System of Mycobacterium tuberculosis,” Archives of Biochemistry and Biophysics, Vol. 427, No. 1, 2004, pp. 41-47. doi:10.1016/j.abb.2004.04.017
- [96] J. H. Zhang, L. W. Sun, L. L. Liu, S. L. An, X. Wang, J. Zhang, J. L. Jin, S. Y. Li and J. H. Xi, “Proteomic Analysis of Interactions between the Generalist Herbivore Spodoptera exigua (Lepidoptera Noctuidae) and Arabidopsis thaliana,” Plant Molecular Biology Reporter, Vol. 28, No. 2, 2010, pp. 324-333. doi:10.1007/s11105-009-0156-6
- [97] H. Li, P. H. Goodwin, Q. Han, L. Huang and Z. Kang, “Microscopy and Proteomic Analysis of the Non-Host Resistance of Oryza sativa to the Wheat Leaf Rust Fungus, Puccinia triticina f. sp. tritici,” Plant Cell Reports, Vol. 31, No. 4, 2012. doi:10.1007/s00299-011-1181-0
- [98] A. K. Mukherjee, M. J. Carp, R. Zuchman, T. Ziv, B. A. Horwitz and S. Gepstein, “Proteomics of the Response of Arabidopsis thaliana to Infection with Alternaria brassicicola,” Journal of Proteomics, Vol. 73, No. 4, 2010, pp. 709-720. doi:10.1016/j.jprot.2009.10.005
- [99] D. Cecconi, S. Orzetti, E. Vandelle, S. Rinalducci, L. Zolla and M. Delledonne, “Protein Nitration during Defese Response in Arabidopsis thaliana,” Electrophoresis, Vol. 30, No. 14, 2009, pp. 2460-2468. doi:10.1002/elps.200800826
- [100] S. Chivasa, J. M. Hamilton, R. S. Pringle, B. K. Ndimba, W. J. Simon, K. Lindsey and A. R. Slabas, “Proteomic Analysis of Differentially Expressed Proteins in Fungal Elicitor-Treated Arabidopsis Cell Cultures,” Journal of Experimental Botany, Vol. 57, No. 7, 2006, pp. 1553- 1562. doi:10.1093/jxb/erj149
- [101] A. M. E. Jones, V. Thomas, M. H. Bennett, J. Mansfield, and M. Grant, “Modifications to the Arabidopsis Defence Proteome Occur Prior to Significant Transcriptional Change in Response to Inoculation with Pseudomonas syringae,” Plant Physiology, Vol. 142, No. 4, 2006, pp. 1603-1620. doi:10.1104/pp.106.086231
- [102] Y. Liang, S. Srivastava, M. H. Rahman, S. E. Strelkov, and N. N. Kav, “Proteome Changes in Leaves of Brassica napus L. as a Result of Sclerotinia sclerotiorum Challenge,” Journal of Agricultural and Food Chemistry, Vol. 56, No. 60, 2008, pp. 1963-1976. doi:10.1021/jf073012d
- [103] N. Sharma, N. Hotte, M. H. Rahman, M. Mohammadi, M. K. Deyholos and N. N. Kav, “Towards Identifying Brassica Proteins Involved in Mediating Resistance to Leptosphaeria maculans: A Proteomics-Based Approach,” Proteomics, Vol. 8, No. 17, 2008, pp. 3516-3535. doi:10.1002/pmic.200701141
- [104] A. Wongpia and K. Lomthaisong “Changes in the 2DE Protein Profiles of Chilli Pepper,” ScienceAsia, Vol. 36, 2010, pp. 259-270.
- [105] G. Segarra, E. Casanova, D. Bellido, M. A. Odena, E. Oliveira and I. Trillas, “Proteome, Salicylic Acid and Jasmonic Acid Changes in Cucumber Plants Inoculated with Trichoderma asperellum Strain T34,” Proteomics, Vol. 7, No. 21, 2007, pp. 3942-3952. doi:10.1002/pmic.200700173
- [106] O. L. Franco, J. L. Pereira, P. H. A. Costa, T. L. Rocha, E. V. S. Albuquerque, M. F. Grossi-de-Sa, R. M. D. G. Carneiro, R. G. Carneiro and A. Mehta, “Methodological Evaluation of 2DE to Study Root Proteomics during Nematode Infection in Cotton and Coffee Plants,” Preparative Biochemistry and Biotechnology, Vol. 40, No. 2, 2010, pp. 152-163. doi:10.1080/10826060903558976
- [107] A. Mehta and Y. B. Rosato, “Differentially Expressed Proteins in the Interaction of Xanthomonas axonopodis pv. citri with Leaf Extract of the Host Plant,” Proteomics, Vol. 1, No. 9, 2001, pp. 1111-1118. doi:10.1002/1615-9861(200109)1:9<1111::AID-PROT1111>3.0.CO;2-7
- [108] N. Buensanteai, G. Y. Yuen and S. Prathuangwong, “Priming, Signaling and Protein Production Associated with Induced Resistance by Bacillus amyloliquefaciens KPS46,” World Journal of Microbiology and Biotechnology, Vol. 25, No. 7, 2009, pp. 1275-1286. doi:10.1007/s11274-009-0014-6
- [109] A. J. Afzal, A. Natarajan, N. Saini, M. J. Iqbal, M. Geisler, H. A. El-Shemy, R. Mungur, L. Willmitzer and D. A. Lightfoot, “The Nematode Resistance Allele at the rhg1 Locus Alters the Proteome and Primary Metabolism of Soyabean Roots,” Plant Physiology, Vol. 151, No. 3, 2009, pp. 1264-1280. doi:10.1104/pp.109.138149
- [110] M. L. Perez-Bueno, J. Rahoutei, C. Sajnani, I. Garcia-Luque and M. Baron, “Proteomic Analysis of the Oxygen-Evolving Complex of Photosystem II under Biotic Stress: Studies on Nicotiana benthamiana Infected with tobamoviruses,” Proteomics, Vol. 4, No. 20, 2004, pp. 418-425. doi:10.1002/pmic.200300655
- [111] G. L. Coaker, B. Willard, M. Kinter, E. J. Stockinger and D. M. Francis, “Proteomic Analysis of Resistance Mediated by Rcm 2.0 and Rcm 5.1, Two Loci Controlling Resistance to Bacterial Canker of Tomato,” Molecular Plant–Microbe Interactions, Vol. 17, No. 9, 2004, pp. 1019-1028. doi:10.1094/MPMI.2004.17.9.1019
- [112] P. M. Houterman, D. Speijer, H. L. Dekker, C. G. De Koster, B. J. C. Cornelissen and M. Rep, “The Mixed Xylem Sap Proteome of Fusarium oxysporum-infected Tomato Plants,” Molecular Plant Pathology, Vol. 8, No. 2, 2007, pp. 215-221. doi:10.1111/j.1364-3703.2007.00384.x
- [113] M. A. Castillejo, A. M. Maldonado, E. Dumas-Gaudot, M. Fernández-Aparicio, R. Susin, R. Diego and J. V. Jorrin, “Differential Protein Expression to Investigate Responses and Resistance to Orobranche crenata in Medicago truncatula,” BMC Genomics, Vol. 10, 2009, pp. 294-298. doi:10.1186/1471-2164-10-294
- [114] F. Delalande, C. Carapito, J. P. Brizard, C. Brugidou and A. Van Dorsselaer, “Multigenic Families and Proteomics: Extended Protein Characterization as a Tool for Paralog Gene Identification,” Proteomics, Vol. 5, No. 2, 2005, pp. 450-460. doi:10.1002/pmic.200400954
- [115] M. Ventelon-Debout, F. Delalande, J. P. Brizard, H. Diemer, A. van Dorsselaer and C. Brugidou, “Proteome Analysis of Cultivar-Specific Deregulations of Oryza sativa indica and O. sativa japonica Cellular Suspension Undergoing rice yellow mottle virus infection,” Proteomics, Vol. 4, No. 1, 2004, pp. 216-225. doi:10.1002/pmic.200300502
- [116] Z. Wei, W. Hu, Q. lin, X. Cheng, M. Tong, L. Zhu, R. Chen and G. He, “Understanding Rice Plant Resistance to the Brown Planthopper (Nilaparvata lugens): A Proteomic Approach,” Proteomics, Vol. 9, No. 10, 2009, pp. 2798-2808. doi:10.1002/pmic.200800840
- [117] S. T. Kim, S. G. Kim, D. H. Hwang, S. Y. Kang, H. J. Kim, B. H. Lee, J. J. Lee and K. Y. Kang, “Proteomic Analysis of Pathogen-Responsive Proteins from Rice Leaves Induced by Rice Blast Fungus, Magnaporthe grisea,” Proteomics, Vol. 4, No. 11, 2004, pp. 3569-3578. doi:10.1002/pmic.200400999
- [118] T. Mahmood, A. Jan, M. Kakishima and S. Komatsu, “Proteomic Analysis of Bacterial-Blight Defence Responsive Proteins in Rice Leaf Blades,” Proteomics, Vol. 6, No. 22, 2006, pp. 6053-6065. doi:10.1002/pmic.200600470
- [119] A. Campos, G. da Costa, A. V. Coelho and P. Fevereiro, “Identification of Bacterial Protein Markers and Enolase as a Plant Response Protein in the Infection of Olea europaea subsp. Pseudomonas savastanoi pv. Savastanoi,” European Journal of Plant Pathology, Vol. 125, No. 4, 2009, pp. 603-616. doi:10.1007/s10658-009-9509-0
- [120] S. T. Tahara, A. Mehta and Y. B. Rosato, “Proteins Induced by Xanthomonas axonopodis pv. passiflorae with leaf extract of the host plant (Passiflorae edulis),” Proteomics, Vol. 3, No. 1, 2003, pp. 95-102.
- [121] P. Diaz-Vivancos, M. Rubio, V. Mesonero, P. M. Periago, A. R. Barcelo, P. Martinez-Gomez and J. A. Hernandez, “The Apoplastic Antioxidant System in Prunus: Response to Long-Term Plum Pox Virus Infection,” Journal of Experimental Botany, Vol. 57, No. 14, 2006, pp. 3813- 3824. doi:10.1093/jxb/erl138
- [122] D. Dahal, D. Heintz, A. Van Dorsselaer, H. P. Braun and K. Wydra, “Pathogenesis and Stress Related, as well as Metabolic Proteins Are Regulated in Tomato Stems Infected with Ralstonia solanacearum,” Plant Physiology and Biochemistry, Vol. 47, No. 9, 2009, pp. 838-846. doi:10.1016/j.plaphy.2009.05.001
- [123] L. Babujee, B. Venkatesh, A. Yamazaki and S. Tsuyumu, “Proteomic Analysis of the Carbonate Insoluble Outer Membrane Fraction of the Soft-Rot Pathogen Dickeya dadantii (syn. Erwinia chrysanthemi) Strain 3937,” Journal of Proteome Research, Vol. 6, No. 1, 2007, pp. 62-69. doi:10.1021/pr060423l
- [124] C. Rampitsch, N. V. Bykova, B. Mccallum, E. Beimcik, and W. Ens, “Analysis of the Wheat and Puccinia triticina (leaf rust) Proteomes during a Susceptible Host Pathogen Interaction,” Proteomics, Vol. 6, No. 6, 2006, pp. 1897-1907. doi:10.1002/pmic.200500351
- [125] W. Zhou, F. Eudes and A. Laroche, “Identification of Differentially Regulated Proteins in Response to a Compatible Interaction between the Pathogen Fusarium graminearum and Its Host, Triticum aestivum,” Proteomics, Vol. 6, No. 16, 2006, pp. 4599-4609. doi:10.1002/pmic.200600052
- [126] S. Campo, M. Carrascal, M. Coca, J. Abian and B. S. Segundo, “The Defence Response of Germinating Maize Embryos against Fungal Infection: A Proteomics Approach,” Proteomics, Vol. 4, No. 2, 2004, pp. 383-396. doi:10.1002/pmic.200300657
- [127] S. Kundu, et al., “Proteomic Analysis of Salicylic Acid Induced Resistance to Mungbean Yellow Mosaic India Virus in Vigna mungo,” Journal of Proteomics, Vol. 74, No. 3, 2011, pp. 337-349. doi:10.1016/j.jprot.2010.11.012
- [128] P. W. Muturi, J. K. Mwololo, S. W. Munyiri, P. Rubaihayo, J. K. Munyua, M. Mgonja, E. Manyasa and N. Kiarie, “A Perspective on Proteomics: Current Applications, Challenges and Potential Uses,” Agriculture and Biology Journal of North America, Vol. 1, No. 5, 2010, pp. 916-918.
- [129] J. M. Dunwell, A. M. Maria and H. Raul, “Transcriptome Analysis and Crop Improvement,” Biological Research, Vol. 34, No. 3-4, 2001, pp. 3-4. doi:10.4067/S0716-97602001000300003
Abbreviations
2D-PAGE: Two dimensional polyacrylamide gel electrophoresis 2D-DIGE: Fluorescent Two-dimensional “Difference Gel Electrophoresis”
AHL: N-acyl homoserine lactone DAMPs/PAMPs: Danger associated molecular patterns/Pathogen associated molecular patterns DIR: Dirigent proteins ESI: Electrospray ionization ETI/PTI: Effector-triggered immunity/PAMPs-triggered immunity GLP3: Germin like proteins 3 GSTs: Glutathione-S-transferases HR: Hypersensitive response ICAT: Isotope-Coded Affinity Tags ITRAQ: Isobarric Tagged for Relative and Absolute Quantitation JA/SA: Jasmonic acid/ Salicylic acid
µLC-ESI-MS/MS: Microcappilary liquid chromatography-electron spray tandem mass spectrometry LC-MS/MS: Liquid chromatography mass spectroscopy
MECS: O-methyltransferase, 2-C-methyl-D-erythritol 2,4 cyclodiphosphate synthase MALDI TOF-MS: Matrix assisted laser desorption/ ionization time-of-flight mass spectrometry MudPIT: Multidimensional Protein Identification Technology NO: Nitric oxide NHR: Non-host resistance PAL: Phenylalanine ammonia lyase PCD: Programmed cell death PR Protein: Pathogenesis related protein PMMoV-S/PMMoV-I: Peeper mild mottle virus-Spanish strain/Peeper mild mottle virus-Italian strain ROS: Reactive oxygen species RILs: Recombinant inbreed lines SAR: Systemic acquired resistance SCX HPLC/RP HPLC: Strong cation exchange high performance liquid chromatography/Reverse phase high performance liquid chromatography TCA: Tricarboxylic acid
NOTES
*The authors contributed equally.
#Corresponding author.