American Journal of Climate Change
Vol.06 No.01(2017), Article ID:74811,19 pages
10.4236/ajcc.2017.61008
Investigation of the CO2 Sequestration by Indirect Aqueous Carbonation of Waste Cement
Myoungwook Mun*, Heechan Cho, Jihoe Kwon, Kihong Kim, Rina Kim
Department of Energy Resources Engineering, Seoul National University, Seoul, Republic of Korea

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: December 6, 2016; Accepted: March 18, 2017; Published: March 21, 2017
ABSTRACT
Mineral carbonation of CO2 with fine-grained waste cement was investigated using NH4Cl as a recyclable extracting agent. The amount of calcium extracted with NH4Cl was not as high as with more commonly used extraction agents such as HCl and CH3COOH. NH4Cl also exhibited high selectivity in the calcium extraction process, such that calcium ions comprised over 99% of the leaching solution. Another positive benefit of using NH4Cl was that precipitation of calcium carbonate by CO2 injection was possible without the addition of basic reagents. Moreover, the NH4Cl regenerated during carbonation can be reused for calcium extraction. However, test results using regenerated NH4Cl solution in a cyclic fashion revealed that the process was not perfectly cyclic, but rather the calcium amount after precipitation increased as the cycle proceeded. The geochemical computer simulation PHREEQC was utilized to gain better insight into the cyclic mineral carbonation processes using NH4Cl solution. The simulation was based on thermodynamic equilibrium so that the amount of Ca in the solution fluctuated between specific values in a periodic fashion, unlike the experimental results of calcium accumulation in the extraction solution. One reason for this phenomenon was the kinetic/thermody- namic balance controlled by the amount of Ca2+ and CO2 present in the solution. However, it was feasible to use a geochemical model to evaluate the mineral carbonation process with the correction factors since the deviation between the experimental and the simulation results remained fairly constant throughout the cycle.
Keywords:
CO2 Sequestration, Mineral Carbonation, Waste Cement, Ammonium Chloride, PHREEQC

1. Introduction
Mineral carbonation offers an alternative option for sequestering CO2. One major benefit of mineral carbonation is the environmentally friendly and virtually permanent trapping of carbon dioxide in the form of carbonate minerals. This option has an additional advantage in that, unlike geological sequestration routes, it provides long-term sequestration without need for post-monitoring processing once the carbon dioxide has been fixed.
CO2 sequestration by mineral carbonation can be viewed as an acid-base reaction where dissolved CO2 reacts with a solid base. Calcium and magnesium are common in nature, which are typically found in silicate minerals such as wollastonite and serpentine. Therefore, these minerals were intensively used in earlier studies on mineral carbonation [1] [2] [3] [4] [5] . However, utilization of these minerals requires large-scale mining operations, which often face criticism over cost and environmental issues related to exploitation of natural resources. In light of this, there has been increasing interest in mineral carbonation using alkaline industrial wastes such as steel-making slag, waste concrete, asbestosmi- ning tailings, and coal fly ash [6] [7] [8] [9] . However, in general, mineral car- bonation is considered to be more expensive than geological storage mainly due to the cost and energy required for pre-treatment, which includes fine grinding and extraction of Mg and Ca from source minerals, as shown in Table 1 [10] .
Therefore, in recent years, various studies have been undertaken seeking diffe- rent routes to enhance the carbonation process while minimizing energy consumption and costs [11] . Two main routes can be distinguished: 1) direct carbo- nation and 2) indirect carbonation. Direct carbonation involves a one-step direct reaction of gaseous carbon dioxide with particulate metal oxide, either as a direct gas-solid reaction or in an aqueous medium. Indirect carbonation is conducted in two steps: extraction of Mg or Ca from the minerals using acids or other solvents, followed by carbonation where the extracted components are reacted with CO2.

Table 1. Costs of CCS: production costs of electricity for different types of generation.
In indirect carbonation, excess acid is required to achieve high extraction efficiency, and in turn, the pH of the extraction solution can be very low [12] [13] . This results in an unfavorable condition for the subsequent carbonation reaction, which is favored by basic pH conditions. Therefore, it is necessary to raise the pH before carbonation. If a caustic reagent is used for this purpose, a considerable amount of reagents is required for the extraction and carbonation steps. Alternately, organic acids such as EDTA are added to enhance the dissolution of alkaline metals from silicates in mild acid conditions [14] . This would reduce the usage of caustic reagent in the second step. However, organic acid is very expensive and makes it more difficult to recover and recycle the acid used in the leaching step. Another approach towards reduction of energy and acid consumption is the use of acetic acid. This is a less severe extraction medium and offers the additional advantage of the ability to recover the acetic acid in the second step [15] [16] .
There have been other attempts to develop an extraction agent with potential for full chemical recycling. Kodama et al. [17] developed a process that uses NH4Cl as an extracting agent. It was reported that NH4Cl could dissolve calcium ions efficiently and selectively from slag. After removing the slag residue, injection of CO2 into the leaching solution resulted in precipitation of pure calcium carbonate with NH4Cl regenerated during carbonation. A caustic reagent was not required for carbonation in this process as the solution pH was stabilized in the alkaline region during extraction. This process seems to be the most promising given the potential for reagent recovery and reuse. Therefore, it was investigated in this study as a preferred method for CO2 sequestration.
A number of potentially suitable types of industrial waste are generated in large quantities in Korea such as slag and waste concrete. Over 20 million tons of steel slag is generated in Korea each year. Since the calcium content of this slag is typically over 40 wt.%, this material has a great potential for CO2 sequestration. However, it contains many potentially hazardous heavy metals that can leach out during extraction. This could complicate the process and the resulting carbona- ted products may not be pure enough to be of value in other industries such as the paper industry. Moreover, residues after extraction can be environmentally hazardous and may require additional treatment. The amount of waste concrete generated in Korea was about 90,000 tons per day in 2010. In order to meet the requirements of the mandatory recycling law, there are about 300 construction waste processing plants in Korea. These plants have typical facilities such as sorting and crushing units to produce mainly recycled aggregates. For every ton of waste concrete, 530 kg of powdery waste cement is produced [18] . This material contains over 40 wt% Ca (as CaO) and therefore is suitable for mineral carbonation due to the availability of fine-grained Ca bearing materials.
The objective was to experimentally analyze mineral carbonation of waste cement in a cyclic manner. To achieve this, the degree of extraction was compared to that of other common acids (acetic acid and hydrochloric acid). Additionally, the potential for direct carbonation with flue gas was evaluated to develop a cost effective method of CO2 sequestration without carbon capture.
2. Materials and Methods
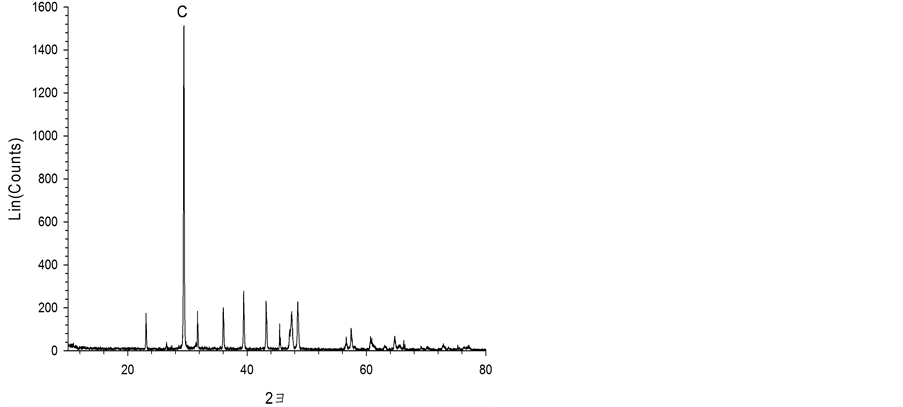
The waste cement used in this study was fine residue recovered from aggregates obtained through crushing demolition concrete waste and size separation. Sieve size analysis indicated particles’ median diameter of 33.4 μm. The specific surface area was 13 m2/g determined by BET gas adsorption method (GEMINI V, Micromeritics). The chemical composition measured by X-ray Fluorescence (XRF) (S4 PIONEER, Bruker AXS) and Powder X-Ray Diffractometry (XRD) (D8 Advance, Bruker) show that waste cement is rich in Si (40.4 wt.% SiO2), Ca (36.7 wt.% CaO), Al (9.8 wt.% Al2O3), Fe (5.1 wt% Fe2O3), K (2.3 wt.% K2O), and Mg (1.9 wt.% MgO) along with minor elements: Na, Ti, and Mn (Figure 1). A Field Emission-Scanning Electron Microscopy (FE-SEM) image (SUPRA 55 VP, Carl Zeiss) of the waste cement particles shows a distinct porous structure that may allow rapid penetration of liquid in solid surface (Figure 2). All of these properties indicate that waste cement is a good material for mineral carbonation.
Ammonium chloride (99.5%, Sigma-Aldrich), acetic acid (1 M, Sigma-Al- drich), and hydrochloric acid (1 M, Sigma-Aldrich) were used to extract Ca ions from the waste cement. The reactions of the waste cement (represented by calcium silicate minerals) with three acids are formulated as shown below:
Hydrochloric acid:
CaSiO3 + 2HCl → CaCl2 + SiO2 + H2O (1)
CaCl2 + CO2 + H2O → CaCO3 + 2HCl (2)
Acetic acid:
CaSiO3 + 2CH3COOH → Ca(CH3COO)2 + SiO2 + H2O (3)
Ca(CH3COO)2 + CO2 + H2O → CaCO3 + 2CH3COOH (4)
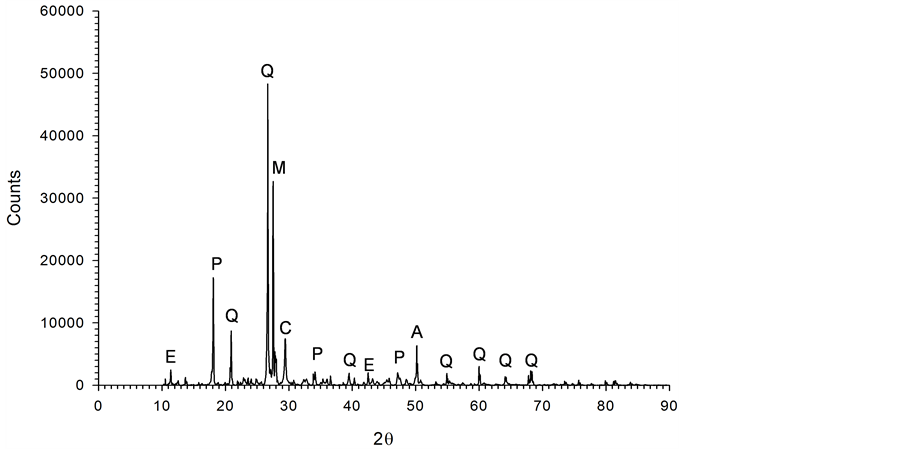
Figure 1. XRD peaks obtained on analysis of waste cement (A: Albite, C: Calcite, E: Ettringite, M: Microcline, P: Portlandite, Q: Qualtz).

Figure 2. Surface morphology of waste cement in FE-SEM.
Ammonium chloride:
2CaSiO3 + 4NH4Cl → 2CaCl2 + 2SiO2 + 4NH3 + 2H2O (5)
2CaCl2 + 2CO2 + 4NH3 + 2H2O → 2CaCO3 + 4NH4Cl (6)
For acetic and hydrochloric acid, it is expected that the dissolution amount of Ca ions will increase as the acidity increases. However, carbonation reaction favors basic pH conditions so that addition of basic reagents is required to raise the pH of the solution to at least 8 - 9 in the precipitation of carbonate minerals. In the case of ammonium chloride, the pH of solution in the extraction step becomes alkaline due to the NH3 generation. This enhances the absorption of CO2 into the solution and precipitation of CaCO3 without addition of basic reagents in the carbonation step. Furthermore, during precipitation of CaCO3, the acidic extraction solution and NH4Cl are regenerated.
Calcium extraction experiments were carried out as follows: 50 g of the waste cement was mixed with 500 mL of solutions containing various amounts of the agents (0.1 - 0.5 M) in a 1 L beaker. The slurry was stirred with a magnetic stirrer at 400 rpm for up to one hour in ambient conditions (25˚C, 1 atm). The liquid samples were obtained at regular time intervals and were analyzed for concentrations of various ions by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) (ICP-730 ES, Varian).
After one hour of extraction, the slurry was immediately filtered through 6 μm filter paper to separate the solid residue. The filtrate was then poured into a 500 mL flask for carbonation experiments. For the filtrates from HCl and CH3COOH leach, the solution pH was adjusted to, and maintained at pH 10 - 12 in ambient condition through the drop addition of 1 M NaOH solution. After pH was stabilized, pure CO2 gas (99.9%) was introduced at a constant flow rate (100 mL/min) into the flask for 2 hours to supply carbon dioxide. The pH of the solution was continuously measured to monitor the progress of the carbonation reaction. After the completion of carbonation, the slurry was filtered and the filtrate was analyzed for Ca and other ions using ICP-OES. The precipitate was collected, washed, and dried before being analyzed by XRD and SEM.
In order to evaluate the recyclability of the NH4Cl solution, additional tests were conducted in a cyclic manner. First, complete extraction and precipitation were conducted in the first cycle using the same procedure described above. Then a second cycle was performed using the filtrate recovered after completion of carbonation in the first cycle as extractant, without adding new reagents. These tests were repeated for several cycles and the intermediate products (precipitated calcium carbonates) were dried, weighed and subjected to chemical analysis.
In addition, carbonation experiments were carried out using synthetic gases of various gas compositions in the same way to evaluate the possibility of carbonation with the flue gas (mixture of CO2-N2-SO2-NOx). The left column of Table 2 shows the compositions of the synthetic gases mixed and tested in this study along with that of the real flue gas emitted from coal power plants in Korea. The syngas was supplied from bottled gases mixed to the designed composition for injection into the flask for carbonation reaction.
3. Results and Discussion
3.1. Extraction of Calcium
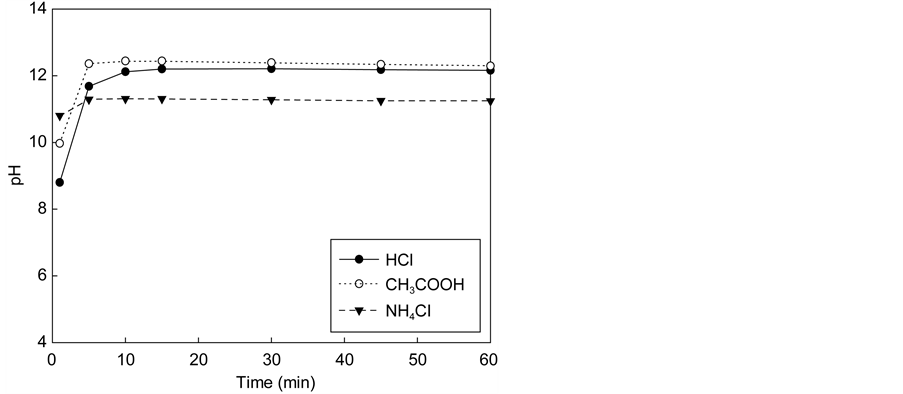
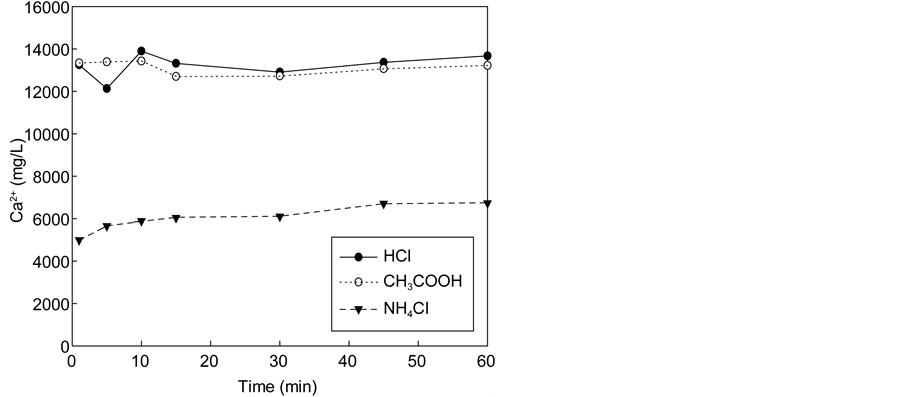
Figure 3 shows the variation of calcium concentration with reaction time using the solution of three acids (HCl, CH3COOH, NH4Cl) containing various amounts of acids. In all cases, the reaction occurred quickly and stabilized after several minutes. When the acid concentration was 0.1 M, the maximum calcium concentration in the extraction solution did not differ greatly among acid types (1900
Table 2. Compositions of flue gas and virtual gas.
 (a) (b)
(a) (b)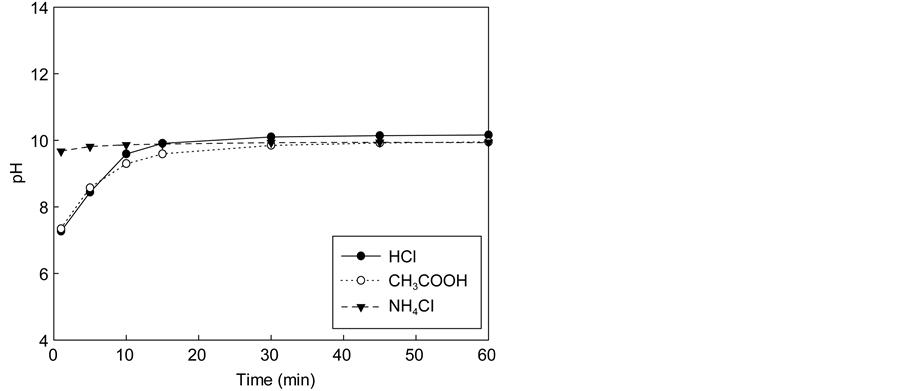
 (c) (d)
(c) (d)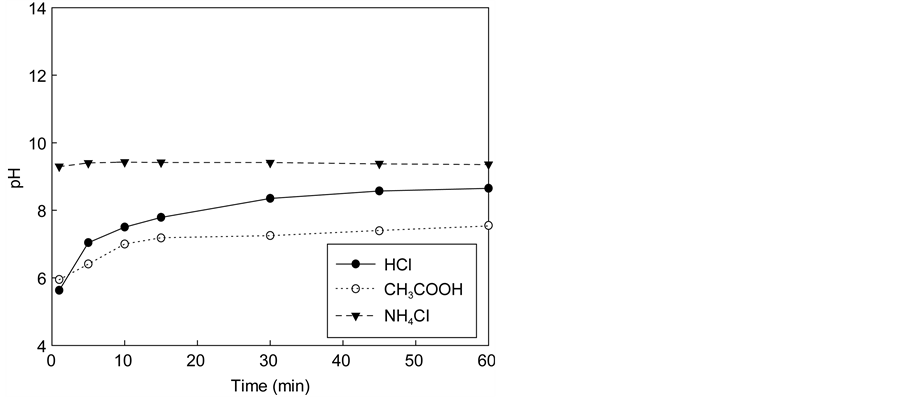
 (e) (f)
(e) (f)
Figure 3. Effect of the dissolution of waste cement in each acid. (a) Calcium concentrations in solution with 0.1 M. (b) pH of solutions after dissolution with 0.1 M. (c) Calcium concentration in solution with 0.3 M. (d) pH of solutions after dissolution with 0.3 M. (e) Calcium concentration in solution with 0.5 M. (f) pH of solutions after dissolution with 0.5 M.
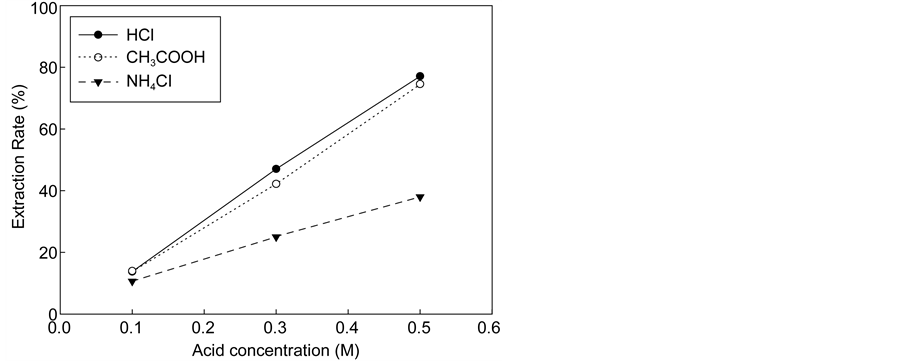
mg/L for NH4Cl versus 2400 mg/L for HCl and CH3COOH). As shown in Figure 4, for the acid concentration levels investigated there seems to be a linear relationship between the maximum extraction yields of calcium in the extraction so- lution with the amount of acid regardless of the acid type. However, the yield of increment depended on the type of acid. When acid concentrations were increased to 0.5 M, the calcium concentration of HCl and CH3COOH solutions increased more than fivefold (~13,000 mg/L), whereas that of NH4Cl increased threefold (~6700 mg/L). If all calcium in the extraction solution was reacted with carbon dioxide completely, NH4Cl could uptake up to 0.1 kg CO2/kg cement while HCl and CH3COOH could absorb 0.14 kg CO2/kg cement.
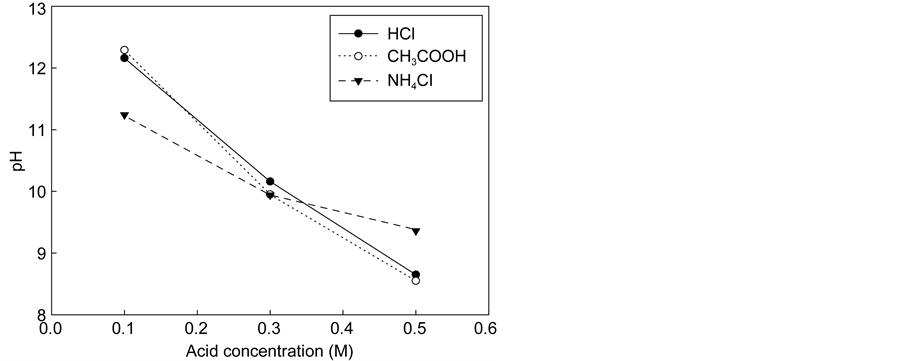
The final equilibrium pH values of the extraction step were 8.65, 8.55, and 9.36 for HCl, CH3COOH, and NH4Cl solutions, respectively. Therefore, the degree of extraction seems to relate to the acidity of the extraction solution. To better understand the chemical reaction involved in the extraction and carbonation of the waste cement with CO2, thermodynamic analysis was conducted with the aid of PHREEQC. This will be discussed further in Section 4.
Table 3 lists the concentrations of the various ions in the final extraction solutions using 0.5 M acids. In all cases, calcium constitutes more than 99% of the extracted ions, indicating that calcium carbonate of high purity could be obtained through the carbonation process. Since the pH of the solution after extraction
 (a) (b)
(a) (b)
Figure 4. Extraction rates of calcium and pH in the extraction solution with acid concentration. (a) Extraction rate of calcium. (b) pH.
Table 3. Composition of the liquid that extracted from the waste cement at 1 hour.
was in the 8 - 10.5 range, extraction of calcium ions could easily react within this pH region that made the calcium concentration high in all acids [17] .
3.2. Response Characteristic of Carbonation
After extraction, the solid residue was separated by filtration and the filtrate re- acted with 99.9% CO2. The extraction solution of HCl and CH3COOH did not produce CaCO3 by injection of CO2 because the solution pH rapidly declined to acidic conditions. Therefore, it was necessary to adjust the pH by adding NaOH in order to induce effective carbonation. However, with the extraction solution of NH4Cl, carbonation was possible without the addition of basic reagents. Moreover, NH4Cl is regenerated during carbonation. Therefore, this route may be more economical, although the amount of Ca extracted using the NH4Cl solution was less than those using HCl or CH3COOH solutions under the same conditions. The amount of Ca converted into carbonate forms was calculated as the difference between the initial and final Ca concentrations in the solution at the carbonation stage. This yield was between that obtained by using HCl and CH3COOH at pH 10 (50%) and pH 12 (95%); NaOH was added to these acid solutions to adjust the pH and induce carbonation. Consequentially, NH4Cl can actually absorb 0.07 kg CO2/kg cement, whereas HCl and CH3COOH can absorb 0.19 kg CO2/kg cement. Although maximum CO2 mineral sequestration capacity in ammonium chloride was lower than that in hydrochloric acid and acetic acid, it was reasonably comparable to when compared with HCl and CH3COOH [19] .
After completion of the carbonation stage, the pH of the solution with 0.5 M NH4Cl stabilized at 6.1, which was close to the initial pH of the NH4Cl solution that was used for Ca extraction from the waste cement. Therefore, this solution can be reused for Ca extraction after recovering the precipitated calcium carbonates by filtration.
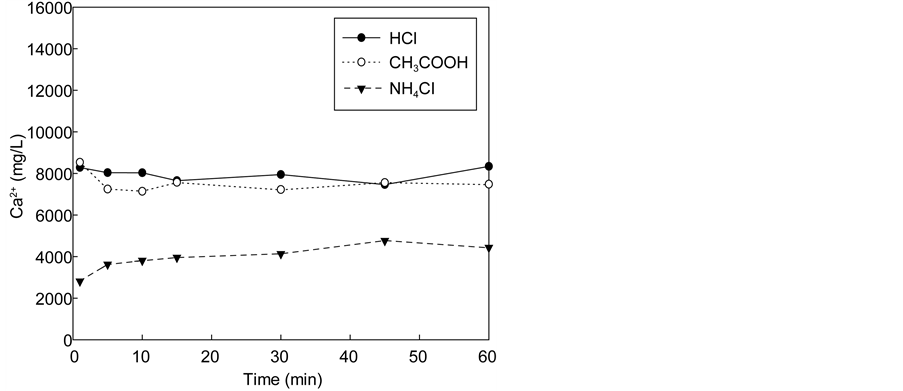
Additional tests were performed in a cyclic manner using 1 M NH4Cl to ensure sufficient Ca extraction, as shown in Figure 5. The calcium concentration
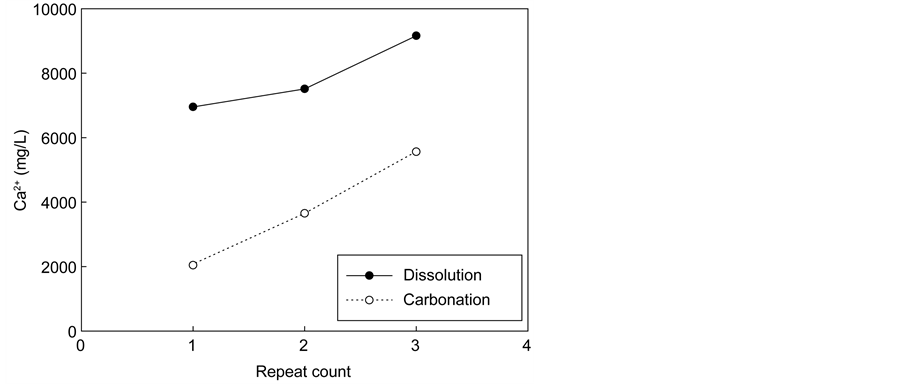
Figure 5. Change in the Ca2+ ion concentration in the solution used in repeated dissolution and carbonation.
in the extraction solution after the first extraction step was about 7000 mg/L. After the carbonation step, the calcium concentration reduced to 2000 mg/L (70% conversion by precipitation). After the completion of the first cycle, the calcium concentration in the extraction solution rose to 7500 mg/L, indicating that additional 5500 mg of calcium was extracted. After the completion of the second cycle carbonation, the calcium concentration reduced to 3700 mg/L, corresponding to 50% conversion by precipitation. In the third cycle, the calcium concentration increased to 9000 mg/L in the extraction stage, and subsequently decreased to 5500 mg/L after carbonation, corresponding to 40% conversion by precipitation. This indicates that the calcium ions continue to accumulate in the recovered NH4Cl solution as the conversion decreases with increasing numbers of cycles. Therefore, it may be necessary to remove residual calcium ions at some point during recycling.
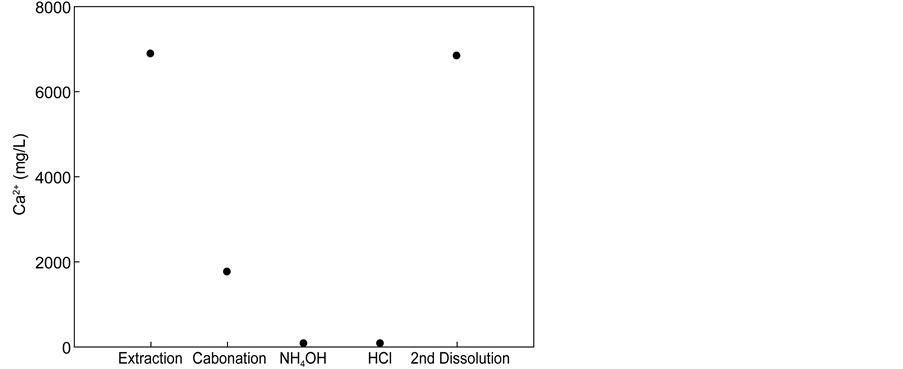
One method used to accomplish this was to increase the pH in the carbonation step to induce an additional carbonation reaction of residual calcium ions. Figure 6 shows the result of such an attempt, where ammonium hydroxide was
 (a)
(a)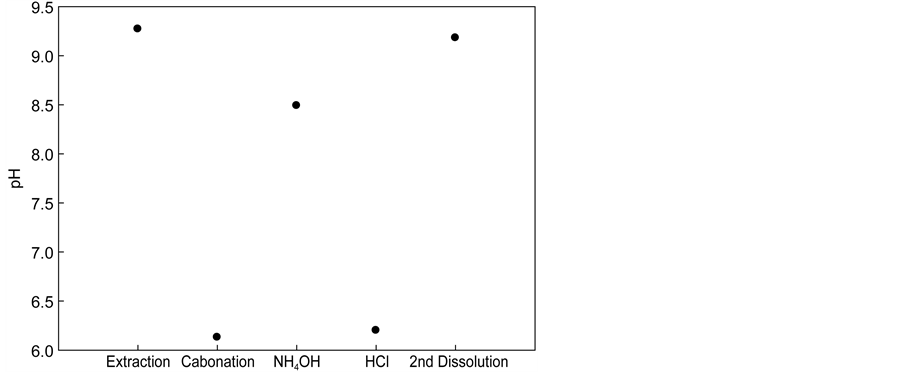 (b)
(b)
Figure 6. Variation in pH with carbonation, NH4OH, and HCl. (a) Calcium concentration after each process; (b) pH of solution after each process.
added after the carbonation step, which resulted in complete removal of the residual calcium ions from the recovered NH4Cl solution. However, by doing so, the pH of the NH4Cl solution changed to 6.2 - 8.5 so that acidic reagents such as HCl were required to bring the pH back to the initial value of 6.2 in order to re- use the solution for Ca extraction. This practice restored the cycle system to the original state and prepared the system to restart a new series of extraction-car- bonation cycles.
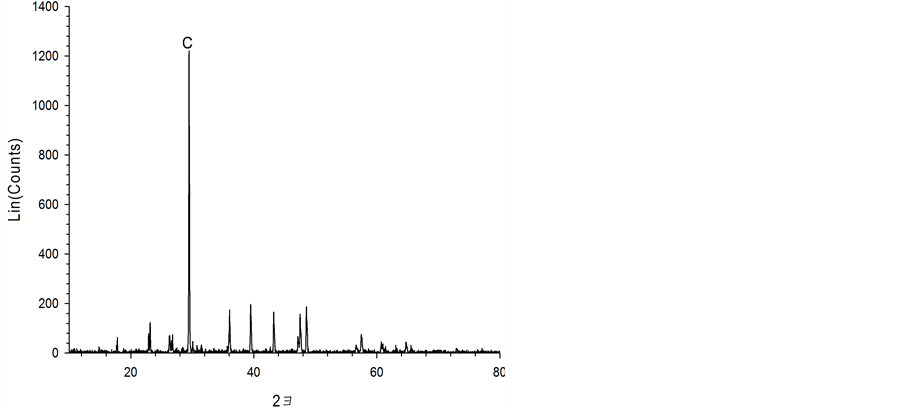
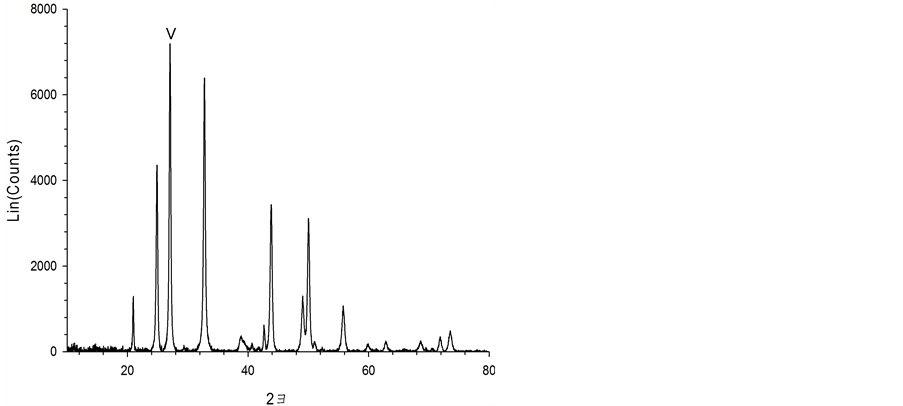
Figure 7 shows the characteristics of the solids resulting from carbonate precipitation of the three acid extracts using XRD and FE-SEM. When the waste cement was dissolved with hydrochloric acid (a) and acetic acid (b), the precipitated solids were pure calcite. However, for the case of the ammonium chloride extraction, flower-like particles were observed. The XRD analysis revealed that these particles were vaterite. Vaterite is the least thermodynamically stable polymorph of CaCO3 and it has been of great interest due to its medical and industrial applications [20] [21] . Therefore, various experimental conditions have been investigated to promote vaterite synthesis. In particular, it was reported that a flower-like vaterite could be synthesized when ammonia was present in the carbonate solution [22] . This may explain the formation of vaterite from the NH4Cl extraction solution since NH3 is generated under this condition (Equation (5)). Since vaterite is of higher commercial value than more common calcite, the NH4Cl extraction has another advantage over other acid extractions.
3.3. Carbonation Characteristics with Flue Gas
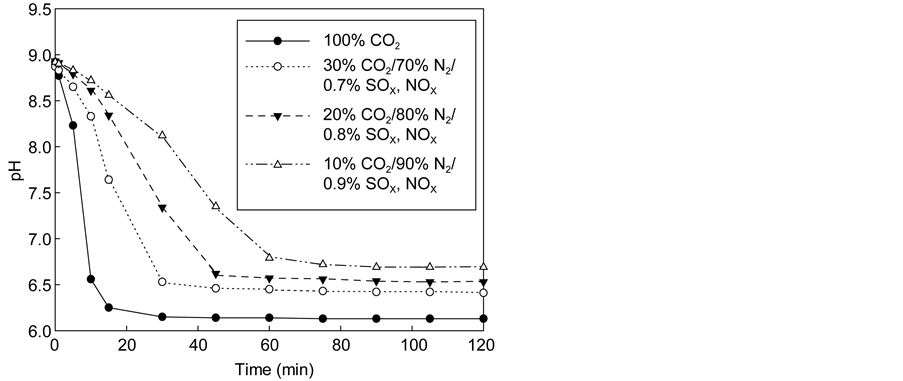
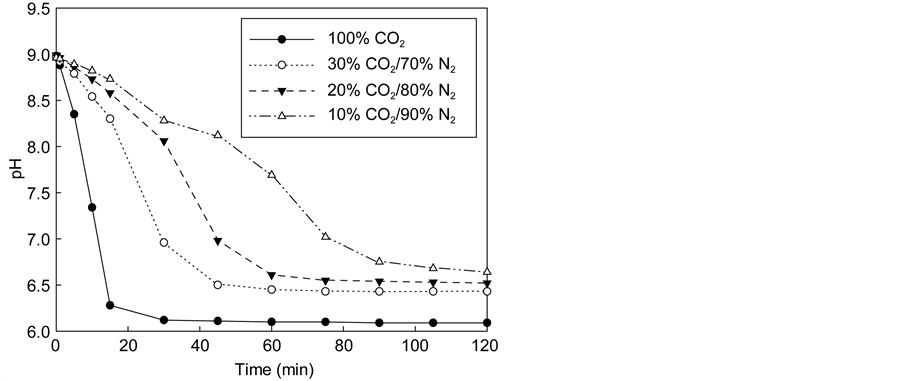
If the flue gas is directly treated for carbonation, carbon capture and storage cost will be considerably reduced as it eliminates the costly carbon capture process. Therefore, in this study, the direct carbonation of flue gas was investigated by reacting synthetic flue gases (mixtures of CO2/N2 and CO2/N2/SOx/NOx) with 0.5 M NH4Cl solution for 1 h. Figure 8(a) shows the pH of the solution measured over time when reacting with N2 and CO2 mixtures. As the proportion of CO2 in the CO2/N2 mixtures was decreased to reduce the rate of reaction, solution pH decreased more slowly over time, taking longer to reach equilibrium. In addition, the final pH increased with decreasing CO2 content in the gas mixtures. Accordingly, the amount of the precipitated CaCO3 was found to be reduced as the CO2 content in the mixture decreased.
Flue gases from plants may contain nitrogen oxides and sulfur oxides. Figure 8(b) shows that although the final pH values are not much different, the kinetics of the carbonation reactions appears to be affected by the gas composition.
Waste cement dissolved in NH4Cl from which the ammonia was released may also take part in the reactions. Ammonia was known as an efficient substance for capturing carbon dioxide from the flue gas. Therefore, the ammonia served to separate the carbon dioxide from the gas mixture in carbonation. It was anticipated that NH4Cl solutions would simultaneously capture and sequester carbon dioxide from the real flue gas.

 (a)
(a)
 (b)
(b)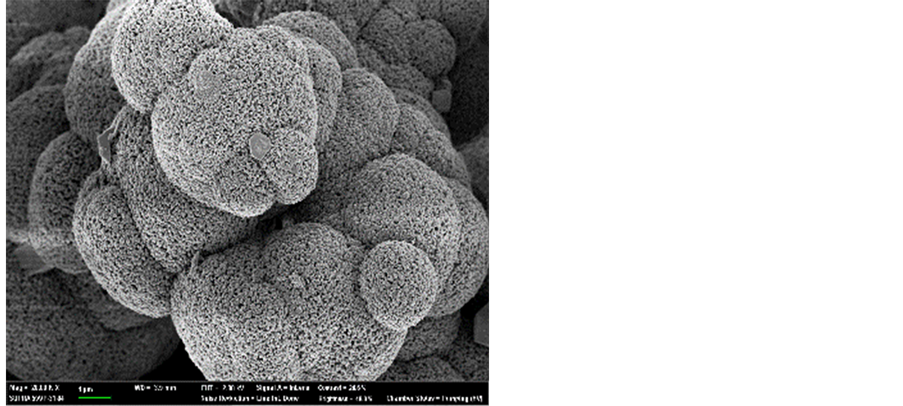
 (c)
(c)
Figure 7. FE-SEM image and XRD data obtained for the by-products of the acid treatments. (a) HCl; (b) CH3COOH; (c) NH4Cl.
 (a) (b)
(a) (b)
Figure 8. Changes in pH of the reaction with the virtual flue gas. (a) CO2/N2; (b) CO2/N2/SOx/NOx.
4. Thermodynamic Analysis of Mineral Carbonation Using PHREEQC
Understanding the chemical reactions involved in any chemical process is important to optimize and control the process’ variables and parameters. Therefore, we wanted to determine if it was feasible to use a geochemical code, PHREEQC [23] , to represent the mineral carbonation processes using NH4Cl solution in a cyclic fashion. PHREEQC is capable of simulating complex reactions between dis- solved gases, aqueous solutions, and minerals. The geochemical program PHREE- QC takes into account the concentration of ions in water, such as calcium, sodium, sulfate, etc., and then calculates how much of these ions and minerals formed are dissolved in the water, and if they will precipitate out of solution to form minerals. Calculations are made using thermodynamic databases, which include a wide range of data for mineral phases and compounds. The PHREEQC simulation is also capable of incorporating kinetic equations for chemical reactions and determining speciation at any designated time.
For the simulation, it is first required to define the mineralogical phases constituting the starting material. Ordinary Portland Cement (OPC) generally consists of calcium silicates (Ca3SiO5 and Ca2SiO4), aluminate (Ca3Al2O6), and ferrite (Ca4(AlxFe1−x)4O10). These components react with water to form hydration products such as amorphous CSH (calcium silicate hydrate), portlandite, ettringite, etc. XRD analysis of the studied material identified the following crystalline constituents: portlandite, calcite, quartz, albite, microcline, and ettringite. However, it is difficult to determine the amount of different phases analytically since XRD only shows materials with crystal structure. Therefore, the amount of amor- phous CSH was determined using thermo-gravimetric analysis (TGA). Figure 9 shows the results of TGA. An abrupt weight loss near 450˚C is associated with the dehydration of portlandite. Weight loss occurring between 700˚C and 800˚C related to the decomposition of calcium carbonate, and a continuous weight loss over the whole range from 105˚C to 500˚C is due to the dehydration of calcium silicate hydrates [24] . When the weight loss in each region was calculated, the content of CSH and portlandite were estimated to be 4.9% and 5.6%, respectively. The content of portlandite was compared with XRD results and normalized to give the estimation of the composition of the sample as shown in Table 4. The CSH composition was assumed to be Ca1.8SiO3.8H2O, which was found to be a good approximation for modeling concrete leaching [25] .
The hydrolysis equilibrium constants of quartz, calcite, microcline, and albite were estimated using the PHREEQC databases. However, CSH, portlandite, and anhydrite were not included in the PHREEQC database, and therefore appropriate values were obtained from research literature [26] [27] and were added to the database. The expected secondary carbonate mineral assemblage due to CO2 gas injection at 1 atm was based on the PHREEQC database. Table 5 gives the complete list of the initial phase and the newly formed phases with their stoichiometry
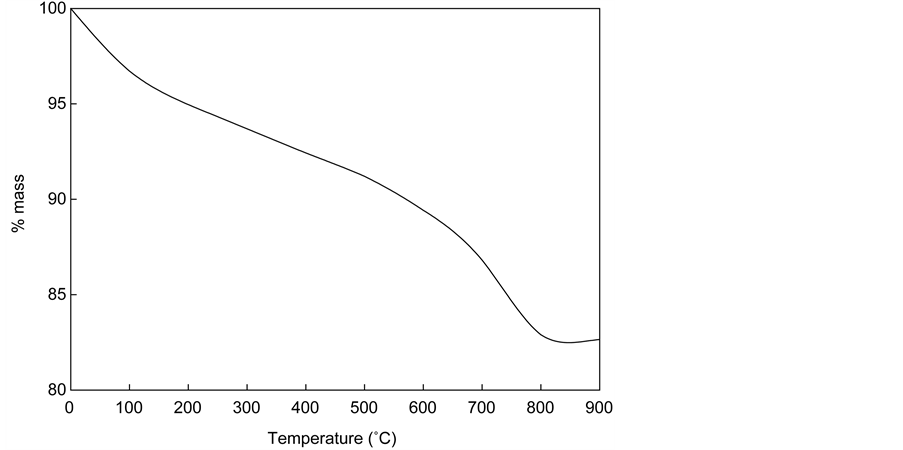
Figure 9. TGA results of waste cement sample.
Table 4. Constituent minerals of the cements resulted obtained by combining TGA in XRD.
Table 5. Phases involved in the reaction.
reactions and equilibrium constants. Assuming that all reactions have reached equilibrium, extraction and carbonation were calculated through EQUILIBRIUM_ PHASES statement.
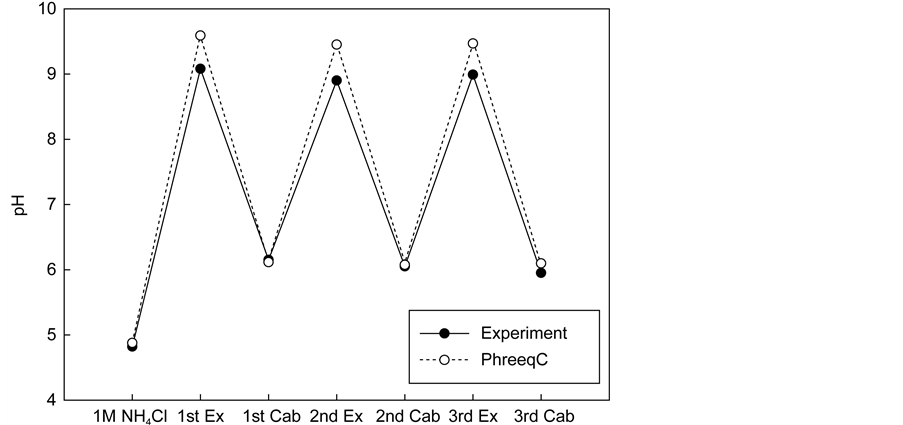
Figure 10 shows the pH and the amount of calcium at different stages along the extraction and carbonation process. After the first stage of extraction, the calcium amount from the simulation was 1.3 times higher than that found in the experimental results. This indicates that not all of the calcium was extracted from the waste cement particles, which is to be expected because the particle co- re may still remain intact. After the first stage of carbonation by injecting CO2 into the solution, the simulation shows 95% calcium removal from the solution by precipitation, whereas in the experiment, only 70% of the calcium was removed.
At the subsequent stages of the extraction and precipitation process, the simulation results showed that the concentration of Ca in the solution rises and falls to the same values in a periodic fashion. At the same time, the experimental results show that the amount of Ca present after the precipitation step increased with each cycle. Correspondingly, the maximum amount of Ca remaining after extraction also increased with each cycle. One possible reason for such aphenomenon is the kinetic/thermodynamic balance as controlled by the amount of Ca2+ and CO2 present in the solution. Calcium carbonate can occur in the form of three anhydrous crystalline polymorphs: vaterite, aragonite, and calcite. Vaterite is not stable under standard conditions and transforms easily into aragonite or calcite when in contact with water. Yet, under particular conditions, vaterite forms and persists without transforming. Brecevic and Kralj [26] reported that vaterite particles were obtained in a very narrow region of Ca2+ and CO2 concentrations at a pH from 9.3 to 9.9. Dickinson et al. [28] reported that the correlation between pCO2 and Ca2+ was one of the most important factors in determining the calcium carbonate precipitation reaction. When they were in complete balance, calcite formed. If the balance was tilted towards high pCO2, vaterite for- mation was induced but the CaCO3 precipitation was kinetically controlled. The
 (a)
(a)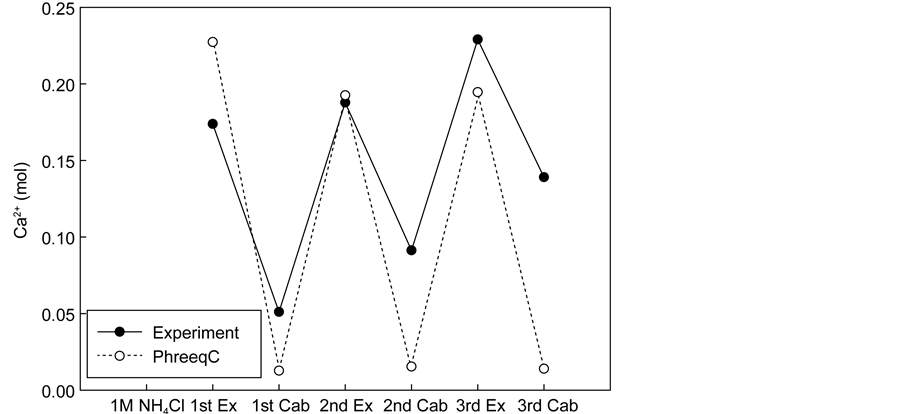 (b)
(b)
Figure 10. pH and amount of calcium from simulation with experiments. (a) pH of solution after each process; (b) Calcium concentration after each process.
precipitation conditions under which the current investigation was conducted were in the range of the aforementioned conditions, which may explain vaterite formations. However, the kinetic aspect was not considered in this simulation, which may be the reason for the discrepancy between the simulation and the experimental results. Currently, databases of reaction kinetics are not sufficient to allow calculation of the vaterite precipitation in detail. However, the results show that it is feasible to use the geochemical model to evaluate the mineral carbonation process with the correction factors since the deviation between the experimental and the simulation results remained fairly constant throughout the cycle. There are two possible reasons for the deviations: 1) kinetics, and 2) lack of thermodynamic data for the formation of different types of calcium carbonate polymorphs, which requires further study.
5. Conclusion
Mineral carbonation was conducted using waste cement with NH4Cl as an extrac- ting agent for preferential carbon dioxide sequestration. The main advantage of using NH4Cl was that it was regenerative, and it did not require the addition of basic reagents for the precipitation of calcium carbonates. Furthermore, it was de- monstrated that NH4Cl solutions could sequester carbon dioxide directly from flue gas without the additional expense of a CO2 capturing step. PHREEQC simulation showed that the calcium extraction and precipitation should operate in a fully periodic fashion. The experimental results showed that the conversion of the precipitation reaction was not fully attained so that the calcium ion concentration increased as the cycle continued. This was possibly due to kinetic/ther- modynamic balance as controlled by the amount of Ca2+ and CO2 present in the solution. However, it is feasible to use the geochemical model to evaluate the mi- neral carbonation process with the correction factors since the deviation between the experimental and the simulation results remained fairly constant throughout the cycle.
Cite this paper
Mun, M., Cho, H., Kwon, J., Kim, K. and Kim, R. (2017) Investigation of the CO2 Sequestration by Indirect Aqueous Carbonation of Waste Ce- ment. American Journal of Climate Change, 6, 132-150. https://doi.org/10.4236/ajcc.2017.61008
References
- 1. Huijgen, W.J.J., Witkamp, G. and Comans, R.N.J. (2006) Mechanisms of Aqueous Wollastonite Carbonation as a Possible CO2 Sequestration Process. Chemical Engineering Science, 61, 4242-4251.
https://doi.org/10.1016/j.ces.2006.01.048 - 2. Huntzinger, D.N., Gierke, J.S., Kawatra, S.K., Eisele, T.C. and Sutter, L.L. (2009) Carbon Dioxide Sequestration in Cement Kiln Dust through Mineral Carbonation. Environmental Science & Technology, 43, 1986-1992.
https://doi.org/10.1021/es802910z - 3. O’Connor, W.K., Dahlin, D.C., Rush, G.E., Gerdemann, S.J., Penner, L.R. and Nilsen, R.P. (2005) Aqueous Mineral Carbonation: Mineral Availability, Pretreatment, Reaction Parametrics, and Process Studies. DOE/ARC-TR-04-002, Albany Research Center, Albany.
- 4. Lee, J.K. and Dong, W.K. (2005) Characteristic Study of Korean Magnesium Silicates for CO2 Mineral Carbonation. 22nd Annual International Pittsburgh Coal Conference, Pittsburgh, 12-15 September 2005, 1874-1883.
- 5. Teir, S., Kuusik, R., Fogelholm, C.J. and Zevenhoven, R. (2007) Production of Magnesium Carbonates from Serpentinite for Long-Term Storage of CO2. International Journal of Mineral Processing, 85, 1-15.
https://doi.org/10.1016/j.minpro.2007.08.007 - 6. Eloneva, S., Teir, S., Salminen, J., Fogelholm, C. and Zevenhoven, R. (2008) Steel Converter Slag as a Raw Material for Precipitation of Pure Calcium Carbonate. Industrial & Engineering Chemistry Research, 47, 7104-7111.
https://doi.org/10.1021/ie8004034 - 7. Teramura, S., Isu, N. and Inagaki, K. (2000) New Building Material from Waste Concrete by Carbonation. Journal of Materials in Civil Engineering, 12, 288-293.
https://doi.org/10.1061/(ASCE)0899-1561(2000)12:4(288) - 8. Wilson, S.A., Dipple, G.M., Power, I.M., Thom, J.M., Anderson, R.G., Raudsep, M., Gabites, J.E. and Southam, G. (2009) Carbon Dioxide Fixation within Mine Wastes of Ultramafic-Hosted Ore Deposits: Examples from the Clinton Creek and Cassiar Chrysotile Deposits, Canada. Economic Geology, 104, 95-112.
https://doi.org/10.2113/gsecongeo.104.1.95 - 9. Montes-Hernandez, G., Perez-Lopez, R., Renard, F., Nieto, J.M. and Charlet, L. (2009) Mineral Sequestration of CO2 by Aqueous Carbonation of Coal Combustion Fly-Ash. Journal of Hazardous Materials, 161, 1347-1354.
https://doi.org/10.1016/j.jhazmat.2008.04.104 - 10. Metz, B., Davidson, O., Coninck, H., Loos, M. and Meyer, L. (2005) Carbon Dioxide Capture and Storage. Working Group III of the Intergovernmental Panel on Climate Change, the United States of America by Cambridge University Press, New York.
- 11. Olajire, A.A. (2013) A Review of Mineral Carbonation Technology in Sequestration of CO2. Journal of Petroleum Science and Engineering, 109, 364-392.
https://doi.org/10.1016/j.petrol.2013.03.013 - 12. MacDowel, N., Florin, N., Buchard, A., Hallett, J., Galindo, A., Jackson, G., Adjiman, C.S., Williams, C.K., Shah, N. and Fennell, P. (2010) An Overview of CO2 Capture Technologies. Energy & Environmental Science, 3, 1645-1669.
https://doi.org/10.1039/c004106h - 13. Gerdemann, S.J., O’Connor, W.K., Dahlin, D.C., Penner, L.R. and Rush, H. (2007) Ex Situ Aqueous Mineral Carbonation. Environmental Science & Technology, 41, 2587-2593.
https://doi.org/10.1021/es0619253 - 14. Park, A.A., Jadhav, R. and Fan, L. (2003) CO2 Mineral Sequestration: Chemically Enhanced Aqueous Carbonation of Serpentine. The Canadian Journal of Chemical Engineering, 81, 885-890.
https://doi.org/10.1002/cjce.5450810373 - 15. Kakizawa, M., Yamasaki, A. and Yanagisawa, Y. (2001) A New CO2 Disposal Process via Artificial Weathering of Calcium Silicate Accelerated by Acetic Acid. Energy, 26, 341-354.
https://doi.org/10.1016/S0360-5442(01)00005-6 - 16. Teir, S., Eloneva, S., Fogelholm, C. and Zevenhoven, R. (2007) Dissolution of Steelmaking Slags in Acetic Acid for Precipitated Calcium Carbonate Production. Energy, 32, 528-539.
https://doi.org/10.1016/j.energy.2006.06.023 - 17. Kodama, S., Nishimoto, T., Yamamoto, N., Yogo, K. and Yamada, K. (2008) Development of a New pH-Swing CO2 Mineralization Process with a Recyclable Reaction Solution. Energy, 33, 776-784.
https://doi.org/10.1016/j.energy.2008.01.005 - 18. Kim, K. (2010) A Study on the Thermally Assisted Liberation Behavior of Aggregates by Free-Fall Impact and Autogenous Milling. PhD in Engineering, Seoul National University, Seoul.
- 19. Bobicki, E.R., Liu, Q., Xu, Z. and Zeng, H. (2012) Carbon Capture and Storage Using Alkaline Industrial Wastes. Progress in Energy and Combustion Science, 38, 302-320.
https://doi.org/10.1016/j.pecs.2011.11.002 - 20. Fuchigami, K., Taguchi, Y. and Tanaka, M. (2009) Synthesis of Calcium Carbonate Vaterite Crystals and Their Effect on Stabilization of Suspension Polymerization of MMA. Advanced Powder Technology, 20, 74-79.
https://doi.org/10.1016/j.apt.2008.10.003 - 21. Hadiko, G., Han, Y., Fuji, M. and Takahashi, M. (2005) Synthesis of Hollow Calcium Carbonate Particles by the Bubble Templating Method. Materials Letters, 59, 2519-2522.
https://doi.org/10.1016/j.matlet.2005.03.036 - 22. Hu, Y., Gao, Z., Sun, W. and Liu, X. (2012) Anisotropic Surface Energies and Adsorption Behaviors of Scheelite Crystal. Colloids and Surfaces A, 415, 439-448.
https://doi.org/10.1016/j.colsurfa.2012.09.038 - 23. Parkhurst, D.L. and Appelo, C.A.J. (2013) Description of Input and Examples for PHREEQC Version 3—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. U.S. Geological Survey Techniques and Methods, Book 6, Chapter A43, 497.
- 24. Pane, I. and Hansen, W. (2005) Investigation of Blended Cement Hydration by Isothermal Calorimetry and Thermal Analysis. Cement and Concrete Research, 35, 1155-1164.
https://doi.org/10.1016/j.cemconres.2004.10.027 - 25. Schiopu, N., Tiruta-Barn, L., Jayr, E., Méhu, J. and Moszkowicz, P. (2009) Modelling and Simulation of Concrete Leaching under Outdoor Exposure Conditions. Science of the Total Environment, 407, 1613-1630.
https://doi.org/10.1016/j.scitotenv.2008.11.027 - 26. Lothenbach, B., Matschei, T., Moschner, G. and Glasser, F.P. (2008) Thermodynamic Modelling of the Effect of Temperature on the Hydration and Porosity of Portland Cement. Cement and Concrete Research, 38, 1-18.
https://doi.org/10.1016/j.cemconres.2007.08.017 - 27. Brecevic, L. and Kralj, D. (2007) On Calcium Carbonates: From Fundamental Research to Application. Croatica Chemica Acta, 80, 457-484.
- 28. Dickinson, S.R., Henderson, G.E. and McGrath, K.M. (2002) Controlling the Kinetic versus Thermodynamic Crystallization of Calcium Carbonate. Journal of Crystal Growth, 244, 369-378.
https://doi.org/10.1016/S0022-0248(02)01700-1