American Journal of Analytical Chemistry
Vol.07 No.01(2016), Article ID:62989,17 pages
10.4236/ajac.2016.71008
Overview of the Analytical Lifecycle of Supercritical Fluid Chromatography Methods
Bertyl Andri, Amandine Dispas, Roland Djang’eing’a Marini, Philippe Hubert
Department of Pharmacy, CIRM, Laboratory of Analytical Chemistry, University of Liege (ULg), Liege, Belgium

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 28 November 2015; accepted 22 January 2016; published 25 January 2016
ABSTRACT
In recent times, the overall interest over Supercritical Fluid Chromatography (SFC) is truly growing within various domains but especially for pharmaceutical analysis. However, in the best of our knowledge modern SFC is not yet applied for drug quality control in the daily routine framework. Among the numerous reported SFC methods, none of them could be found to fully satisfy to all steps of the analytical method lifecycle. Thereby, the present contribution aims to provide an overview of the current and past achievements related to SFC techniques, with a targeted attention to this lifecycle and its successive steps. The included discussions were therefore structured accordingly and emphasizing the analytical method lifecycle in accord with the International Conference on Harmonisation (ICH). Recent and important scientific outputs in the field of analytical SFC, as well as instrumental evolution, qualification strategies, method development methodologies and discussions on the topic of method validation are reviewed.
Keywords:
Supercritical Fluid Chromatography, Ultra High Performance Supercritical Fluid Chromatography, Analytical Method Lifecycle, Quality by Design

1. Introduction
The use of supercritical fluid as mobile phase was firstly suggested by Lovelock in 1958 to extend applications of gas chromatography to solid or/and ionic compounds [1] . In 1962, while working on the separation of porphyrins, Klesper, Corwin and Turner made the first demonstration of Dense Gas Chromatography which is nowadays known as Supercritical Fluid Chromatography (SFC) [2] [3] . Since that, this technique faced a quite tortuous existence, mostly overshadowed by HPLC success [4] . Indeed, a few years ago, SFC instruments were still suffering from drawbacks such as lack of robustness, poor UV sensitivity and inadequate dynamic range [5] that didn’t allow the application of SFC in some areas of interest like e.g. medicines Quality Control (QC). In this context, sensitivity and reproducibility were sought by the instruments manufacturers to design more performing SFC instrument. This finally led to the development and marketing of modern SFC instruments in the 2010’s [6] . Such instruments are addressing the past issues and enable analysis in the Ultra High Performance SFC range (UHPSFC) while operated with sub-2 µm particles packed columns [7] [8] . In addition, these modern instruments allow benefiting from the valuable advantages of the SFC: 1) orthogonal selectivity with liquid chromatography, 2) high chromatographic efficiency, 3) high throughput, 4) compatibility with a large range of detectors, 5) short equilibration time, 6) low cost of analysis per sample, 7) low solvent consumption and, 8) low waste generation [9] . Moreover, this technique is recognized and well placed as an alternative to LC [6] [10] -[14] with a particular advantage to be a useful tool in the framework of green analytical chemistry [4] . However, it appears that the overall greenness is not the sole argument leading to the adoption of SFC [15] . In fact, the interest is mostly economic and goes along with the drastic reduction of waste for which handling and treatment are expensive [16] . In this case, greenness as safe environment contribution is associated with cheapness and both are part of sustainability that takes into account the economic aspects of techniques or processes [17] .
Given all these progress and advantages, a resurgence of interest was devoted to SFC in the last decade. Indeed, its applications are expanding inside several domains and especially in the pharmaceutical field where numerous researches were published. Thus, SFC was demonstrated to be useful for drug analysis [18] -[29] , drug discovery [30] -[35] , impurity profiling [26] [27] [31] [36] -[39] , quantitative applications [40] , bioanalysis [41] - [48] , metabolic/metabolomics studies [49] -[55] , chiral separations [56] [57] , drug screening and preparative purposes at various stages [58] -[60] . However, the interest of SFC is not limited to pharmaceutical applications, other fields of application being polymer sciences [61] [62] , petroleum industry [63] -[65] , food analyses [66] - [69] , and for various industrial processes [70] -[72] .
To now, despite this wide range of applications, it appears that the description of the whole lifecycle of a SFC method was never published yet. The analytical method lifecycle encompasses the successive steps that analytical methods have to follow from their development/onset to their routine use. Given that, it is really interesting to notice that only few steps of this cycle are successfully achieved for most of the papers related to SFC, which somehow may suggest only a very limited routine use of SFC. However, this statement has to be balanced with the novelty of the modern SFC instruments and the restrictions of confidentiality in industrial frameworks [73] .
This review focuses on the analytical method lifecycle with an attention to pharmaceutical achiral applications. Considering that routine use is suggested but not reported, method development and method validation will be mainly highlighted. By trying to emphasize the key question “is modern SFC ready to pass through the whole analytical procedure lifecycle?”, the present contribution aims reviewing the literature in order to overview the SFC implementation in the framework of pharmaceutical routine analysis. The discussions will rely on the method lifecycle steps as shown in the Figure 1, mainly through the ones leading to potential routine use of SFC methods although it is still not described.
2. Prior Knowledge in SFC
2.1. 30 Years of Global Literature Overview
The SFC scientific interest growth is correlated with an increasing number of publications as can be seen in the Figure 2(a) that provides an overview of the yearly issued papers. Since 2012, the number of publications increased to more than 120 papers per year against circa 80 for the previous decade (2002-2011). Over a 30 years period, the cumulative numbers of SFC papers also shows a steady rate of progression (Figure 2(b)). However, in the recent years, the rate/slope of progression of SFC literature increased a little, somehow stressing the resurgence of interest devoted to the supercritical fluid chromatography. Indeed, on the stacked plot, a larger black portion corresponding to the yearly issued papers is observed for the last four years.
2.2. Particular Literature Overview
Some important reviews of the field were published by Taylor in 2008 [58] , in 2009 [59] and in 2010 [74] . In
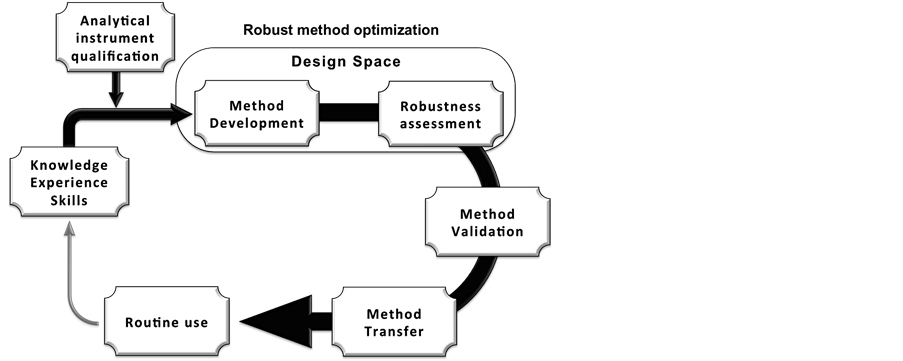
Figure 1. Simplified analytical procedure lifecycle, proposed in accord to ICH requirements of Quality by Design applied into the pharmaceutical industry [74] .

Figure 2. (a) Cumulative number of issued papers yielded by the exact search term “supercritical fluid chromatography”, inside Scopus© (25th November 2015); (b) Annual progress of SFC’s literature over a 30 years period.
these works, the author provides a yearly overview of the literature dedicated to SFC, focusing on various topics such as analytical scale SFC for both chiral and achiral, hyphenation with mass-spectrometry, instrumental improvements, trends in method development and preparative SFC. In 2012, he also reviewed the different papers dealing with the use of water-rich modifiers in SFC [75] .
In 2011, Guiochon et al. proposed an interesting review dedicated to preparative SFC [60] with more than 500 references that allowed covering virtually any aspects of the SFC. Indeed, their article discusses many empirical and theoretical features of the SFC at both analytical and preparative scale. Very recently, Lesellier & West also reviewed the field of SFC, giving critical insights on the characteristics of the achiral analytical SFC as well as its broad applicability [9] . They also points out several theoretical features such as the condition of the fluid, the dimensions and the properties of the stationary phases, the detectors, the method development… as well as their consequences and their interests in modern SFC.
Kalikova et al. published a review dedicated to chiral analysis in SFC and focused on the papers issued from January 2000 to August 2013 [56] . The authors proposed a summary of the different SFC methods employed (including columns and analytes) for both preparative and analytical chiral SFC. They also discussed the potential mechanism leading to some chiral selectivity. Nováková et al. recently presented a tutorial providing a guideline and the explanations for the choice and the use of modern UHPSFC equipment [6] . This work based on current knowledge allowed examining many specific features of the nowadays SFC among which can be cited organic modifier composition, the injection solvent composition, column sizes and the analysis of different kind of compounds. Desfontaine et al. recently discussed the experimental conditions and the benefits related to the application of modern SFC in the pharmaceutical field [73] . Some special attention was also dedicated to the analysis of formulations and biological materials issued from the pharmaceutical field.
Another review in the field of SFC published by Grand-Guillaume Perrenoud et al. is focused on the introduction of sub-2 µm particles packed columns to SFC [76] . The different thermodynamic aspects and kinetic benefits are discussed as well as some recent applications specifically achieved in the Ultra High performance range (UHPSFC). The different instruments able to perform in the UHPSFC performance range are also reviewed in the same way. The authors also proposed a comprehensive summary of the current knowledge and comparisons of LC, SFC, UHPLC and UHSPFC.
3. Analytical Instrument Qualification in SFC
3.1. Scientific and Regulatory Context
The analytical instrument’s qualification is a mandatory assessment of the proper working order of the equipment. It typically involves three levels: 1) Installation Qualification (IQ), 2) Operational Qualification (OQ) and 3) Performance Qualification (PQ), which assess, document and report the adequacy of the instrument for its intended use [77] .
This has a particular importance in the field of pharmaceutical industry, which is among the most regulated industries in the world due to public health issues. Thus, to guarantee the pharmaceutical quality control (QC), several regulatory authorities provide guidelines to maintain and enhance the overall quality of the products [78] . Prior to any analytical test, the instrument qualification becomes a mandatory step required by these guidelines in order to rely on the analytical data generated. This is even emphasized by the USP [79] , which considers that a qualified instrument is the cornerstone of the components of data quality. Indeed, to obtain reliable, reproducible and valuable data, it is necessary to use properly the correct instrument but also to ensure that its performances are achieved through qualification while demonstrated them by mean of System Suitability Tests (SSTs) on a regular basis. Such requirements are to be done into scientific fields to ensure the reproducibility of data provided by the instruments and potential method transfer.
3.2. The Current Case of SFC
In the framework of SFC, several documents are reporting the qualification of instruments for highly regulated industrial environments. Even if most of these documents have been published for years ago, they are still useful and may provide helpful bases for current SFC systems. Anton & Siffrin [80] demonstrated that three packed columns SFC systems were tools with suitable performances for drug product and drug substances analysis, meeting the 15 years ago “cGMP” (current Good Manufacturing Practices) and ICH (International Conference on Harmonisation) specifications. The authors also presented general qualification protocols and tables suggesting specifications to meet for method validation according to ICH criteria (LOD, LOQ, Precision, Linearity…). The different instruments used to verify those specifications (flow meter, pressure gauge, temperature sensor…) are also reviewed in the same way. Another contribution was the observation report made on the high compressibility and the differences in flow rate measured on the instruments, which are able to affect the reproducibility of performances while transferring methods.
In 2011, Sun reported the qualification of SFC instruments for the pharmaceutical product development [81] , demonstrating the ability of SFC to meet nowadays regulatory requirements. Moreover, as LC inspires modern SFC, it is not surprising to notice that several protocols presented in that paper are derived from HPLC qualification, e.g. UV detector calibration, injector qualification, temperature monitoring, gradient composition verification…. Huber also authored a primer providing guidelines for the qualification of SFC equipment, in accord to current regulations (GxP, ISO, FDA, USP…) [82] . The documents aforementioned provide helpful instructions to qualify SFC instruments, demonstrating the ability of any modern packed SFC instrument as a potential tool suitable for highly regulated environments. Hicks et al. recently updated this work, assessing the performances of Waters UPC2 and Agilent 1260-Aurora A5 fusion systems for chiral analysis under GMP industrial environments [83] . However, unlike previous works, they didn’t propose any specific procedures to qualify/monitor performances of nowadays (UHP) SFC systems.
3.3. Flow Rate Measurement in SFC
Following the above discussion about SFC instrument qualification for GMP purposes, Sun reported another way to evaluate the SFC flow rate [81] . Indeed, due to its nature (compressible, dense, not viscous), SFC mobile phase flow rate may render more difficult to measure and thus to qualify compared to classical LC. So, the authors replaced the column by a calibrated sample loop (1 or 5 ml depending on the flow rate), injected acetone and performed UV detection. The flow rate is then calculated after the data are acquired. The interest of such approach stands in its overall easiness of implementation and the absence of a possible instrumental modification. However its limitation is the impossibility to measure the true condition of the flow rate while performing an analysis. In this regard, the workgroup of Tarafder & Guiochon [84] highlighted the importance to change the flow rates mobile phase measurement and expression. They suggested expressing the flow rate as a mass flow rate instead of volumetric flow rate because it remains almost constant in any part of the SFC system, while volumetric flow rate is subject to variation along the channels of the apparatus. Fluctuations of these parameters are mainly explained by high compressibility of the fluid but also, regarding volumetric flow rate, by room temperature and instrument’s design. Therefore, the same workgroup issued a second paper [85] always on the same topic reporting the adaptation of a Coriolis Flow Meter (CFM) to their SFC instruments (TharSFC, Waters UPC2 and Jasco). This adaptation of SFC instrumentation [80] provided on-line accurate measurement of mass flow rate. However, the maximal operating pressure of the device (150 bars) imposed the installation of the flow meter before the pump, measuring the CO2 consumption. Consequently, the signal recorded was very noisy due to the reciprocation of the pump’s piston. Thus, the signal had to be averaged and the flow rate measured was found to be lower than the one reported by the instrument’s software.
This observation was also made by Enmark et al. [86] while using the same kind of CFM to monitor the mobile phase flow, thus corroborating the statement of Tarafder et al. Although these results are coherent, the implementation of a pulsation damper between the flow meter and the pump could be required to avoid noisy signal and data treatment. However, this flow meter should be verified through qualification especially to evaluate the potential bias induced by the pulsations of the pump or leaks in the various connections.
Moreover, in GMP frameworks, re-qualification (IQ, OQ, PQ) of the instrument must be performed after such modification of the instrument.
4. Method Development
4.1. Quality by Testing and Quality by Design
Method development takes place at the onset of the lifecycle of analytical methods. During this step, analysts have to answer to a given analytical problem (e.g. separation of two compounds, identification of compounds…), while dealing with constraints and requirements (e.g. matrix effect and composition, runtime reduction, sample degradation, limit of detection...).
Nowadays, two major trends are applied to methods development in analytical sciences: Quality by Testing (QbT) and the recently introduced Quality by Design (QbD) methodology. These two strategies have opposite approaches of the analytical problem, being respectively univariate and multivariate strategies. Indeed, the QbT is the common “trial and errors” methodology that was employed by almost all the analysts for years as confirmed by numerous published examples of SFC methods. This terminology was introduced in order to reference what was not QbD which strategy focuses on the quality of the final product since the onset of its development, using chemometric tools and taking into account risk management.
4.2. QbT Methodology and SFC
In the context of QbT approach, a set of “standard” starting conditions is usually employed for SFC method development. For instance, most back-pressure regulators are typically set to 130 - 150 bars, while column temperature is usually set to 40˚C [6] [9] . Obviously, these values are set to match the supercritical parameters of neat carbon dioxide (73.8 bars and 31.1˚C) and lower compressibility of the fluid. In addition, the other usually screened method parameters are the column chemistry, flow rate, density (pressure and temperature), composition of the mobile phase (organic modifier and/or additives) and the elution mode (isocratic or gradient mode). Afterwards, these factors are individually tested targeting an achievement during the method development, which may reveal itself tedious due to the interdependence of parameters (pressure, temperature, mobile phase composition…). Generally, the experiments stopped when the observed chromatograms are satisfying (e.g. an adequate separation of the peaks, an acceptable run time, a correct peak shape...). Unfortunately, in this case only a limited knowledge is gained on the method, making difficult to exactly know any possible failures or potential improvements of the analytical method. Indeed, small modifications to parameters may greatly affect the quality of the results. So, with univariate methodology, a satisfactory compromise may be hard to really set up, potentially leading to lengthy method developments. In addition, robustness of the method has to be assessed at the end of the development process and prior method validation, thus even requiring additional experiments.
On the contrary, the QbD strategy offers an interesting improvement over the traditional QbT approach since the knowledge gained during the experiments process allows predicting optimal parameters of the analytical method. Moreover, the confidence in observed responses is greater and the overall number of experiments may be lowered including both development and robustness. However, since the implementation of the traditional QbT strategy is much easier/intuitive than the multivariate QbD methodology, it is still widely employed for the scouting of experimental parameters at the origin of method development. For instance, Alexander et al. used a supplementary quaternary HPLC pump to directly generate different mobile phase modifiers [24] . This pump was located upstream of the binary SFC pump, which was then fed with a large panel of modifiers (binary and ternary mixtures of methanol and additives). This implementation allowed higher flexibility, accuracy and productivity while scouting for SFC organic modifiers.
In the same way Xia et al. went one step further by developing an autoblender for their UHPSFC equipment [87] . This team adapted a computer controlled multi-position proportioning valve on the modifier pump. This device allowed them to easily obtain mixtures of modifiers and additives, overcoming the issues (mainly time and solvent consumption) related to the binary design (a pump for carbon dioxide and B pump for one pre- mixed blend of solvent at a time) of SFC pumps.
4.3. QbD Methodology and SFC
As described above, the other strategy applied to the analytical method development is the Quality by Design. The Food and Drug Administration (FDA) introduced this strategy for the development and manufacturing of new pharmaceutical product. This evolution came from the recognition that testing final products is not sufficient to raise its quality [88] . Upon this, the International Conference on Harmonization (ICH) has soon adopted the QbD for the pharmaceutical industry. In this regard, Borman et al. reported the analogies between manufacturing process and analytical method, which enhance the use of a QbD approach for analytical method development [89] .
The QbD is defined by ICH as “A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management” [90] . Thus, QbD enables the establishment of a Design Space (DS), which for analytical method is defined as “The multidimensional space which includes any combination of the variables that have been demonstrated to provide assurance of quality of the data produced by the method” [91] . According to its definition, the DS can be represented in Figure 3 as a subspace of the experimental domain in which the quality of the method has been proved with a defined probability, i.e. a zone of robustness [90] [91] . In this way, ICH affirms: “working within the design space is not considered as a change to the procedure” [90] . According to this assertion, the experiments performed outside of the DS may be likely to provide significant changes in the overall quality offered by the method. In accord with several authors, Rozet et al. suggested the appellation of Method Operable Design Region (MODR), while referring to DS. This suggests that MODR is in close relationship with the analytical procedure in light with its definition that is “multivariate range of values of operating parameters where the analytical method provides quality outputs with adequate probability [92] ”. Notice that operating parameters also called operating factors may be controllable analytically.
Given the multivariate considerations of the DS/MODR, its construction benefits from experimental data obtained from the use of the Design of Experiments (DoE). The application of DoE in analytical chemistry is a rather known practice and is associated with significant benefits, allowing great understanding of the procedure from a minimum of experiments [93] . Indeed, the use of the DoE allows the identification and characterization of the key parameters ruling the method investigated. These parameters are the factors that impact the most on the separation quality. In addition, if uncertainty related to the process is taken into account when performing the DoE, this allows robust optimization of the method within the same step [94] . So, thanks to robust optimization, further robustness assessment is no longer required. Thereby, this strategy allows significant savings in time and resources. Another cornerstone of the QbD approach is the consideration of the Critical Quality Attributes (CQAs) that are defined by ICH as “physical, chemical, biological or microbiological property or characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality” [90] . In chromatography, a CQA might be the separation between two peaks often of interest [95] , the analysis time [96] , a peak efficiency… These attributes are representatives of quality and must satisfy to the corresponding acceptance limits previously defined. Ideally, these CQAs are representative of the multiple conditions being included in the DS so as to be characterized as robust CQAs [97] .

Figure 3. Probability surfaces for e.g. the separation of two peaks forming a critical pair. The Design Space is located in the white zone on the upper left of the map. It may be observed that using a back-pressure and a temperature of 135 bars and 55˚C respectively, it exists a quality level of at least 82% to e.g. separate the two peaks, meaning that a mobile phase of lower density favours this separation.
It is also useful to know that the conversion of QbT methodology to QbD philosophy is possible. It obviously involves some experiments but still lesser than a complete QbD development. In this case, the older analytical method may serve as a basic starting point for a new method development/optimization. Thereby, it doesn’t cost much effort to convert methods from QbT to QbD. Even more if the benefits associated to QbD are considered. This case was recently reported and discussed by Hubert et al. in the framework of HPLC [95] and could be applicable to the case of SFC.
Concerning the field of SFC, to the best of our knowledge, it appears that only Dispas and co-workers reported the development of SFC method in agreement with ICH Q8 (R2) guidelines [90] . They demonstrated its ability to separate very polar compounds within only 66 experiments [96] while including both the experimental parameters screening and the optimization designs of the method. In addition, such an achievement is a proof of the relevance of the QbD in the framework of SFC, especially since the separation of very polar compounds is often considered as a “worst case” for SFC [75] . In a recent article, the same work team demonstrated the transfer of a robust SFC method between two laboratories and two different brands SFC instruments [94] . It appeared that the transfer between two SFC instruments that had different designs and two columns with different particles sizes was possible without significant changes in terms of selectivity. These successful outcomes pointed out the robustness of the method in one hand, and also emphasized the benefits provided by robust method optimization in another hand. In SFC, these two aspects are even more important because the instrument design and the particle size are recognized to greatly impact on the density of the fluid, which may lead to changes in retention and/or selectivity. By focusing on these two aspects, this recent work highlighted that robust method optimization is a really helpful strategy applicable to SFC. Furthermore, while keeping in mind the analytical method lifecycle, the following benefits appears as really valuable to speed up the cycle leading to routine application: enhanced knowledge of the method and eased transfer of the method between laboratories…
4.4. Method Development and SFC
Nowadays, great interests are devoted to analytical SFC method developments. Indeed, the development of the analytical method is an important step, which may require significant amount of time and condition the method’s life. So, it is worth to mention several examples of development and/or optimization using DoE and response surface methodology without considering QbT and QbD as such [12] [94] [96] [98] -[102] . In this way, these papers report the use of factorial (full or fractional) and Box-Behnken experimental designs in SFC. These designs allowed development of the analytical methods, the optimization of hyphenation of SFC to MS detector or the study of the impact of experimental parameters.
Recently, several authors suggested different ways to perform the SFC method development. For example generic method development was proposed by De Klerck et al. to fasten the method development of (UHP) SFC chiral analysis [57] , Delahaye et al. reported the application of stationary phase optimized selectivity predictions for isocratic SFC separations [103] , by demonstrating the applicability of this tool to predict the retention of solutes on different columns under isopycnic conditions.
We can mention that De Pauw et al. also theoretically investigated the benefits of constant pressure against constant flow rate gradient elution [104] . So, they developed a model of the density of the fluid during gradient elution. Simulating constant pressure analysis, this model predicted up to 40% of time saving while compared to constant flow gradient elution. This observation is mainly due to higher mobile phase velocity at the onset of the gradient when low modifier concentrations are used. So, at that moment, the reduced viscosity of the fluid requires the use of higher flow rates to maintain constant pressure in the system. This approach, derived from latest developments in HPLC, is promising faster SFC analysis but the selectivity may be affected by the higher constant density of the fluid. So, constant pressure gradient elution must be difficult to apply to compounds/se- paration impacted by the mobile phase density (e.g. apolar compounds on silica based stationary phase).
5. Method Validation
5.1. Method Validation in the Field of SFC
As a result of the past technical issues of SFC, a review of the literature revealed that only a limited number of papers dealing with method validation. It also appears that most of the nowadays applications of SFC are essentially qualitative or preparative and only a few are quantitative [60] [105] [106] . It seems that SFC methods are easily scaled-up from analytical to preparative scale [107] -[109] . So, in this case, it would be reasonable to consider that a validation step is not mandatory for preparative purposes.
Many efforts were devoted to improve robustness and sensitivity of the SFC equipments [4] [27] [42] [110] [111] . As a result, very low quantification levels were reached for example 0.05% of the active ingredient― matching ICH requirements―for traces analysis of organic impurities. However, taking into account its sensitivity, some authors described a lower sensibility in SFC-UV while compared to HPLC/UHPLC [36] [40] and recommend the use of more concentrated samples [38] . This suggestion may be actually impossible to apply if products are available in limited quantities (e.g. some specified impurities) and/or poorly soluble in the injection solvents. So, in the case of SFC, the observation of only a few validated methods is not a real surprise. However, the situation is changing and quantitative capabilities of the technique are challenged in the various areas where SFC is currently applied [10] [13] [23] [36] [40] [47] [48] [105] [106] [112] -[114] .
5.2. Validated SFC Methods
In the field of SFC, as well as in other analytical fields, it appears that some works reported results without actual conclusions on the method’s validity while dealing with methods validation. Furthermore, only limited discussion was proposed, stressing the current lack of consensus regarding acceptance criteria [40] .
Desai et al. reported the validation of an SFC method on drug tablets. However, concerns should be made about the relative standard deviation (RSD) values obtained between intra and inter-days precision for some concentration levels, intra-series values being higher than inter-series ones. On another note, the authors showed, by means of a Student t-test, that there was no significant difference between the results obtained with the SFC and the HPLC method. In such manner, they concluded on the adequacy of the SFC for the quality control of marketed drugs. Méjean et al. also reported the validation of the quantification of retinoid compounds [114] . However in this case, the reproducibility was considered to be satisfying by the authors, in spite of quite high RSD values. This is even more concerning because the RSD is generally around 5%, while the specification for a finished drug product lies within 95% - 105% of the API. So, it should be difficult to use this method to adequately appreciate the quality of the pharmaceutical product. In the case of retinoid, the authors mentioned that sensitivity wasn't an issue, which in the framework of SFC-UV, is a rare statement. This is although not surprising given the chemical structure of the compounds, which provides high UV absorption.
Alexander et al. also described a method for the impurity profiling of pharmaceutical compounds [27] . Even if validation was not the purpose of the work, some of the validation criteria were assessed in order to prove the ability of that method to quantify impurities. This is compliant with the ICH Q2 (R1) definition of method validation that stipulates, “The objective of validation of an analytical procedure is to demonstrate that it is suitable for its intended purpose”. However, considering some of the results published, the RSD values associated to the area of some impurities were found somewhat quite high (>10%) but acceptable for drug substance assay [80] . This observation may be related to the small amount of the impurity (<0.1% of the active) or the difficulty to totally separate some impurities.
Though the early situation mentioned in paragraph 5.1, the first “SFC-Validation paper”, published in 2001 [115] , highlights the robustness issue. Indeed, triplicate injection of the same solution yielded high fluctuation of peak area across two series. The worst case is a RSD value going from 0.3% to 6.67% during the second series. Regarding these figure cases, it is obvious that the error was too high to allow drug analysis in a daily routine framework. A posteriori, a significant part of the problem is supposed to be related to the injection sequence [5] [116] . In this case, the use of an internal standard, in a GC-like fashion, should have lowered the error generated during the injection.
On another note, some methods determining chiral compounds were also subjected to limited validation. However, in the case of chiral compounds, it often appears that the validation targeted some molecules or the assessment of some validation criteria. In this regard, Xiang et al. reported the chiral separation of 9 amide drugs [117] . In spite of that, the validation was performed for scarcely one of those compounds. Moreover, only the linearity, reproducibility and Limit of Quantification (LOQ) were assessed. In the same way, Wang et al. only described and discussed the orthogonality of the SFC method in comparison to current RP-LC method [36] . For that purpose, selectivity of the method was the main criteria considered in the work. Some other validation criteria were claimed as assessed but unfortunately they were reported with limited details. Marley et al. recently reported a analytical method for the determination of R-timolol in pharmaceutical forms [10] . The method precision was evaluated at repeatability and intermediate precision levels with 3 different standards solutions daily upon two trial days, each solution being injected and measured six times to comply with ICH Q2 (R1) guideline. Also according to that guideline, accuracy was assessed on the basis of 6 determinations at 100% of the test concentration (1.0% of R-timolol regarding to S-timolol) in presence of S-timolol at the same concentration (1.0% too) as bracketing standard. This allowed the validation of the quantitative impurity assay, meeting the specifications described by the European Pharmacopeia (max 1.0% of R-timolol in the presence of S-timolol). In the same way, Mukherjee also reported the chiral separation of a compound that is still under its early development step [118] . The author assessed several validation criteria in harmony to ICH and FDA guidelines. However, for intra-days repeatability assay, high RSD values of 9% were reported for some concentration levels. Moreover, improper evaluation of inter-days accuracy is stated (mean value of intra-days values…).
Recently, Nováková et al. reported the assessment of LOD, LOQ and linearity while screening for doping agent in UHPSFC-MS/MS [47] [48] . They also described quantitative analysis and method validation of topical formulations containing oestrogens. Indeed, the authors described the validation criteria according to ICH Q2 (R1) guideline, also suggesting routine use of their method for the determination of oestrogen steroids in real samples. Ganzera also reported the use of UHPSFC for the determination of isoflavone in dietary supplements [106] . The performances of the method were also evaluated according to ICH guidelines. Though the author didn’t present much of the development output, its method shows interesting features (accuracy, inter & intra-day precision…) and is able to separate isoflavones from different vegetal matrix such as herbal products.
5.3. Application of the Total Error Approach to SFC Methods
Recently, Dispas et al. published a paper reporting fully validated UHPSFC and UHPLC methods for the analysis of amoxicillin powder in capsules [40] . They also demonstrated the potential of UHPSFC as a reproducible and quantitative technique, in contrast with outcomes in some reports [119] . Moreover, Dispas et al. showed the interests of applying robust optimization strategy and total error approach for method validation [120] -[123] , which consequently allowed them to rapidly develop and safely validate the UHPLC and UHSFC methods. Obviously, from an industrial perspective (where high throughput is important) this demonstration is highly valuable.
6. Outcomes and Perspectives
An increasing number of publications and communications highlight the current renaissance of SFC as can be retained from the novel SFC fundamental works and the applicative demonstrations currently found in the nowadays-scientific literatures.
In the present contribution, SFC papers were reviewed while keeping in mind the lifecycle of an analytical method. Thereby many evidences highlighted the capabilities of the SFC to support various steps of this lifecycle. In this regard, the SFC proved being able to fulfill the current pharmaceutical requirements such as European Medicine Agency (EMA), GxP and ICH. Moreover, it appeared that few documents suggesting procedures and specifications for the qualification of the SFC instruments were readily available in the literature.
Concerning the question of analytical method development, it seems that the empirical QbT approach is still predominantly used to find suitable analytical conditions of a method. However, the QbD approach was recently demonstrated to be successful and helpful for the development of analytical method (SFC method included), providing real advantages over the empirical methodologies. Recently, several examples of method development by means of DoE were also described. Though it’s not the most common strategy, this observation marks a clear evolution in the way that chromatographic methods are developed and optimized.
Along with the instrumental improvements, the quantification of compounds by means of the SFC is undergoing a growing interest among the analysts. In this regard, the modern SFC proved to be a quantitative instrument suitable for the highly regulated environments of the pharmaceutical industry. Thus, it is not surprising to observe an increasing number of reports related to its quantitative capabilities. Considering this, the observation of partial method validation (e.g. assessing only a few criteria), inconsistent results (e.g. high RSD values) and improper manipulations (e.g. non independent validation standards) are lowering the quality of the validated method. In this sense, many efforts should be done to render the results presented more consistent and reliable. So, the regulatory authorities should also propose acceptance guidelines of the already established validation criteria. Indeed, if no references are defined, anyone may appreciate on its own results of the validation. Moreover, there is no official basis for a discussion on the results and performances yielded by the method. In this sense, the method validation based on total error and accuracy profile as a decision tool is a valuable approach. Indeed, it allows the assessment and the conclusion of the validity (or not) of the analytical method, thus providing a useful basis for further discussions.
Concerning the robustness testing, it appears that only a few examples are available from the writings [124] . This may be related to the global lack of robustness previously associated to the technique. However, given the fact that improved instruments are available in the market, it would be really interesting to update these scientific studies.
To the best of our knowledge, the review of the SFC documents also pointed out that the transfer and routine use of an analytical SFC method are not yet reported in the scientific literature. Hopefully, thanks to modern instrument and the fact that SFC is commonly agreed as a good alternative of the Normal Phase HPLC (NP-HPLC) [10] [27] , the figure may change soon. Indeed, due to the advantages of SFC over the NP-HPLC, it is reasonable to think that some analytical applications are on the way of being switched from NP-HPLC to SFC. In addition, the SFC was compared several times with other techniques such as RP-HPLC or UHPLC [27] [29] [40] , being considered as references in the pharmaceutical industry. It thereby appears that the SFC, although a bit less versatile and sensitive than the latter [8] [18] [29] , didn’t exclude from these comparison.
From another point of view, the SFC is to date included in the framework of green analytical chemistry [4] . Considering this and the amount of works dedicated to the technique, it wouldn’t be objective to present SFC only as a “green analytical tool”. Indeed, SFC is now considered as a truly mature technique, which is complementary to gas chromatography and liquid chromatography [19] [32] [51] [58] [60] [107] .
The review of the scientific literature permitted to assess the situation of the SFC. It was pointed out that still significant works remain to be done [9] . In addition, this review permitted to evidence several achievements already made as well as some current trends. Thus at the moment, it is reasonable to claim that an analytical SFC method has never ever been closer to accomplish the complete lifecycle of analytical method.
Acknowledgements
Research grants from the Walloon Region: Project First International (Convention In-SCrit, no. 1217860) to B. Andri are gratefully acknowledged.
Cite this paper
BertylAndri,AmandineDispas,Roland Djang’eing’aMarini,PhilippeHubert, (2016) Overview of the Analytical Lifecycle of Supercritical Fluid Chromatography Methods. American Journal of Analytical Chemistry,07,75-91. doi: 10.4236/ajac.2016.71008
References
- 1. King, J.W. (1990) Introduction: Historical Development of SFC. In: Lee, M.L. and Markides, K.E., Eds., Analytical Supercritical Fluid Chromatography and Extraction, Chromatography Conferences, Inc., Provo, Utah, USA.
- 2. Wenclawiak, B.W. (1992) Ernst Klesper, the “Father of Supercritical Fluid Chromatography”. Fresenius’ Journal of Analytical Chemistry, 344, 425.
http://dx.doi.org/10.1007/BF00323737 - 3. Klesper, E., Corwin, A.H. and Turner, D.A. (1962) High Pressure Gas Chromatography above Critical Temperatures. Journal of Organic Chemistry, 27, 700-701.
- 4. Saito, M. (2013) History of Supercritical Fluid Chromatography: Instrumental Development. Journal of Bioscience and Bioengineering, 115, 590-599.
http://dx.doi.org/10.1016/j.jbiosc.2012.12.008 - 5. Berger, T.A. and Fogelman, K. (2009) Improving Signal-To-Noise Ratio and Dynamic Range in Supercritical Fluid Chromatography with UV Detection. LC GC North America: The Peak, 9, 17-33.
- 6. Nováková, L., Grand-Guillaume Perrenoud, A., Francois, I., West, C., Lesellier, E. and Guillarme, D. (2014) Modern Analytical Supercritical Fluid Chromatography Using Columns Packed with Sub-2 μm Particles: A Tutorial. Analytica Chimica Acta, 824, 18-35.
http://dx.doi.org/10.1016/j.aca.2014.03.034 - 7. Sarazin, C., Sassiat, P.R., Vial, J. and Thiébaut, D. (2011) Feasibility of Ultra High Performance Supercritical Neat Carbon Dioxide Chromatography at Conventional Pressures. Journal of Separation Science, 34, 2773-2778.
http://dx.doi.org/10.1002/jssc.201100332 - 8. Grand-Guillaume Perrenoud, A., Guillarme, D. and Veuthey, J.L. (2012) Comparison of Ultra-High Performance Supercritical Fluid Chromatography and Ultra-High Performance Liquid Chromatography for the Analysis of Pharmaceutical Compounds. Journal of Chromatography A, 1266, 158-167.
http://dx.doi.org/10.1016/j.chroma.2012.10.005 - 9. Lesellier, E. and West, C. (2015) The Many Faces of Packed Column Supercritical Fluid Chromatography A Critical Review. Journal of Chromatography A, 1382, 2-46.
http://dx.doi.org/10.1016/j.chroma.2014.12.083 - 10. Marley, A. and Connolly, D. (2014) Determination of (R)-timolol in (S)-timolol Maleate Active Pharmaceutical Ingredient: Validation of a New Supercritical Fluid Chromatography Method with an Established Normal Phase Liquid Chromatography Method. Journal of Chromatography A, 1325, 213-220.
http://dx.doi.org/10.1016/j.chroma.2013.12.011 - 11. Kamarei, F., Gritti, F., Guiochon, G. and Burchell, J. (2014) Accurate Measurements of Frontal Analysis for the Determination of Adsorption Isotherms in Supercritical Fluid Chromatography. Journal of Chromatography A, 1329, 71-77.
http://dx.doi.org/10.1016/j.chroma.2013.12.033 - 12. Grand-Guillaume Perrenoud, A., Veuthey, J.L. and Guillarme, D. (2014) Coupling State-of-the-Art Supercritical Fluid Chromatography and Mass Spectrometry: From Hyphenation Interface Optimization to High-Sensitivity Analysis of Pharmaceutical Compounds. Journal of Chromatography A, 1339, 174-184.
http://dx.doi.org/10.1016/j.chroma.2014.03.006 - 13. Zhou, Y., Du, Z. and Zhang, Y. (2014) Simultaneous Determination of 17 Disperse Dyes in Textile by Ultra-High Performance Supercritical Fluid Chromatography Combined with Tandem Mass Spectrometry. Talanta, 127, 108-115.
http://dx.doi.org/10.1016/j.talanta.2014.03.055 - 14. Shaaban, H. and Górecki, T. (2015) Current Trends in Green Liquid Chromatography for the Analysis of Pharmaceutically Active Compounds in the Environmental Water Compartments. Talanta, 132, 739-752.
http://dx.doi.org/10.1016/j.talanta.2014.09.050 - 15. Cudiamat, G. (2013) SFC Market Profile. LC GC North America, 31-6.
http://www.chromatographyonline.com/market-profile-supercritical-fluid-chromatography-sfc - 16. De la Guardia Cirugeda, M. (2014) Greening Analytical Science. The Analytical Scientist, #0214.
https://theanalyticalscientist.com/issues/0214/greening-analytical-science/ - 17. Turner, C. (2013) Sustainable Analytical Chemistry More Than Just Being Green. Pure and Applied Chemistry, 85, 2217-2229.
http://dx.doi.org/10.1351/pac-con-13-02-05 - 18. Pinkston, J.D., Wen, D., Morand, K.L., Tirey, D.A. and Stanton, D.T. (2006) Comparison of LC/MS and SFC/MS for Screening of a Large and Diverse Library of Pharmaceutically Relevant Compounds. Analytical Chemistry, 78, 7467-7472.
http://dx.doi.org/10.1021/ac061033l - 19. Farrell, W.P., Aurigemma, C.M. and Masters-Moore, D.F. (2009) Advances in High Throughput Supercritical Fluid Chromatography. Journal of Liquid Chromatography & Related Technologies, 32, 1689-1710.
http://dx.doi.org/10.1080/10826070902956394 - 20. Hühnerfuss, H. and Shah, M.R. (2009) Enantioselective Chromatography A Powerful Tool for the Discrimination of Biotic and Abiotic Transformation Processes of Chiral Environmental Pollutants. Journal of Chromatography A, 1216, 481-502.
http://dx.doi.org/10.1016/j.chroma.2008.09.043 - 21. Xiao, Y., Ng, S.C., Tan, T.T.Y. and Wang, Y. (2012) Recent Development of Cyclodextrin Chiral Stationary Phases and Their Applications in Chromatography. Journal of Chromatography A, 1269, 52-68.
http://dx.doi.org/10.1016/j.chroma.2012.08.049 - 22. Wang, R.-Q., Ong, T.-T., Ng, S.-C. and Tang, W.H. (2012) Recent Advances in Pharmaceutical Separations with Supercritical Fluid Chromatography Using Chiral Stationary Phases. TrAC Trends in Analytical Chemistry, 37, 83-100.
http://dx.doi.org/10.1016/j.trac.2012.02.012 - 23. Desai, P.P., Patel, N.R., Sherikar, O.D. and Mehta, P.J. (2012) Development and Validation of Packed Column Supercritical Fluid Chromatographic Technique for Quantification of Chlorzoxazone, Paracetamol and Aceclofenac in Their Individual and Combined Dosage Forms. Journal of Chromatographic Science, 50, 769-774.
http://dx.doi.org/10.1093/chromsci/bms059 - 24. Alexander, A.J. (2012) SFC Instrument Modification to Allow Greater Flexibility in Method Development by Generating Mixtures of Solvents and Modifiers On-Line for Mobile Phase B Optimization. Chromatographia, 75, 1185-1190.
http://dx.doi.org/10.1007/s10337-012-2291-8 - 25. McClain, R.T., Li, Y., Hyun, M.H. and Welch, C.J. (2013) Design, Synthesis and Evaluation of Stationary Phases for Improved Achiral Supercritical Fluid Chromatography Separations. Journal of Chromatography A, 1302, 163-173.
http://dx.doi.org/10.1016/j.chroma.2013.06.038 - 26. Regalado, E.L., Schafer, W.A., McClain, R.T. and Welch, C.J. (2013) Chromatographic Resolution of Closely Related Species: Separation of Warfarin and Hydroxylated Isomers. Journal of Chromatography A, 1314, 266-275.
http://dx.doi.org/10.1016/j.chroma.2013.07.092 - 27. Alexander, A.J., Zhang, L., Hooker, T.F. and Tomasella, F.P. (2013) Comparison of Supercritical Fluid Chromatography and Reverse Phase Liquid Chromatography for the Impurity Profiling of the Antiretroviral Drugs Lamivudine/ BMS-986001/Efavirenz in a Combination Tablet. Journal of Pharmaceutical and Biomedical Analysis, 78-79, 243-251.
http://dx.doi.org/10.1016/j.jpba.2013.02.019 - 28. Toribio, L., Bernal, J.L., Martín, M.T. and del Nozal, M.J. (2014) Effects of Organic Modifier and Temperature on the Enantiomeric Separation of Several Azole Drugs Using Supercritical Fluid Chromatography and the Chiralpak AD Column. Biomedical Chromatography, 28, 152-158.
http://dx.doi.org/10.1002/bmc.3013 - 29. Gourmel, C., Veuthey, J.L., Rudaz, S., Perrenoud, A.G.G., Waller, L., Reginato, E., et al. (2013) Evaluation and Comparison of Various Separation Techniques for the Analysis of Closely-Related Compounds of Pharmaceutical Interest. Journal of Chromatography A, 1282, 172-177.
http://dx.doi.org/10.1016/j.chroma.2013.01.095 - 30. De Klerck, K. and Mangelings, D. (2012) Supercritical Fluid Chromatography for the Enantioseparation of Pharmaceuticals. Journal of Pharmaceutical and Biomedical Analysis, 69, 77-92.
http://dx.doi.org/10.1016/j.jpba.2012.01.021 - 31. Alexander, A.J. and Staab, A. (2006) Use of Achiral/Chiral SFC/MS for the Profiling of Isomeric Cinnamonitrile/ Hydrocinnamonitrile Products in Chiral Drug Synthesis. Analytical Chemistry, 78, 3835-3838.
http://dx.doi.org/10.1021/ac060326b - 32. Bolanos, B., Farrell, W.P., Aurigemma, C.M., Greig, M., Ventura, M., Li, H., et al. (2004) SFC/MS in Drug Discovery at Pfizer, La Jolla. International Journal of Mass Spectrometry, 238, 85-97.
http://dx.doi.org/10.1016/j.ijms.2003.11.021 - 33. McClain, R.T., Dudkina, A., Barrow, J., Hartman, G. and Welch, C.J. (2009) Evaluation and Implementation of a Commercially Available Mass-Guided SFC Purification Platform in a High Throughput Purification Laboratory in Drug Discovery. Journal of Liquid Chromatography & Related Technologies, 32, 483-499.
http://dx.doi.org/10.1080/10826070802671325 - 34. Brunelli, C., Dunkle, M., Morris, S. and Sandra, P. (2009) Development of a Generic Gradient SFC Method for Application in Drug Discovery and Development. Chromatography Today, February/March, 5-8.
- 35. Goetz, G.H., Farrell, W.P., Shalaeva, M., Sciabola, S., Anderson, D., Yan, J., et al. (2014) High Throughput Method for the Indirect Detection of Intramolecular Hydrogen Bonding. Journal of Medicinal Chemistry, 57, 2920-2929.
http://dx.doi.org/10.1021/jm401859b - 36. Wang, Z., Zhang, H., Liu, O. and Donovan, B. (2011) Development of an Orthogonal Method for Mometasone Furoate Impurity Analysis Using Supercritical Fluid Chromatography. Journal of Chromatography A, 1218, 2311-2319.
http://dx.doi.org/10.1016/j.chroma.2011.02.027 - 37. Alsante, K.M., Baertschi, S.W., Coutant, M., Marquez, B.L., Sharp, T.R. and Zelesky, T.C. (2011) Degradation and Impurity Analysis for Pharmaceutical Drug Candidates. Separation Science and Technology, 10, 59-169.
http://dx.doi.org/10.1016/B978-0-12-375680-0.00003-6 - 38. Alexander, A.J., Hooker, T.F. and Tomasella, F.P. (2012) Evaluation of Mobile Phase Gradient Supercritical Fluid Chromatography for Impurity Profiling of Pharmaceutical Compounds. Journal of Pharmaceutical and Biomedical Analysis, 70, 77-86.
http://dx.doi.org/10.1016/j.jpba.2012.05.025 - 39. Schou-Pedersen, A.M.V., Johansson, M., Ostergaard, J., Dubant, S., Frederiksen, R.B. and Hansen, S.H. (2014) Evaluation of Supercritical Fluid Chromatography for Testing of PEG Adducts in Pharmaceuticals. Journal of Pharmaceutical and Biomedical Analysis, 88, 256-261.
http://dx.doi.org/10.1016/j.jpba.2013.08.039 - 40. Dispas, A., Lebrun, P., Ziemons, E., Marini, R., Rozet, E. and Hubert, P. (2014) Evaluation of the Quantitative Performances of Supercritical Fluid Chromatography: From Method Development to Validation. Journal of Chromatography A, 1353, 78-88.
http://dx.doi.org/10.1016/j.chroma.2014.01.046 - 41. Coe, R.A., Rathe, J.O. and Lee, J.W. (2006) Supercritical Fluid Chromatography-Tandem Mass Spectrometry for Fast Bioanalysis of R/S-Warfarin in Human Plasma. Journal of Pharmaceutical and Biomedical Analysis, 42, 573-580.
http://dx.doi.org/10.1016/j.jpba.2006.05.025 - 42. Matsubara, A., Uchikata, T., Shinohara, M., Fukusaki, E., Nishiumi, S., Yoshida, M., et al. (2012) Highly Sensitive and Rapid Profiling Method for Carotenoids and Their Epoxidized Products Using Supercritical Fluid Chromatography Coupled with Electrospray Ionization-Triple Quadrupole Mass Spectrometry. Journal of Bioscience and Bioengineering, 113, 782-787.
http://dx.doi.org/10.1016/j.jbiosc.2012.01.017 - 43. Uchikata, T., Matsubara, A., Nishiumi, S., Yoshida, M., Fukusaki, E. and Bamba, T. (2012) Development of Oxidized Phosphatidylcholine Isomer Profiling Method Using Supercritical Fluid Chromatography/Tandem Mass Spectrometry. Journal of Chromatography A, 1250, 205-211.
http://dx.doi.org/10.1016/j.chroma.2012.05.083 - 44. Uchikata, T., Matsubara, A., Fukusaki, E. and Bamba, T. (2012) High-Throughput Phospholipid Profiling System Based on Supercritical Fluid Extraction-Supercritical Fluid Chromatography/Mass Spectrometry for Dried Plasma Spot Analysis. Journal of Chromatography A, 1250, 69-75.
http://dx.doi.org/10.1016/j.chroma.2012.06.031 - 45. Geng, Y., Zhao, L., Zhao, J., Guo, B., Ma, P., Li, Y., et al. (2014) Development of a Supercritical Fluid Chromatography-Tandem Mass Spectrometry Method for the Determination of Lacidipine in Beagle Dog Plasma and Its Application to a Bioavailability Study. Journal of Chromatography B, Analytical Technologies in the Biomedical and Life Sciences, 945-946, 121-126.
http://dx.doi.org/10.1016/j.jchromb.2013.11.029 - 46. Taguchi, K., Fukusaki, E. and Bamba, T. (2013) Simultaneous and Rapid Analysis of Bile Acids including Conjugates by Supercritical Fluid Chromatography Coupled to Tandem Mass Spectrometry. Journal of Chromatography A, 1299, 103-109.
http://dx.doi.org/10.1016/j.chroma.2013.05.043 - 47. Nováková, L., Perrenoud, A.G.G., Nicoli, R., Saugy, M., Veuthey, J.L. and Guillarme, D. (2015) Ultra High Performance Supercritical Fluid Chromatography Coupled with Tandem Mass Spectrometry for Screening of Doping Agents. I: Investigation of Mobile Phase and MS Conditions. Analytica Chimica Acta, 853, 637-646.
http://dx.doi.org/10.1016/j.aca.2014.10.004 - 48. Nováková, L., Rentsch, M., Perrenoud, A.G.G., Nicoli, R., Saugy, M., Veuthey, J., et al. (2015) Ultra High Performance Supercritical Fluid Chromatography Coupled with Tandem Mass Spectrometry for Screening of Doping Agents. II: Analysis of Biological Samples. Analytica Chimica Acta, 853, 647-659.
http://dx.doi.org/10.1016/j.aca.2014.10.007 - 49. Issaq, H.J., Abbott, E. and Veenstra, T.D. (2008) Utility of Separation Science in Metabolomic Studies. Journal of Separation Science, 31, 1936-1947.
http://dx.doi.org/10.1002/jssc.200700601 - 50. Bamba, T. and Fukusaki, E. (2009) Separation of Hydrophobic Metabolites Using Monolithic Silica Column in High-Performance Liquid Chromatography and Supercritical Fluid Chromatography. Journal of Separation Science, 32, 2699-2706.
http://dx.doi.org/10.1002/jssc.200900124 - 51. Bamba, T., Lee, J.W., Matsubara, A. and Fukusaki, E. (2012) Metabolic Profiling of Lipids by Supercritical Fluid Chromatography/Mass Spectrometry. Journal of Chromatography A, 1250, 212-219.
http://dx.doi.org/10.1016/j.chroma.2012.05.068 - 52. Yamada, T., Uchikata, T., Sakamoto, S., Yokoi, Y., Nishiumi, S., Yoshida, M., et al. (2013) Supercritical Fluid Chromatography/Orbitrap Mass Spectrometry Based Lipidomics Platform Coupled with Automated Lipid Identification Software for Accurate Lipid Profiling. Journal of Chromatography A, 1301, 237-242.
http://dx.doi.org/10.1016/j.chroma.2013.05.057 - 53. Jones, M.D., Rainville, P.D., Isaac, G., Wilson, I.D., Smith, N.W. and Plumb, R.S. (2014) Ultra High Resolution SFC-MS as a High Throughput Platform for Metabolic Phenotyping: Application to Metabolic Profiling of Rat and Dog Bile. Journal of Chromatography B, Analytical Technologies in the Biomedical and Life Sciences, 966, 200-207.
http://dx.doi.org/10.1016/j.jchromb.2014.04.017 - 54. Laboureur, L., Ollero, M. and Touboul, D. (2015) Lipidomics by Supercritical Fluid Chromatography. International Journal of Molecular Sciences, 16, 13868-13884.
http://dx.doi.org/10.3390/ijms160613868 - 55. Lísa, M. and Holcapek, M. (2015) High-Throughput and Comprehensive Lipidomic Analysis Using Ultrahigh Performance Supercritical Fluid Chromatography-Mass Spectrometry. Analytical Chemistry, 87, 7187-7195.
http://dx.doi.org/10.1021/acs.analchem.5b01054 - 56. Kalíková, K., Slechtová, T., Vozka, J. and Tesarová, E. (2014) Supercritical Fluid Chromatography as a Tool for Enantioselective Separation: A Review. Analytica Chimica Acta, 821, 1-33.
http://dx.doi.org/10.1016/j.aca.2014.02.036 - 57. De Klerck, K., Vander Heyden, Y. and Mangelings, D. (2014) Generic Chiral Method Development in Supercritical Fluid Chromatography and Ultra-Performance Supercritical Fluid Chromatography. Journal of Chromatography A, 1363, 311-322.
http://dx.doi.org/10.1016/j.chroma.2014.06.011 - 58. Taylor, L. (2008) Supercritical Fluid Chromatography. Analytical Chemistry, 80, 4285-4294.
http://dx.doi.org/10.1021/ac800482d - 59. Taylor, L. (2009) Supercritical Fluid Chromatography for the 21st Century. The Journal of Supercritical Fluids, 47, 566-573.
http://dx.doi.org/10.1016/j.supflu.2008.09.012 - 60. Guiochon, G. and Tarafder, A. (2011) Fundamental Challenges and Opportunities for Preparative Supercritical Fluid Chromatography. Journal of Chromatography A, 1218, 1037-1114.
http://dx.doi.org/10.1016/j.chroma.2010.12.047 - 61. Takahashi, K. (2013) Polymer Analysis by Supercritical Fluid Chromatography. Journal of Bioscience and Bioengineering, 116, 133-140.
http://dx.doi.org/10.1016/j.jbiosc.2013.02.001 - 62. Takahashi, K., Takahashi, R., Horikawa, Y., Matsuyama, S., Kinugasa, S. and Ehara, K. (2013) Optimization of Experimental Parameters for Separation of Nonionic Surfactants by Supercritical Fluid Chromatography. The Journal of Supercritical Fluids, 82, 256-262.
http://dx.doi.org/10.1016/j.supflu.2013.08.007 - 63. Adam, F., Thiébaut, D., Bertoncini, F., Courtiade, M. and Hennion, M.C. (2010) Supercritical Fluid Chromatography Hyphenated with Twin Comprehensive Two-Dimensional Gas Chromatography for Ultimate Analysis of Middle Distillates. Journal of Chromatography A, 1217, 1386-1394.
http://dx.doi.org/10.1016/j.chroma.2009.11.092 - 64. Thiébaut, D. (2012) Separations of Petroleum Products Involving Supercritical Fluid Chromatography. Journal of Chromatography A, 1252, 177-188.
http://dx.doi.org/10.1016/j.chroma.2012.06.074 - 65. Dutriez, T., Thiébaut, D., Courtiade, M., Dulot, H., Bertoncini, F. and Hennion, M.C. (2013) Application to SFC-GC × GC to Heavy Petroleum Fractions Analysis. Fuel, 104, 583-592.
http://dx.doi.org/10.1016/j.fuel.2012.04.048 - 66. Bernal, J.L., Martín, M.T. and Toribio, L. (2013) Supercritical Fluid Chromatography in Food Analysis. Journal of Chromatography A, 1313, 24-36.
http://dx.doi.org/10.1016/j.chroma.2013.07.022 - 67. Berger, T.A. and Berger, B.K. (2013) Separation of 9 Sulfonamide Drugs in a Parts per Thousand 4 Min by Ultra-High Performance Supercritical Fluid Chromatography (UHPSFC): With a Feasibility Study for Detection in Milk. Chromatographia, 76, 1631-1639.
http://dx.doi.org/10.1007/s10337-013-2539-y - 68. Ishibashi, M., Izumi, Y., Sakai, M., Ando, T., Fukusaki, E. and Bamba, T. (2015) High-Throughput Simultaneous Analysis of Pesticides by Supercritical Fluid Chromatography Coupled with High-Resolution Mass Spectrometry. Journal of Agricultural and Food Chemistry, 63, 4457-4463.
http://dx.doi.org/10.1021/jf5056248 - 69. Bijttebier, S., D’Hondt, E., Noten, B., Hermans, N., Apers, S., Exarchou, V., et al. (2014) Automated Analytical Standard Production with Supercritical Fluid Chromatography for the Quantification of Bioactive C17-Polyacetylenes: A Case Study on Food Processing Waste. Food Chemistry, 165, 371-378.
http://dx.doi.org/10.1016/j.foodchem.2014.05.093 - 70. Farrell, W.P., Aurigemma, C.M. and Masters-Moore, D.F. (2013) Advances in High Throughput Supercritical Fluid Chromatography. Journal of Liquid Chromatography and Related Techniques, 32, 1689-1710.
- 71. Miller, L. and Mahoney, M. (2012) Evaluation of Flash Supercritical Fluid Chromatography and Alternate Sample Loading Techniques for Pharmaceutical Medicinal Chemistry Purifications. Journal of Chromatography A, 1250, 264-273.
http://dx.doi.org/10.1016/j.chroma.2012.06.029 - 72. Van Anda, J. (2010) Use of SFC/MS in the Purification of Achiral Pharmaceutical Compounds. American Pharmaceutical Review, 13, 111-115.
- 73. Desfontaine, V., Guillarme, D., Francotte, E. and Nováková, L. (2015) Supercritical Fluid Chromatography in Pharmaceutical Analysis. Journal of Pharmaceutical and Biomedical Analysis, 113, 56-71.
http://dx.doi.org/10.1016/j.jpba.2015.03.007 - 74. ICH (2009) Pharmaceutical Development. Q8(R2).
- 75. Taylor, L. (2010) Supercritical Fluid Chromatography. Analytical Chemistry, 82, 4925-4935.
http://dx.doi.org/10.1021/ac101194x - 76. Taylor, L. (2012) Packed Column Supercritical Fluid Chromatography of Hydrophilic Analytes via Water-Rich Modifiers. Journal of Chromatography A, 1250, 196-204.
http://dx.doi.org/10.1016/j.chroma.2012.02.037 - 77. Perrenoud, A.G.G., Veuthey, J.L. and Guillarme, D. (2014) The Use of Columns Packed with Sub-2 μm Particles in Supercritical Fluid Chromatography. TrAC Trends in Analytical Chemistry, 63, 44-54.
http://dx.doi.org/10.1016/j.trac.2014.06.023 - 78. European Commission for Health and Food Safety (2015) EudraLex—Volume 4: EU Guidelines for GMP for Medicinal Products for Humans and Veterinary Use. Annex 15: Qualification and Validation, 1-16.
http://ec.europa.eu/health/files/eudralex/vol-4/2015-10_annex15.pdf - 79. Huber, L. (2002) Good Laboratory Practice and Current Good Manufacturing Practice—A Primer. Agilent Technologies Deutschland GmbH.
https://www.agilent.com/cs/library/primers/Public/59886197.pdf - 80. USP (2014) <1058> Analytical Instrument Qualification.
https://hmc.usp.org/sites/default/files/documents/HMC/GCs-Pdfs/c1058.pdf - 81. Anton, K. and Siffrin, C. (1999) Packed-Column SFC in the Pharmaceutical Industry: cGMP Aspects. Analusis, 27, 691-700.
http://dx.doi.org/10.1051/analusis:1999270691 - 82. Sun, J. (2011) Critical Attributes and Technical Challenges of Successful SFC Instrument Qualification. American Pharmaceutical Review, 14, 79-83.
- 83. Huber, L. (2011) Qualification and Validation for Supercritical Fluid Chromatography. Agilent Technologies Inc.
https://www.agilent.com/cs/library/primers/Public/5990-9148EN.pdf - 84. Hicks, M.B., Regalado, E.L., Tan, F., Gong, X. and Welch, C.J. (2015) Supercritical Fluid Chromatography for GMP Analysis in Support of Pharmaceutical Development and Manufacturing Activities. Journal of Pharmaceutical and Biomedical Analysis, 117, 316-324.
http://dx.doi.org/10.1016/j.jpba.2015.09.014 - 85. Tarafder, A. and Guiochon, G. (2013) Accurate Measurements of Experimental Parameters in Supercritical Fluid Chromatography. I. Extent of Variations of the Mass and Volumetric Flow Rates. Journal of Chromatography A, 1285, 148-158.
http://dx.doi.org/10.1016/j.chroma.2013.01.080 - 86. Tarafder, A., Vajda, P. and Guiochon, G. (2013) Accurate On-Line Mass Flow Measurements in Supercritical Fluid Chromatography. Journal of Chromatography A, 1320, 130-137.
http://dx.doi.org/10.1016/j.chroma.2013.10.041 - 87. Enmark, M., Forssén, P., Fornstedt, T. and Samuelsson, J. (2013) Determination of Adsorption Isotherms in Supercritical Fluid Chromatography. Journal of Chromatography A, 1312, 124-133.
http://dx.doi.org/10.1016/j.chroma.2013.09.007 - 88. Xia, B., Feng, M., Ding, L. and Zhou, Y. (2014) Fast Separation Method Development for Supercritical Fluid Chromatography Using an Autoblending Protocol. Chromatographia, 77, 783-791.
http://dx.doi.org/10.1007/s10337-014-2684-y - 89. Lionberger, R.A., Lee, S.L., Lee, L., Raw, A. and Yu, L.X. (2008) Quality by Design: Concepts for ANDAs. The American Association of Pharmaceutical Scientists Journal, 10, 268-276.
http://dx.doi.org/10.1208/s12248-008-9026-7 - 90. Borman, P., Chatfield, M., Nethercote, P., Thompson, D. and Truman, K. (2007) The Application of Quality by Design to Analytical Methods. Pharmaceutical Technology, 31, 142-152.
- 91. Vogt, F.G. and Kord, A.S. (2010) Development of Quality-by-Design Analytical Methods. Journal of Pharmaceutical Sciences, 100, 797-812.
http://dx.doi.org/10.1002/jps.22325 - 92. Rozet, E., Debrus, B., Boulanger, B., Lebrun, P. and Hubert, P. (2013) Design Spaces for Analytical Methods. TrAC Trends in Analytical Chemistry, 42, 157-167.
http://dx.doi.org/10.1016/j.trac.2012.09.007 - 93. Orlandini, S., Pinzauti, S. and Furlanetto, S. (2013) Application of Quality by Design to the Development of Analytical Separation Methods. Analytical and Bioanalytical Chemistry, 405, 443-450.
http://dx.doi.org/10.1007/s00216-012-6302-2 - 94. Dispas, A., Lebrun, P., Andri, B., Rozet, E. and Hubert, P. (2014) Robust Method Optimization Strategy—A Useful Tool for Method Transfer: The Case of SFC. Journal of Pharmaceutical and Biomedical Analysis, 88, 519-524.
http://dx.doi.org/10.1016/j.jpba.2013.09.030 - 95. Hubert, C., Lebrun, P., Houari, S., Ziemons, E., Rozet, E. and Hubert, P. (2014) Improvement of a Stability-Indicating Method by Quality-by-Design versus Quality-by-Testing: A Case of a Learning Process. Journal of Pharmaceutical and Biomedical Analysis, 88, 401-409.
http://dx.doi.org/10.1016/j.jpba.2013.09.026 - 96. Dispas, A., Lebrun, P., Sassiat, P.R., Ziemons, E., Thiébaut, D., Vial, J., et al. (2012) Innovative Green Supercritical Fluid Chromatography Development for the Determination of Polar Compounds. Journal of Chromatography A, 1256, 253-260.
http://dx.doi.org/10.1016/j.chroma.2012.07.043 - 97. Debrus, B., Guillarme, D. and Rudaz, S. (2013) Improved Quality-by-Design Compliant Methodology for Method Development in Reversed-Phase Liquid Chromatography. Journal of Pharmaceutical and Biomedical Analysis, 84, 215-223.
http://dx.doi.org/10.1016/j.jpba.2013.06.013 - 98. Li, J.-R., Li, M., Xia, B., Ding, L.-S., Xu, H.-X. and Zhou, Y. (2013) Efficient Optimization of Ultra-High Performance Supercritical Fluid Chromatographic Separation of Rosa Sericeaby Response Surface Methodology. Journal of Separation Science, 36, 2114-2120.
http://dx.doi.org/10.1002/jssc.201300289 - 99. Kaul, N., Agrawal, H., Paradkar, A.R. and Mahadik, K.R. (2007) Effect of System Variables Involved in Packed Column Supercritical Fluid Chromatography of Stavudine Taken as Model Analyte Using Response Surface Methodology along with Study of Thermodynamic Parameters. Journal of Pharmaceutical and Biomedical Analysis, 43, 471-480.
http://dx.doi.org/10.1016/j.jpba.2006.07.038 - 100. Ibanez, E., Tabera, J., Reglero, G. and Herraiz, M. (1995) Optimization of Fat-Soluble Vitamin Separation by Supercritical Fluid Chromatography. Chromatographia, 40, 448-452.
http://dx.doi.org/10.1007/BF02269911 - 101. Végh, K., Alberti, á., Riethmüller, E., Tóth, A., Béni, S. and Kéry, á. (2014) Supercritical Fluid Extraction and Convergence Chromatographic Determination of Parthenolide in Tanacetum parthenium L.: Experimental Design, Modeling and Optimization. The Journal of Supercritical Fluids, 95, 84-91.
http://dx.doi.org/10.1016/j.supflu.2014.07.029 - 102. Asberg, D., Enmark, M., Samuelsson, J. and Fornstedt, T. (2014) Evaluation of Co-Solvent Fraction, Pressure and Temperature Effects in Analytical and Preparative Supercritical Fluid Chromatography. Journal of Chromatography A, 1374, 254-260.
http://dx.doi.org/10.1016/j.chroma.2014.11.045 - 103. Delahaye, S. and Lynen, F. (2014) Implementing Stationary-Phase Optimized Selectivity in Supercritical Fluid Chromatography. Analytical Chemistry, 86, 12220-12228.
http://dx.doi.org/10.1021/ac503313j - 104. De Pauw, R., Desmet, G. and Broeckhoven, K. (2013) Theoretical Evaluation of the Advantages and Limitations of Constant Pressure versus Constant Flow Rate Gradient Elution Separation in Supercritical Fluid Chromatography. Journal of Chromatography A, 1312, 134-142.
http://dx.doi.org/10.1016/j.chroma.2013.09.008 - 105. Nováková, L., Chocholous, P. and Solich, P. (2014) Ultra-Fast Separation of Estrogen Steroids Using Subcritical Fluid Chromatography on Sub-2-Micron Particles. Talanta, 121, 178-186.
http://dx.doi.org/10.1016/j.talanta.2013.12.056 - 106. Ganzera, M. (2015) Supercritical Fluid Chromatography for the Separation of Isoflavones. Journal of Pharmaceutical and Biomedical Analysis, 107, 364-369.
http://dx.doi.org/10.1016/j.jpba.2015.01.013 - 107. Rajendran, A. (2012) Design of Preparative-Supercritical Fluid Chromatography. Journal of Chromatography A, 1250, 227-249.
http://dx.doi.org/10.1016/j.chroma.2012.05.037 - 108. Brunelli, C., Pereira, A., Dunkle, M., David, F. and Sandra, P. (2010) Green Chromatography (Part 2): The Role of GC and SFC. LC-GC Europe, 23, No. 8.
http://www.chromatographyonline.com/green-chromatography-part-2-role-gc-and-sfc - 109. Tarafder, A., Hudalla, C., Iraneta, P. and Fountain, K.J. (2014) A Scaling Rule in Supercritical Fluid Chromatography. I. Theory for Isocratic Systems. Journal of Chromatography A, 1362, 278-293.
http://dx.doi.org/10.1016/j.chroma.2014.08.009 - 110. Helmy, R., Biba, M., Zang, J., Mao, B., Fogelman, K., Vlachos, V., et al. (2007) Improving Sensitivity in Chiral Supercritical Fluid Chromatography for Analysis of Active Pharmaceutical Ingredients. Chirality, 19, 787-792.
http://dx.doi.org/10.1002/chir.20451 - 111. Berger, T.A. and Berger, B.K. (2011) Minimizing UV Noise in Supercritical Fluid Chromatography. I. Improving Back Pressure Regulator Pressure Noise. Journal of Chromatography A, 1218, 2320-2326.
http://dx.doi.org/10.1016/j.chroma.2011.02.030 - 112. del Carmen Salvatierra-Stamp, V., Ceballos-Magana, S.G., Gonzalez, J., Ibarra-Galván, V. and Muniz-Valencia, R. (2015) Analytical Method Development for the Determination of Emerging Contaminants in Water Using Supercritical-Fluid Chromatography Coupled with Diode-Array Detection. Analytical and Bioanalytical Chemistry, 407, 4219- 4226.
http://dx.doi.org/10.1007/s00216-015-8581-x - 113. Berger, T.A. (2014) Minimizing Ultraviolet Noise Due to Mis-Matches between Detector Flow Cell and Post Column Mobile Phase Temperatures in Supercritical Fluid Chromatography: Effect of Flow Cell Design. Journal of Chromatography A, 1364, 249-260.
http://dx.doi.org/10.1016/j.chroma.2014.08.067 - 114. Méjean, M., Vollmer, M., Brunelle, A. and Touboul, D. (2013) Quantification of Retinoid Compounds by Supercritical Fluid Chromatography Coupled to Ultraviolet Diode Array Detection. Chromatographia, 76, 1097-1105.
http://dx.doi.org/10.1007/s10337-013-2508-5 - 115. Ashraf-Khorassani, M., Taylor, L., Williams, D.G., Roston, D.A. and Catalano, T.T. (2001) Demonstrative Validation Study Employing a Packed Column Pressurized Fluid Chromatography Method That Provides Assay, Achiral Impurities, Chiral Impurity, and IR Identity Testing for a Drug Substance. Journal of Pharmaceutical and Biomedical Analysis, 26, 725-738.
http://dx.doi.org/10.1016/S0731-7085(01)00441-1 - 116. Coym, J.W. and Chester, T.L. (2003) Improving Injection Precision in Packed-Column Supercritical Fluid Chromatography. Journal of Separation Science, 26, 609-613.
http://dx.doi.org/10.1002/jssc.200390083 - 117. Xiang, Y., Dunetz, J.R. and Lovdahl, M. (2013) Chiral Separation of Amides Using Supercritical Fluid Chromatography. Journal of Chromatography A, 1293, 150-158.
http://dx.doi.org/10.1016/j.chroma.2013.03.048 - 118. Mukherjee, P.S. (2007) Validation of Direct Assay of an Aqueous Formulation of a Drug Compound AZY by Chiral Supercritical Fluid Chromatography (SFC). Journal of Pharmaceutical and Biomedical Analysis, 43, 464-470.
http://dx.doi.org/10.1016/j.jpba.2006.07.026 - 119. Fornstedt, T. and Majors, R.E. (2015) Modern Supercritical Fluid Chromatography—Possibilities and Pitfalls. LC-GC Europe, 33, No. 3.
http://www.chromatographyonline.com/modern-supercritical-fluid-chromatography-possibilities-and-pitfalls-0 - 120. Hubert, P., Nguyen-Huu, J.J., Boulanger, B., Chapuzet, E., Chiap, P., Cohen, N., et al. (2004) Harmonization of Strategies for the Validation of Quantitative Analytical Procedures: A SFSTP Proposal, Part I. Journal of Pharmaceutical and Biomedical Analysis, 36, 579-586.
http://dx.doi.org/10.1016/j.jpba.2004.07.027 - 121. Hubert, P., Nguyen-Huu, J.J., Boulanger, B., Chapuzet, E., Chiap, P., Cohen, N., et al. (2007) Harmonization of Strategies for the Validation of Quantitative Analytical Procedures. A SFSTP Proposal, Part II. Journal of Pharmaceutical and Biomedical Analysis, 45, 70-81.
http://dx.doi.org/10.1016/j.jpba.2007.06.013 - 122. Hubert, P., Nguyen-Huu, J.J., Boulanger, B., Chapuzet, E., Cohen, N., Compagnon, P.A., et al. (2007) Harmonization of Strategies for the Validation of Quantitative Analytical Procedures. A SFSTP Proposal, Part III. Journal of Pharmaceutical and Biomedical Analysis, 45, 82-96.
http://dx.doi.org/10.1016/j.jpba.2007.06.032 - 123. Hubert, P., Nguyen-Huu, J.J., Boulanger, B., Chapuzet, E., Cohen, N., Compagnon, P.A., et al. (2008) Harmonization of Strategies for the Validation of Quantitative Analytical Procedures: A SFSTP Proposal. Part IV. Examples of Application. Journal of Pharmaceutical and Biomedical Analysis, 48, 760-771.
http://dx.doi.org/10.1016/j.jpba.2008.07.018 - 124. Dejaegher, B. and Vander Heyden, Y. (2007) Ruggedness and Robustness Testing. Journal of Chromatography A, 1158, 138-157.
http://dx.doi.org/10.1016/j.chroma.2007.02.086