American Journal of Analytical Chemistry
Vol.4 No.6(2013), Article ID:33445,13 pages DOI:10.4236/ajac.2013.46041
Stability Indicating RP-HPLC Method for the Determination of Dronedarone Hydrochloride and Its Potential Process-Related Impurities in Bulk Drug and Pharmaceutical Dosage Form
Research and Development, Megafine Pharma (P) Ltd., Nashik, India
Email: *drvtmathad@yahoo.co.in, vt.mathad@megafine.in
Copyright © 2013 Shashikant B. Landge et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 15, 2013; revised April 20, 2013; accepted May 1, 2013
Keywords: Dronedarone; RP-LC; LC-MS; Forced Degradation; Validation; Stability-Indicating
ABSTRACT
Simple, sensitive and accurate stability indicating analytical method for dronedarone has been developed and validated using RP-HPLC technique. Developed method is used to evaluate the assay and related substances of dronedarone drug substance and tablets (Multaq®). The drug substance was subjected to the stress conditions such as hydrolysis (acid and base), oxidation, photolysis and thermal degradation as per International Conference on Harmonization (ICH) prescribed stress conditions to show the stability-indicating the nature of the method. Significant degradation was observed during acid and base hydrolysis, and peroxide degradation. The major degredants were identified by LC-MS, FTIR and 1H NMR spectral analysis. The chromatographic conditions were optimized using an impurity-spiked solution and the samples generated from forced degradation studies. In the developed HPLC method, the resolution between dronedarone, process-related impurities, (namely Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11) and degradation products were found to be greater than 1.5. The eleven potential process related impurities were separated on an Ascentis® Express C18 column (4.6 × 10 cm i.d., particle size 2.7 µm) at a flow rate of 1.2 mL∙min–1. The LC method employed a linear gradient elution and the detection wavelength at 220 nm. The chromatographic behavior of all the impurities was examined under variable compositions of different solvents, temperatures and pH values.
1. Introduction
Dronedarone, a benzofuran derivative, N-{2-butyl-3-[4-(3-dibutylaminopropoxy)benzoyl]-benzofuran-5-yl}methanesulfonamide, is a potent drug mainly used for the indication of cardiac arrhythmias [1]. It has been developed by Sanofi Aventis to overcome iodineassociated adverse effects of the commonly used antiarrhythmic drug, amiodarone, with which it is structurally related [2]. Dronedarone is a multi-ion channel blocker, inhibiting the potassium currents involved in cardiac repolarisation including IKr, IKs, IKur, and IK(Ach) and has been shown to be effective in the treatment of cardiovascular hospitalization in patients with paroxysmal or persistent atrial fibrillation (AF) or atrial flutter (AFL) [1-4]. This drug is currently being marked under the brand name of MULTAQ® contains 400 mg of active ingredient (expressed as base). Dronedarone HCl is a white fine powder that is practically insoluble in water and freely soluble in methylene chloride and methanol.
Impurity profiling studies of drug substances and drug products in pharmaceutical industries play a vital role during drug/process development. Gathering of analyticcal data on impurities is important for early stage of pharmaceutical development. During the chemical process development of bulk drug, several impurities aroused from precursors, side products formed in the reaction and impurities related to the reagents used may be present at low levels [5,6]. Separation of all these impurities including degradation impurities in a single analytical HPLC method is a challenging job. Moreover, stringent ICH guidelines are forcing to monitor and control the level of impurities to the specified limit in drug substances and drug products and hence there is a practical and scientific need to develop a suitable and efficient analytical method for analysis.
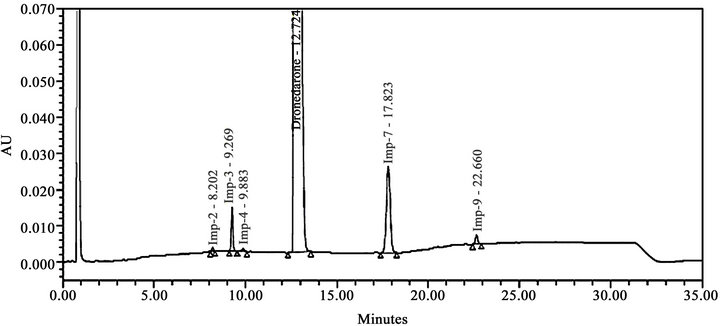
Extensive efforts have been found in literature towards the development of analytical method for the determination dronedarone hydrochloride and its related impurities in biological samples, bulk drug sample and in pharmaceutical dosage form. A literature survey also revealed that the analytical methods were available for the investigation of pharmacokinetics of dronedarone where the content of dronedarone individually and with its active metabolite de-butyldronedarone in human plasma was studied by liquid chromatography-tandem mass spectrometry [7] and HPLC-UV [8] methods respectively. Stability indicating HPLC method reported [9] for dronedarone bulk drugs samples and pharmaceutical dosage forms has been found to be non-selective at our end. In addition, few other methods for the quantitative determination of dronedarone in pharmaceutical dosage forms have also been reported [10,11]. However, these papers were restricted to the determination of dronedarone and failed to discuss the details of process-related impurities and degradation impurities formed under the stress conditions employed.
As per the requirements of various regulatory authorities, the impurity profile study of drug substance and drug product has to be carried out using a suitable analytical method in the final product [12,13]. While studying the synthetic process for the preparation of dronedarone hydrochloride in our laboratory [14], we identified eleven process/degradation related impurities (Imp-1 to Imp-11) of dronedarone hydrochloride as shown in Figure 1. The present study was carried out to describe detailed degradation studies as per ICH guidelines and develop a suitable stability indicating LC method for the separation and estimation of both process related impurities and degradation impurities. Interestingly the author observed two major degradation products in acid and peroxide degradation of dronedarone. The major possible degradation products were synthesized and confirmed by chromatographic retention time and using advanced spectral techniques such as IR, LC-MS and NMR. The developed LC method was validated with respect to specificity, LOD, LOQ, linearity, precision, accuracy and robustness. These studies were performed in accordance with established ICH guidelines [15,16]. The method could also be applied for pharmacokinetic study of dronedarone apart from estimating the impurities and assay in bulk samples and drug products.
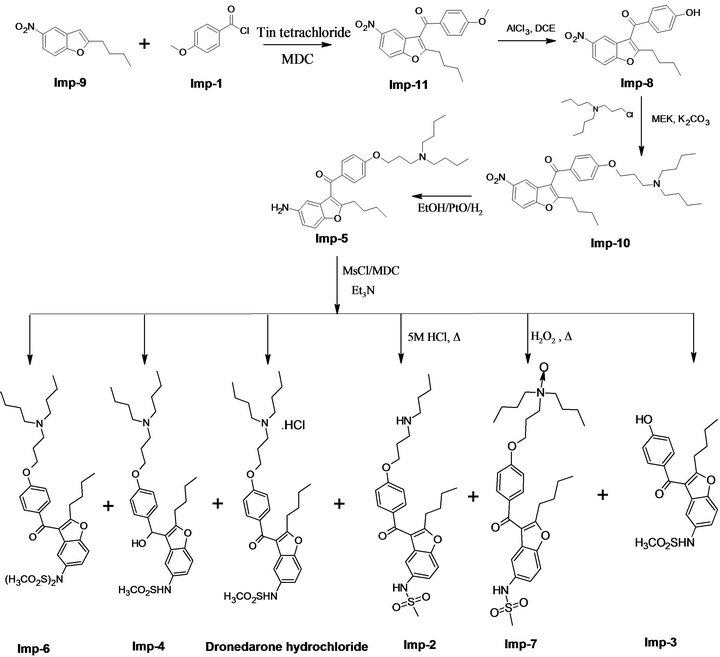
Figure 1. Synthetic schemes of dronedarone hydrochloride: generation of process related and degradation related impurities.
2. Experimental
2.1. Materials and Reagents
HPLC grade acetonitrile and methanol procured from Merck (Mumbai, India) were used. Ammonium dihydrogen orthophosphate, hydrochloric acid, sodium hydroxide and hydrogen peroxide were all of AR grade, procured from Merck (Mumbai, India). HPLC grade water obtained from Millipore system (Millipore Inc., USA) was used throughout the analysis. The investigated samples of dronedarone hydrochloride and its potential process related impurities (Imp-1 to Imp-11) were received from synthetic laboratory of Megafine Pharma (P) Ltd., Nashik, India.
2.2. Instrumentation and Chromatographic Conditions
A Waters HPLC (Milford, MA, USA) equipped with Alliance 2695 separations module and 2996 photodiode array detector was used for method development, forced degradation studies, and method validation. The column Ascentis® Express C18, 10 cm × 4.6 mm, 2.7 µm (SUPELCO Analytical, USA), thermostated at 35˚C was used for the analysis/study. The mobile phase-A consisting a mixture of buffer (0.05M Ammonium dihydrogen orthophosphate) and methanol in the ratio of 80:20 v/v and mobile phase-B consisting a mixture of acetonitrile, methanol and water in the ratio of 45:45:10 v/v. The flow rate and injection volumes were 1.2 mL·min–1 and 10 µl respectively. The analysis was carried out under gradient conditions as follows, time (min)/A (v/v): B (v/v); T0.01/ 65:35, T7.0/45:55, T17.0/45:55, T24.0/30:70, T28.0/30:70, T29.0/65:35, T35.0/65:35. The data was processed by Empower data handling system (Waters, USA). For the analysis of forced degradation samples, the photodiode array detector was used in the scan mode from 200 nm to 400 nm. The peak homogeneity was expressed in terms of peak purity values.
2.3. Solution Preparation and Analytical Procedure
A mixture of water and acetonitrile in the ratio of 30:70 (v/v) was used as diluent in the preparation of analytical solutions. A solution of dronedarone was prepared at a concentration of 500 µg·mL–1 and 50 µg·mL–1 in the diluent for related substances determination and assay determination, respectively. The individual stock solutions of all the related substances (150 µg·mL–1) and dronedarone hydrochloride (50 µg·mL–1) were prepared by dissolving known amount of the substances in 5 ml of acetonitrile, made up to the mark with diluent and these solutions were further diluted adequately to study the validation attributes. The specification limits used for validation studies was 0.15% for the known related substances viz., Imp-1 to Imp-11 and 0.10% for unknown related substances. Dronedarone hydrochloride working reference standard solution (500 µg·mL–1) spiked with all impurities at a specified level (w/w) was used as resolution mixture solution (RMS). The system suitability solution of all impurities was prepared at specified level by diluting above stock solutions.
Sample of drug product was prepared by; powdering five tablets of Multaq, dissolving an equivalent of 25 mg of active ingredient in diluent in a 50-mL volumetric flask and ultra sonicating for about 15 min. The volume was made up to the mark with the diluent, and filtered through Merck Nylon syringe filter having pore size 0.45 µm. The clear liquid was collected and used for the determination of related substances in the pharmaceutical dosage forms. This solution was ten times diluted for the assay determination in pharmaceutical dosage forms.
10.0 µL of blank, RMS, six replicate injections of system suitability solution and test sample solution were separately chromatographed. A resolution of not less than 1.5 between Imp-7 and Imp-8 was set as a system suitability requirement in RMS. The relative standard deviation (RSD) of not more than 5.0% for all related substances peak areas obtained from six replicate injections of system suitability solution was used to verify the system precision. All the known related substances in test sample were determined against mean area of respective impurities obtained from replicate injections of system suitability solution. As well as all the unknown related substances in test sample were determined against mean area of dronedarone obtained from replicate injections of system suitability solution.
2.4. Characterization of Impurities
2.4.1. 1H NMR Spectroscopy
The 1H NMR spectra were recorded on Bruker AV400 (400 MHz) spectrometer using deuteriated chloroform as solvent and tetramethylsilane (TMS) as internal standard.
2.4.2. Mass Spectrometry (MS)
Mass spectra were recorded on Waters Micro massQuattro micro API mass spectrometer (Waters Corporation, Milford, MA, USA) equipped with a quadrupole mass analyzer. Ions were detected in electron spray ionization with positive ion mode. Spectra were acquired from m/z 60 to 1100 in 0.1 amu steps with 10 numbers of scans.
2.5. Method Development and Optimization
Literature reported methods [7-9] were examined at our end to evaluate their efficiency for eluting all the eleven impurities before taking up the development of a new method. As shown in Figures 2-4, the reported methods failed to separate the peaks of Imp-4 and Imp-5, Imp-6 and Imp-7, and Imp-3 and Imp-9 from each other. The wavelength selected (288 nm) was also found to be not suitable for the determination of dronedarone and specified impurities. Methods [8,9], were also found to be nonspecific for said impurities determination Thus we felt a need for the development of a new chromatographic method that detect and determine all the potential process and degradation related impurities viz., Imp-1 to Imp-11 present in bulk samples of dronedarone hydrochloride and to achieve the separation of all eleven impurities (Figure 1) from the analyte peak. The new method developed in our laboratory showed separation of all the eleven impurities of dronedarone hydrochloride satisfactorily as shown in Figure 5(b).

Figure 2. HPLC chromatograms of system suitability solution; (conditions: Mobile phase-A: mix. of 0.01 M Potassium dihydrogen phosphate (KH2PO4) + 0.01 M Tetra n-butyl ammonium hydrogen sulfate, pH 3.2 with potassium hydroxide, Mobile phase-B: Acetonitrile, Gradient elution as time (min)/% solution B: 0/35, 25/80, 25.1/35 and 30/35, flow 1.5 mL·min–1, 25˚C, 220 nm, Agilent Zorbax RX C8 column (150 × 4.6 mm, 5 μm), 5 µm, run time 30 min).
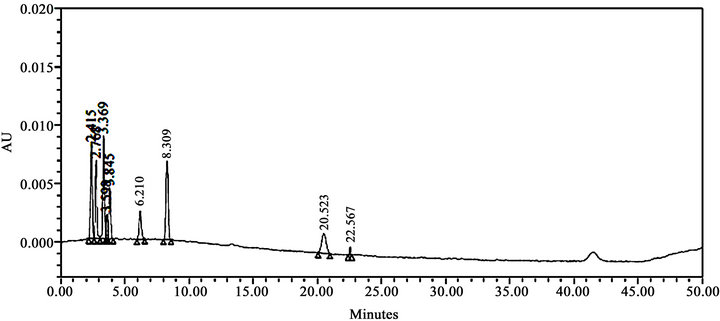
Figure 3. HPLC chromatograms of system suitability solution; (conditions: Buffer (0.05 M KH2PO4 solution, pH 3.0 with OPA): Acetonitrile, 42:58% v/v, flow 1.1 mL·min–1, 30˚C, 220 nm, Hypersil ODS 3 V column, (250 mm × 4.6 mm i.d., 5 µm), run time 50 min).
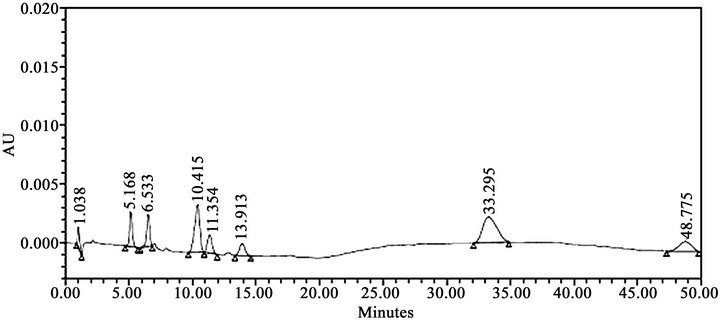
Figure 4. HPLC chromatograms of system suitability solution; (conditions: MeOH:Buffer, 60:40 v/v, Buffer: 0.05 M KH2PO4 + 0.1% Triethylamine, pH = 2.5 with Ortho phosphoric acid, flow 1.0 mL·min–1, 30˚C, 220 nm, waters symmetry C8 (100 × 4.6 mm i.d., 5 μm), run time 50 min).
 (a) Unspiked test sample
(a) Unspiked test sample 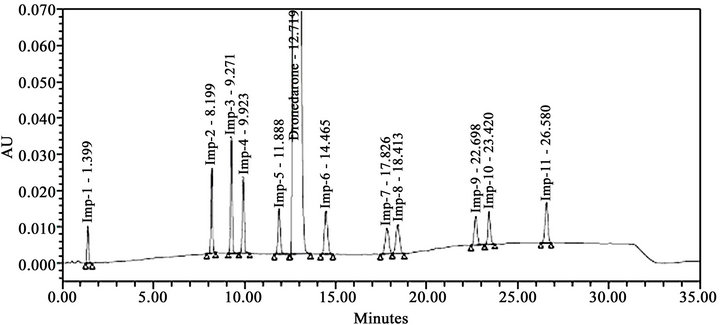 (b) Spiked test sample
(b) Spiked test sample
Figure 5. Typical RP-HPLC chromatograms of: a) dronedarone hydrochloride unspiked test preparation, b) dronedarone hydrochloride spiked test preparation with known impurities (Imp-1 to Imp-11).
Standard solution for dronedarone hydrochloride and all the 11 impurities was prepared in diluent at a concentration of 10 ppm and scanned in UV-visible spectrophotometer. All the eleven impurities and dronedarone hydrochloride shown good response at UV maxima of 219 nm, thus detection at 220 nm was selected for method development purpose. Ammonium dihydrogenorthophospate (0.05 M) and acetonitrile (60:30, v/v) as a mobile phase and C18 e column (250 × 4.6 mm, 5 μm) as a stationary phase with flow rate of 1 mL·min–1 were selected for initial trials. Spike sample analysis revealed that the principal peak (dronedarone hydrochloride) eluted at 27 minutes and other impurities (Imp-1 to Imp-11) were found to be not resolved properly. By decreasing acetonitrile percentage in mobile phases the resolution between Imp-4 and imp-5 was slightly improved but no improvement was observed in the retention of dronedarone, Imp-6 and Imp-7. The careful examination of all the impurities with respect to their physical property revealed that they possess varying polarities i.e. Imp-1 to Imp-5 are highly polar in nature whereas Imp-6, Imp-7 and dronedarone are moderately polar and Imp-8 to Imp-11 are highly non-polar in nature, thus isocratic conditions may not be suitable for separation of closely eluting impurities. More specifically the Imp-11 was not eluted within the 50 min, hence further method development trials were preformed on a gradient method.
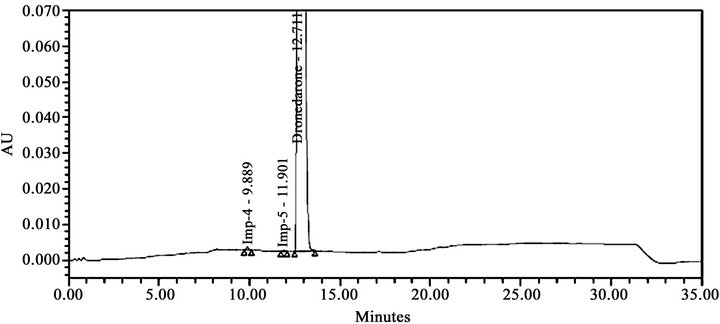
During the optimization of chromatographic conditions, different stationary phases such as C8, C18, Cyano and Phenyl; different mobile phases containing buffers such as phosphate, sulphate and acetate with different pH between 3.0 and 7.0; and various organic modifiers like acetonitrile, methanol and tetrahydrofuran in the mobile phase with different ratios were used. Apart from the co-elution of impurities poor peak shapes for some impurities and dronedarone were noticed during the above trails. Further optimizations to resolve the above shortcomings were explored by introducing various amount of methanol both in mobile phase-A and mobile phase-B. With the introduction of methanol, satisfactory chromatographic separation was achieved for all impurities with dronedarone peak with excellent peak shapes on Ascentis® Express C18 (10 cm × 4.6 mm, 2.7 µm) column. The composition of the mobile phase consisting a mixture of buffer (0.05 M ammonium dihydrogen orthophosphate in water) and methanol in the ratio of 80:20 v/v as solvent A and a mixture of acetonitrile, methanol and water in the ratio of 45:45:10 v/v as solvent B. All analytical system suitability criteria’s were found satisfactory under these optimized chromatographic conditions as described in Section 2.2. In the optimized conditions the dronedarone and all the eleven impurities were well separated with a resolution greater than 1.5 confirming specificity and selectivity of the developed method. The typical retention times of Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, dronebedarone, Imp-6, Imp-7, Imp-8, Imp-9, Imp- 10 and Imp-11 were about 1.4, 8.2, 9.3, 9.9, 11.9, 12.7, 14.5, 17.8, 18.4, 22.7, 23.4, and 26.6 min respectively (Figure 5(a)), and the developed HPLC method was found to be specific for dronedarone and its eleven specified impurities in bulk drug and pharmaceutical dosage form. The system suitability result of developed LC method was given in Table 1.
2.6. Stress Studies/Specificity
2.6.1. Design of Forced Degradation and Specificity Study
Specificity is the ability of the method to measure the analyte response in the presence of its potential impurities [17]. The specificity of the developed LC method for dronedarone hydrochloride was determined in the presence of its impurities and degradation products (namely Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-8, Imp-9, Imp-10, and Imp-11). Forced degradation studies were also performed on dronedarone hydrochloride to
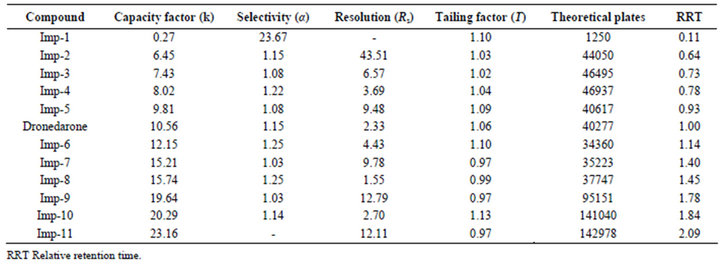
Table 1. System suitability test results.
provide an indication of the stability-indicating property and specificity of the proposed method [7-12]. The stress conditions employed for the degradation study included light (carried out as per ICH Q1B), heat (105˚C), acid hydrolysis (Conc.HCl), base hydrolysis (5 M NaOH), Water hydrolysis (Refluxed for 12 h) and oxidation (5% H2O2). For heat and light studies, the samples were exposed for 10 days, whereas the samples were treated for 12 h for acid hydrolysis, base hydrolysis and for oxidation. The peak purity of the dronedarone hydrochloride stressed samples was checked by using a Waters 2996 photo diode array detector (PDA). The purity angle was within the purity threshold limit in all of the stressed samples, demonstrating the homogeneity of the analyte peak. Assays were carried out for the stress samples against a qualified reference standard. The mass balance (% assay + % of impurities + % of degradation products) was calculated for all of the samples.
2.6.2. Identification of Degradation Product by LC-MS Analysis
A LC-MS study was carried to determine the m/z value of the major degradation product formed under stressed conditions. LC-MS analysis was performed by using triple quadrupole mass spectrometer (API 2000, PE SCIEX) coupled with a Shimadzu HPLC equipped with SPD 10A VP UV-vis detector and LC AT VP pumps [Foster city, CA, USA]. Analyst software was used for data acquisition and data processing. The turbo ion spray voltage was maintained at 5.5 kV and temperature was set at 375˚C. High pure nitrogen gas was used as auxiliary gas and curtain gas. Zero air was used as nebulizer gas. LC-MS spectra were acquired from m/z 50 to 1200 in 0.1 amu steps with 2.0 s dwell time. Dronedarone hydrochloride stressed sample was subjected to LC-MS analysis. The analysis was carried out on Agilent Zorbax SB-Phenyl (250 × 4.6 mm, 5 μm) column under gradient elution by a binary mixture of solvent-A and B. Solvent-A was composed of mixture of 0.1% trifluoroacetic acid solution, whereas solvent-B contains acetonitrile. Detection was carried out at 220 nm and flow rate was kept at 1.2 mL·min–1. Water and acetonitrile mixture in the ratio of 50:50 (v/v) was used as diluent. Data acquisition time was 40 min. The gradient program was set as follows: time (min)/A (v/v): B (v/v); T0.0/55:45, T3.0/55:45, T20.0/ 20:80, T30.0/20:80, T33.0/55:45 and T40.0/55:45.
2.7. Method Validation
The proposed method was validated per ICH guide lines [14,15].
2.7.1. Precision
The precision of the related substance method was investigated by injecting six individual preparations of dronedarone hydrochloride (500 μg·mL–1) spiked with 0.15% each of Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp- 7, Imp-9, Imp-10 and Imp-11. The %RSD for percentage of each impurity was calculated. The intermediate precision (ruggedness) of the method was evaluated by different analyst using different column, different day and different analyst in the same laboratory.
The precision of the assay method was evaluated by carrying out six independent assays of a test sample of dronedarone hydrochloride at 50 μg·mL–1 concentration against a qualified reference standard. The %RSD of six obtained assay values was calculated.
2.7.2. Limit of Detection (LOD) and Limit of Quantification (LOQ)
The LOD and LOQ of all the eight impurities were determined using calibration curve method according to ICH Q2R1 recommendations by establishing the lowest concentration that can be measured. Precision study was also carried out at the LOQ level by injecting six individual preparations of all the impurities and calculating RSD of the area.
2.7.3. Linearity
Linearity test solutions for the assay method were prepared from a stock solution at five concentration levels from 50 to 150% of the assay analyte concentration (25, 37.5, 50, 62.5, and 75 μg·mL–1). The peak area versus concentration data was analyzed with least-squares linear regression. Linearity test solutions for the related substance method were prepared by diluting the impurity stock solution (Section 2.3) to the required concentrations. The solutions were prepared at eight concentration levels from the LOQ to 200% of the specification level (LOQ, 0.05%, 0.10%, 0.15%, 0.20%, 0.25% and 0.3%). The slope and y-intercept of the calibration curve are reported.
2.7.4. Accuracy
The accuracy of an analytical procedure expresses the closeness of agreement between the value, which is accepted either as a conventional true value or an accepted reference value and the value found. Standard addition and recovery experiments were conducted to determine the accuracy of the related substance method for the quantification of all eleven known impurities in the drug substance as well as in the drug product. The study was carried out in triplicate at four concentration levels i.e. LOQ, 0.075%, 0.15% and 0.225% of the analyte concentration (1000 µg·mL–1). The percentage of recoveries for Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-9, Imp-10 and Imp-11 were calculated by using following formula: % Recovery = Amount recovered/Amount added × 100.
2.7.5. Robustness
To determine the robustness of the developed method, the experimental conditions were altered and the resolution between two adjacent peaks was evaluated. The flow rate of the mobile phase was 1.2 ml/min. To study the effect of the flow rate on the resolution, the flow rate was changed by 0.1 units (1.1 and 1.3 ml/min). The effect of methanol in mobile phase-A on the resolution of the impurities was studied by varying the organic solvent by ± 2 mL (methanol: buffer, 82:18 v/v and 78:22 v/v). The effect of the column temperature on the resolution was studied at 32˚C and 38˚C instead of 35˚C. In all these varied conditions, the components of the mobile phase remained constant, as outlined in Subsection 2.2.
2.7.6. Solution Stability and Mobile Phase Stability
The solution stability of dronedarone hydrochloride and its impurities in the related substance method was carried out by leaving a spiked sample solution in a tightly capped volumetric flask at room temperature for 48 h. Impurity content was determined for every 6 h interval up to the study period. Mobile phase stability was performed for 48 h by injecting the freshly prepared sample solutions for every 12 h interval. The content of impurities was determined in the test solutions. The prepared mobile phase remained constant during the study period. The solution stability of dronedarone hydrochloride in the assay method was carried out by leaving both the sample and reference standard solutions in tightly capped volumetric flasks at room temperature for 48 h. The same sample solutions were assayed for in 12 h intervals over the study period. The mobile phase stability was also examined by assaying the freshly prepared sample solutions against freshly prepared reference standard solutions for 12 h intervals up to 48 h. The prepared mobile phase remained constant during the study period. The %RSD of the dronedarone hydrochloride assay was calculated for the mobile phase and solution stability experiments.
3. Results and Discussion
3.1. Structural Elucidation
All impurities (Imp-1 to Imp-11) were characterized with the help of MS and NMR techniques. The mass spectral data and 1H NMR chemical shift values of these impurities are presented in Table 2.

Table 2. 1H NMR chemical shift values and mass spectra data.
3.2. Results of Forced Degradation Studies
Degradation was not observed in dronedarone hydrochloride stressed samples subjected to light, heat, and base and water hydrolysis. Significant degradation of the drug substance and product was detected under acid and peroxide stressed degradation, leading to the formation of two major unknown degradation products one is at 0.93 RRT (due to acid hydrolysis) and another one is at 1.39 RRT (due to peroxide degradation) as shown in Figure 6. The data on degradation studies revealed that the degradation products were well separated from the dronedarone and known related substances, moreover peak purity data of dronedarone hydrochloride indicated that the compound is spectrally pure. The data on forced degradation studies is given in Table 3 shows that the mass balance of stressed samples was close to 99.9% confirming the stability-indicating power of the developed method. The acid and peroxide stressed samples were subjected to LC-MS analysis for identification of degradation peak. The m/z of detected peaks in acid stressed sample were 501.3[(M+H)+] and 479.5[(M+H)+] and in peroxide stressed sample was 1041.2[(M+H)+]. Based on these observed m/z, starting material and reactants used in the synthetic scheme (Figure 1) the schematic diagram of possible structures of degradation has been drawn (see Figure 7). The same predicted impurities (Imp-5, and Imp-7) were synthesized and co-injected with dronedarone and confirmed by chromatographic retention times.
 (a) Peroxide treated test sample
(a) Peroxide treated test sample 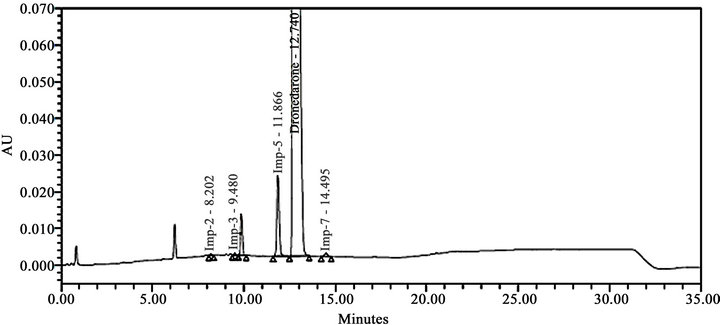 (b) Acid treated test sample
(b) Acid treated test sample
Figure 6. Typical RP-HPLC chromatograms of forced degradation: (a) peroxide treated test sample; (b) acid treated test sample.
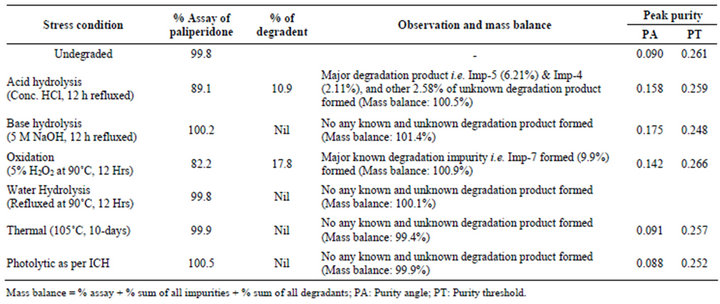
Table 3. Forced degradation results.
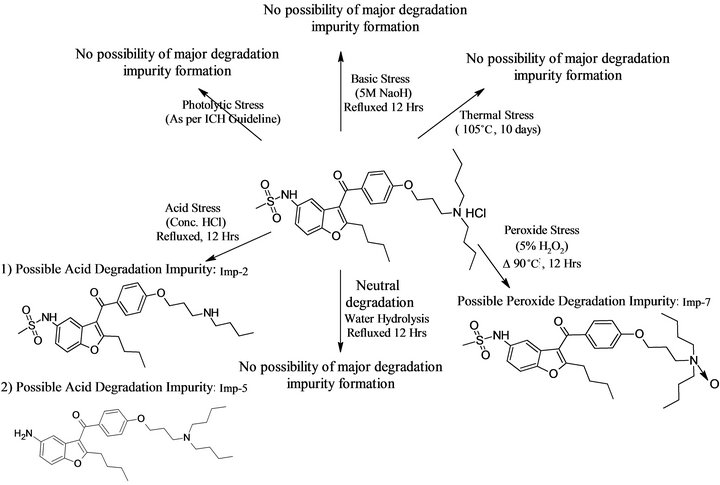
Figure 7. Schematic diagram showing possible formation of degradation related impurities.
3.3. Method Validation
3.3.1. Precision
The % RSD of dronedarone hydrochloride during the assay method precision study was within 0.5% and the % RSD values of the area of Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11 in the related substance method precision study were within 1.0%. The %RSD of the results obtained in the intermediate precision study was within 0.8% and the %RSD of the areas of Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp- 6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11 were well within 1.8%, revealing the high precision of the method.
3.3.2. Limit of Detection and Limit of Quantification
The limits of detection and quantification of dronedarone hydrochloride, Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp- 6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11 (analyte concentration of 500 µg·mL–1) for a 10 µl injection volume are given in Table 4. The precision at the LOQ concentration for Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11 was below 2.2%.
3.3.3. Linearity
The linear calibration plot for the assay method was obtained over the tested calibration range (12.5-37.5 µg·mL–1) and the obtained correlation coefficient was greater than 0.999. The results revealed an excellent correlation between the peak area and analyte concentration. The linear calibration plot for the related substance method was determined over the calibration ranges (LOQ to 0.3%) for Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11, and a correlation coefficient of greater than 0.99 was obtained. The RF of each impurity was determined using the slope of the dronedarone hydrochloride plot against the each impurity plot.
3.3.4. Accuracy
The percentage recovery of dronedarone hydrochloride in the drug substance and product ranged from 98.5 to 101.6 and from 96.2 to 101.6, respectively. The percentage recoveries of Imp-1, Imp-2, Imp-3, Imp-4, Imp-5, Imp-6, Imp-7, Imp-8, Imp-9, Imp-10 and Imp-11 in the drug substance and product ranged from 95.9 to 102.1 and from 96.3 to 102.5 respectively. The HPLC chromatograms of spiked samples at the 0.15% level of all eleven impurities in the dronedarone hydrochloride drug substance sample are shown in Figure 5. The accuracy results are depicted in Tables 5 and 6.
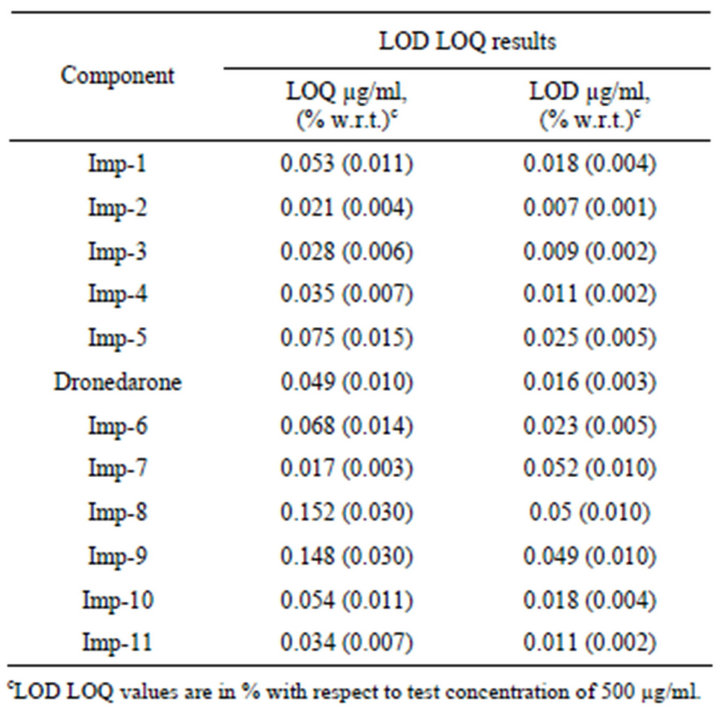
Table 4. Linearity, limit of detection (LOD) and limit of quantitation (LOQ) data for dronedarone hydrochloride and related substances.
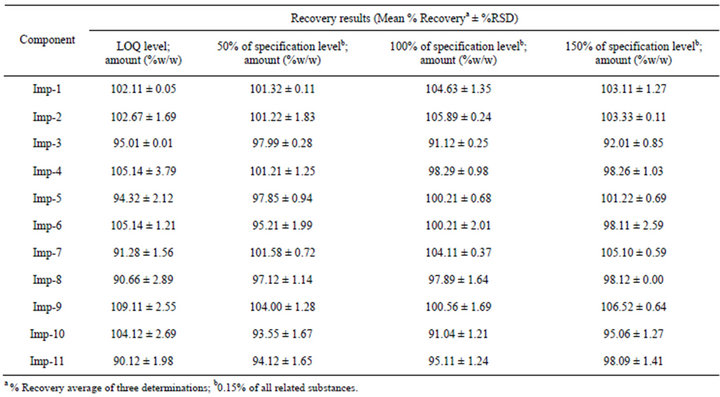
Table 5. Accuracy data of related substances.
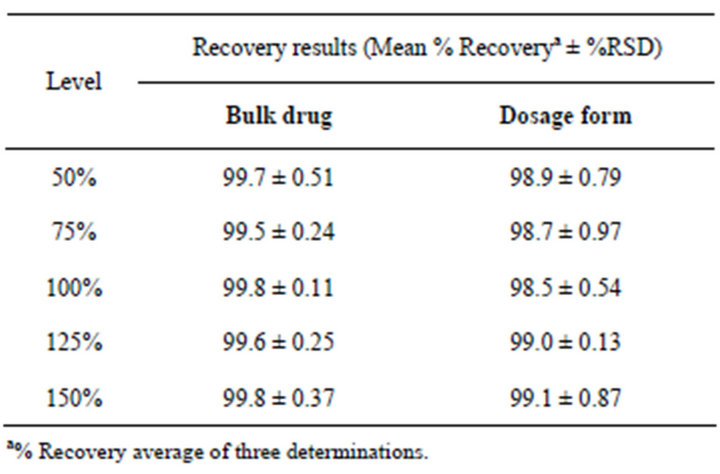
Table 6. Accuracy data of assay of dronedarone hydrochloride in bulk drug and pharmaceutical form.
3.3.5. Robustness
In all the deliberate varied chromatographic conditions (flow rate, column temperature and pH of buffer) the resolution between Imp-7 and -8 was greater than 1.5, while other impurities were greater than 2.0 illustrating the robustness of the method.
3.3.6. Solution Stability and Mobile Phase Stability
The RSD of assay of dronedarone hydrochloride during solution stability and mobile phase stability experiments were within 0.8%. There is no significant changes were observed in the content of any of the impurity during solution stability and mobile phase stability experiments. The accuracy of the assay at each time point against the initial value is between 99.8% and 100.1%. The accuracy of the content of each impurity against the initial value is between 90.7% and 105.2%. The solution stability and mobile phase stability experimental data confirms that sample solutions and mobile phase used were stable up to 48 h. It is an advantage that from the same run, the assay results and impurities quantification can be derived. This helps to reduce the analysis time and number of samples that can be analyzed till 48 h in the same sequence in the quality control during the regular analysis.
4. Application of the Method
The analysis of commercial formulation sample and bulk drug sample indicated that the method is specific and selective for determination of related substances and assay in the formulation and bulk drug samples (Table 7). The developed method is capable for quantitative analysis of dronedarone in the bulk drug and in a pharmaceutical dosage form.
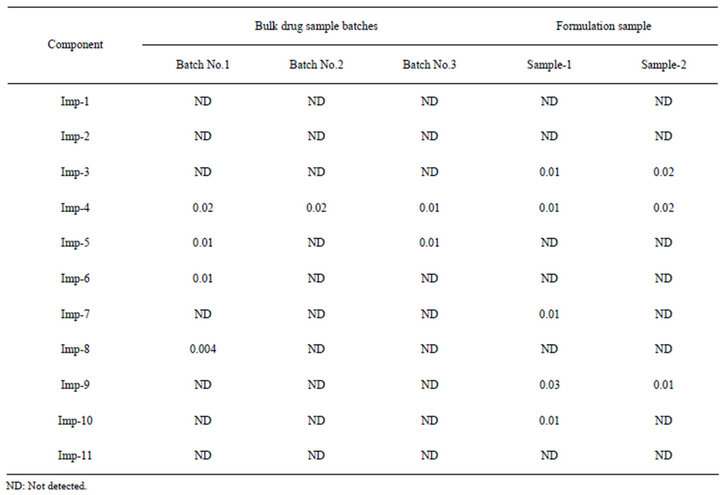
Table 7. Results of formulated tablet analysis and bulk drug batches sample analysis.
5. Conclusion
The method presented in this communication describes the development of a selective, accurate and sensitive gradient RP-HPLC method for dronedarone hydrochloride that separates all related substances with good resolution. Eleven related substances are separated from its dronedarone peak and quantitative determination has been performed by this method. The developed method is validated to ensure the compliance in accordance with ICH guidelines. The method is found to be simple, selective, precise, accurate and robust hence, this method can be used for routine testing as well as stability analysis of dronedarone hydrochloride drug substance and drug products. All statistical results (percentage, mean, R.S.D., percentage difference and recovery %) are within the acceptance criteria.
6. Acknowledgements
The authors wish to thank the management of Megafine group for supporting this work. We would also like to thank colleagues in the division of Research and Development of Megafine Pharma (P) Ltd for their cooperation in carrying out this work.
REFERENCES
- “Australian Public Assessment Report for Dronedarone Hydrochloride,” Submission No: PM-2008-3045-3, 2010. http://www.tga.gov.au/pdf/auspar/auspar-multaq.pdf
- Assessment Report for Multaq, “Evaluation of Medicines for Human Use,” European Medicines Agency, Procedure No. EMEA/H/C/001043, 2009. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_Public_assessment_report/human/001043/WC500044538.pdf
- Center for Drug Evaluation and Research, “Chemistry Review(s),” Application Number: 22-425, 2009. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022425s000_ChemR.pdf
- FDA, “FDA Approves Multaq to Treat Heart Rhythm Disorder,” 2009.
- S. B. Bari, B. R. Kadam, Y. S. Jaiswal and A. A. Shirkhedkar, “Impurity Profile: Significance in Active Pharmaceutical Ingredient,” Eurasian Journal of Analytical Chemistry, Vol. 2, No. 1, 2007, pp. 177-189.
- S. Gorog, “The Importance and the Challenges of Impurity Profiling in Modern Pharmaceutical Analysis,” Trends in Analytical Chemistry, Vol. 25, No. 8, 2006. doi:10.1016/j.trac.2006.05.011
- C. Xie, S. L. Yang, D. F. Zhong, X. J. Dai and X. Y. Chen, “Simultaneous Determination of Dronedarone and Its Active Metabolite Debutyldronedarone in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry: Application to a Pharmacokinetic Study,” Journal of Chromatography B, Vol. 879, No. 28, 2011, pp. 3071-3075.
- D. Patel and A. Choudhury, “Development and Validation of Dronedarone HCl In Plasma by RP-HPLC Method Coupled with UV-Detector,” Inventi Impact: Biomedical Analysis, Vol. 2012, 2012, Article ID: Inventi:pba/47/12. http://www.inventi.in/Article/pba/47/12.aspx
- N. Tondepu1, S. S. Sait, K. V. Surendranath, R. K. Kaja1 and S. Kumar, “A Stability Indicating HPLC Method for Dronedarone in Bulk Drugs and Pharmaceutical Dosage Forms,” American Journal of Analytical Chemistry, Vol. 3, 2012, pp. 544-551. doi:10.4236/ajac.2012.38072
- A. Patel and J. Akhtar, “RP-HPLC Method Development and Validation of Dronedarone HCl in Its Pure Form and Tablet Dosage Form,” Journal of Chemical and Pharmaceutical Research, Vol. 4, No. 4, 2012, pp. 2173-2179.
- B. Dabhi, Y. Jadeja, M. Patel, H. Jebaliya, D. Karia and A. Shah, “Method Development and Validation of StabilityIndicating RP-HPLC Method for Quantitative Analysis of Dronedarone Hydrochloride in Pharmaceutical Tablets,” Scientific Pharmacology, in press, pp. 1209-1215.
- US Food and Drug Administration, “ICH Guidelines on Impurities in New Drug Substances Q3A(R2),” Federal Register, FDA, Silver Spring, 2006.
- US Food and Drug Administration, “ICH Guidelines on Impurities in New Drug Substances Q7A,” Federal Register, FDA, Silver Spring, 2005.
- A. C. Mali, S. S. Ippar, M. S. Bodke, N. S. Patil and V. T. Mathad, “An Improved, and Efficient Process for the Production of Dronedarone Hydrochloride, an Antiarrhythmic Drug,” Organic Process Research & Development, Vol. 17, No. 5, 2013, pp. 863-868.
- The United States Pharmacopeia, “Validation of Compendial Methods,” 2008.
- US Food and Drug Administration, “ICH Guidelines on Validation of Analytical Procedures, Text and Methodology Q2(R1),” Federal Register, FDA, Silver Spring, 2005.
- J. T. Carstensen and C. T. Rhodes, “Drug Stability Principles and Practices,” 3rd Edition, Marcel Dekker, New York, 2000.
NOTES
*Corresponding author.