American Journal of Analytical Chemistry
Vol. 3 No. 2 (2012) , Article ID: 17288 , 9 pages DOI:10.4236/ajac.2012.32020
Quantification of Flavonoids, Naphthopyranones and Xanthones in Eriocaulaceae Species by LC-PDA
1UEMS, Course of Chemistry, State University of Mato Grosso do Sul, Dourados, Mato Grosso do Sul, Brazil
2Department of Biological Sciences, Faculty of Pharmaceutical Sciences, UNESP-Universidade Estadual Paulista, Araraquara, Sao Paulo, Brazil
3Institute of Biosciences, Department of Botany, University of Sao Paulo, Sao Paulo, Brazil
4Campus Experimental do Litoral Paulista, UNESP-Universidade Estadual Paulista, Sao Vicente, Sao Paulo, Brazil
5Institute of Chemistry, Department of Organic Chemistry, UNESP-Universidade Estadual Paulista, Araraquara, Sao Paulo, Brazil
Email: *loursant@iq.unesp.br
Received November 19, 2011; accepted December 21, 2011; accepted January 5, 2012
Keywords: LC-PDA; Naphthopyranone; Flavonoid; Xanthone
ABSTRACT
The linearity, stability, accuracy and inter-day precisions of the assay method were evaluated in methanolic aerial-part extracts of Paepalanthus giganteus and Syngontnhus nitens from the Eriocaulaceae family. Their small capitulae hinder morphological analysis, and thus complicate taxonomic studies of these species, which present anti-ulcer, antimutagenic and antioxidant activities. Taxonomic studies of these plants revealed that the Paepalanthus genus presents flavonols and naphthopyranones while the Syngontnhus genus has flavone and xanthone as majority compounds. The prepared samples were analyzed quantitatively by High Performance Liquid Chromatography with PDA detection for the presence of quercetin, luteolin, 3,6-dimethoxy-1,5,7-tri-hydroxyxanthone and paepalantine. The substances were recovered from these samples at rates from 98.01% to 99.99%. The coefficient of variation in the quantitative analysis of the sample compounds was under 5%. The linearity of the method was determined by linear regression. The analysis of the samples spiked with known amounts of analyte demonstrated that the response was proportional to the concentrations of the samples with respective determination coefficients of r2 = 0.9999 (luteolin and 3,6-dimethoxy-1,5,7-tri-hydroxyxanthone) and r2 = 0.9998 (quercetin and paepalantine) for the linear range of the analytical calibration curves of the samples. The detection limits were 0.07 µg·mL–1 for quercetin and luteolin, 0.06 µg·mL–1 for 3,6-dimethoxy-1,5,7-tri-hydroxyxanthone and 0.10 µg·mL–1 for paepalantine. The quantification limits were 0.23 µg·mL–1 for quercetin and luteolin, 0.20 µg·mL–1 for 3,6-dimethoxy-1,5,7-tri-hydroxyxanthone and 0.33 µg·mL–1 for paepalantine by LC. The method was considered sensitive for quantification of the quercetin, luteolin, 3,6-dimethoxy-1,5,7-tri-hydroxyxanthone and paepalantine in plant samples.
1. Introduction
Eriocaulaceae is an unusual monocotyledonous plant family that shares with the Asteraceae the presence of a capitula in the inflorescence, a fact that accounts for the phrase “the Compositae of the monocotyledons”, which has been employed to emphasize this morphological convergence [1,2].
Our interest in naphthopyranones and xanthones has now led us to investigate the methanolic extract from the capitula and scape of two species, Paepalanthus giganteus and Syngonanthus nitens.
Syngonanthus nitens (Bong.) Ruhland is a grass-like species of Eriocaulaceae [3], a pantropical, predominantly herbaceous monocotyledonous family that comprises around 1100 species in 11 genera [1,2]. The plants are clonal, and fertile rosettes can annually produce from 1 to 10 scapes (flower stalks or stems), with each scape bearing a capitulum flower [4]. The flower stems of S. nitens are bright and assume a beautiful golden color when dried, acquiring a phenomenal aesthetic similarity to spun gold, hence their common name “capim dourado” (golden grass). This grass-like species occurs throughout the Brazilian Cerrado, in areas of intermediate humidity within the humid grasslands [4].
The chemical studies performed by Pacifico [5] identified significant differences between capitulae and the scapes of S. nitens. Xanthones and flavones were identified in the capitulae, while the scapes presented mostly flavones. This study also served to establish taxonomic relationships between the genera Leiothrix and Syngonanthus.
Paepalanthus giganteus Sano is a species belonging to the family Eriocaulaceae, genus Paepalanthus. It constitutes the largest genus in the family, comprising about 450 species [6]. This genus has as its main feature great morphological variability [7], which hampers morphological classification.
The present work includes the first known chemical investigation of the species P. giganteus. The LC-PDA profile of the methanolic extract showed UV spectra typical of flavonols and naphthopyranones (Table 1, Figures 1(a) and 1(b)).
Given the difficulty of obtaining sufficient mass of polar extracts for the purification of compounds due to the smallness of the capitulae in the species Eriocaulaceae, new methodologies must be sought to simplify its chemical study. Therefore, the present study aims to evaluate the sensitivity, linearity, accuracy and inter-day precision of flavonoids, xanthones and naphthopyranones in methanol extracts of capitulae and scapes from S. nitens and P. giganteus, two member species of Eriocaulaceae.
2. Materials and Methods
2.1. Reagents
Spectroscopy grade acetonitrile and methanol were purchased from Merck (Darmstadt, a collection in our laboratory. Stock mixtures of these standards were constituted from individual solutions, dissolved in methanol and then used as external standards. Standards of quercetin and luteolin were purchased from Sigma Aldrich. Standards of the 3,6-dimethoxy-1,5,7-tri-hydroxyxanthone and paepalantine were obtained from a collection purified by chromatogramphy in our laboratory and revealed a purity of 98.5%. These stock mixtures were used as external standards.
2.2. Plant Material
Capitulae and scapes of S. nitens and P. giganteus were collected, respectively, in December 2009 in Jalapão city, Tocantins state, and February, 2010 at Serra do Cipó, Minas Gerais state, Brazil, and authenticated by Professor Dr. Paulo Takeo Sano of the São Paulo University (USP), Sao Paulo state, Brazil. Voucher specimens of S. nitens (SPF 189975) and P. giganteus (3736 SPF) were deposited at the Herbarium of the IB-USP.
2.3. Extraction
Dried and powdered capitulae (416 g) of S. nitens and

Table 1. Compounds identified in methanolic extracts from S. nitens and P. giganteus.
(256 g) of P. giganteus and scapes (176.7 g) of P. giganteus and (410 g) of S. nitens were separately and successsively extracted for one week, at room temperature, with methanol. The crude methanolic extracts were obtained after filtration and evaporated to dryness in a vacuum at a temperature no greater than 40˚C; the respective masses and recuperation rates obtained from S. nitens were: 11.8 g from capitulae (2.87%), and 16.0 g from scapes (3.90%) while those from P. giganteus were: 15.0 g from capitulae (5.86%), and 6.81 g from scapes (3.85%).
2.4. Clean Up of Extracts
In the clean-up step of the methanolic extracts, a small mass (1 mg) of sample for each 10 mg of the C-18 reversed-phase was applied to the SPE cartridges. In C-18, the cartridge had been previously activated with methanol (4 mL) and then environed with water (5 mL). The material to be applied in support of SPE had been previously dissolved in approximately 0.5 mL of methanol with the aid of ultrasound and 2 mL of H2O was added into the solution. The elution in C-18 followed the general order (methanol/water, 5:95 v/v, methanol/water, 1:1 v/v, and methanol); however, adjustments were made during the optimizations. All the fractions were injected into the LC-PDA and the fractions with the highest number of peaks are represented in Figures 1(a) and 1(b).
2.5. Preparation of Standards for Analysis by LC-PDA
The samples of standards (quercetin, luteolin, 3.6-dimethoxy-1,5,7-trihydroxyxanthone and paepalantine (0.1 g)) were dissolved in 3 mL of methanol, separately, filtered through a 0.45 mm Millex filter and the resulting solutions were diluted separately in methanol in a volumetric flask (5 mL each) for analysis by LC-PDA.
2.6. Fingerprint of the Methanolic Extract of the Capitulae and Scapes from P. giganteus by LC-PDA
The chromatographic profiles of methanolic extracts of the capitulae and scapes from P. giganteus were obtained in a LC-(Jasco ®) equipped with a photodiode array detector (PDA), utilizing a C-18 reversed-phase column, Phenomenex ® Luna (2) (250 × 4.6 mm id, 5 mm). The mobile phase composition was: water acidified with 0.1 % TFA, pH 2.5 (eluent A) and methanol with 0.1% TFA, pH 5.5 (eluent B). The gradient program was as follows: 5% - 100% B (55 min) and flow rate 1.0 mL·min–1 monitored at λ = 200 - 400 nm.
 (a)
(a)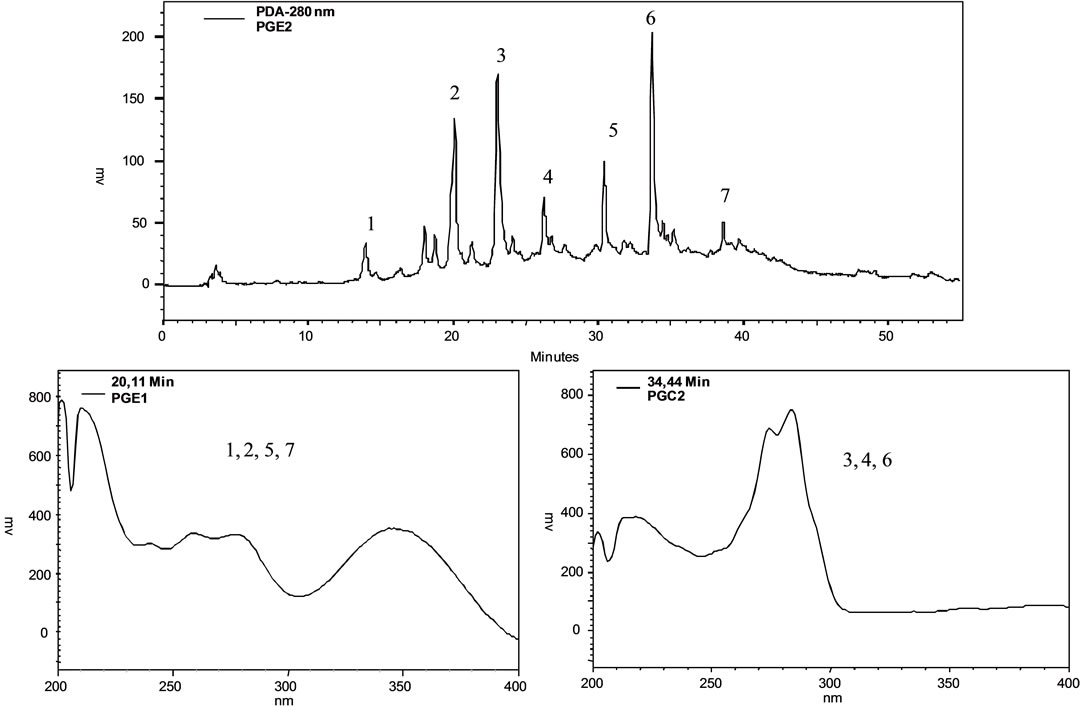 (b)
(b)
Figure 1. (a) Fingerprint by LC-PDA of the methanolic extract from P. giganteus capitulae. For chromatographic conditions, see the experimental section; (b) Fingerprint by LC-PDA of the methanolic extract from P. giganteus scapes. For chromatographic conditions, see the experimental section.
2.7. Preparation of Methanolic Extracts for Quantification by LC-PDA
Samples of capitulae from P. giganteus (1 g) and S. nitens (1 g) and scapes S. nitens (1 g) and P. giganteus (1 g) were separately extracted with 8 mL of methanol for 60 min in ultrasound, filtered with a 0.45 mm Millex filter after which the solutions were diluted with methanol in a volumetric flask (10 mL) for LC analysis. The validation procedure was performed according to the International Conference on Harmonization Guidlines (Validation of Analytical Procedures, Text on Validation of Analytical Procedures Q2A and Q2B) [8].
2.8. LC-PDA Analysis of Standards and Methanolic Extracts of Capitulae and Scapes from S. nitens and P. giganteus
The methanolic extracts obtained from the samples were analyzed on an analytical LC (Varian 210) system with a ternary solvent delivery system equipped with an autosampler, a photodiode array detector (PDA) and the software Star WS (Workstation), which was used to measure the peak areas of the chromatogram. The LC column was a C-18 (25 cm × 4.6 mm; particle size, 5 mm) (Luna, Phenomenex, Torrance, CA, USA), with a small pre-column (2.5 cm × 3 mm) containing the same packing, used to protect the analytical column. The flow rate of 1.0 mL/min and injected volume of 10 μL were used for each analysis. All chromatographic analyses were performed at 22˚C. Elution was carried out using the following solvent-gradient program for standards (quercetin and paepalantine) and capitulae and scapes from S. nitens: water:acetonitrile (85:15, v/v) taking 40 minutes to reach 45% water and 55% acetonitrile, then returning in exactly 5 minutes to the initial conditions (Figures 2(a) and (b)). For the standards (luteolin and 3,6-dimethoxy- 1,5,7-trihydroxyxanthone) and capitulae and scapes from P. giganteus: water: acetonitrile (80:20, v:v) taking 60 minutes to reach 100% acetonitrile, returning after 5 minutes to the initial conditions (Figures 3(a) and (b)).
2.9. Method Validation
2.9.1. Determination of the Detection and Quantification Limits
The detection limits were determined by injecting (n = 5) solutions of quercetin, luteolin, 3,6-dimethoxy-1,5,7-trihydroxyxanthone and paepalantine of known concentration (10 µL each) and then decreasing the concentrations of the samples until detection of a peak with a signal /noise ratio of 3. The corresponding concentration was considered the minimal detectable concentration. The quantification limits were determined by performing the same methodology and thus, the quantification limit was defined as the chromatographic peak having a signal/ noise ratio of 10.
2.9.2. Extraction Recovery
The extraction efficiency (recovery) was determined by analyzing aliquots of each sample spiked with the standards corresponding to low, medium and high concentrations. The spiked samples were submitted to the same procedure as described in Section 2.7.
2.9.3. Linearity
The content estimation of the quercetin (0.20 - 40.00 µg·mL–1), luteolin (0.23 - 40.00 µg·mL–1), paepalantine (0.20 - 40.00 µg·mL–1) and 3,6-dimethoxy-1,5,7-trihydroxyxanthone (0.20 - 40.00 µg·mL–1) in the samples was performed by external calibration. The compounds in the study were dissolved separately in spectroscopy grade methanol in order to obtain stock solutions, which were appropriately diluted for each of the substances. Aliquots of 10 µL dilutions for each standard were analyzed via LC with each determination being carried out five times. For each standard, the corresponding chromatogram was obtained and a graph was constructed from the mean of the chromatogram areas plotted against the concentration of each substance. A linear least-square regression of the peak areas as a function of the concentrations was performed to determine the correlation coefficients. The equation parameters (slope and intercept) of each standard curve were used to obtain the concentration values for the samples. Specimens with an analytic concentration exceeding the analytical curve were re-assayed upon appropriate dilution of the samples.
2.9.4. Accuracy and Precision
The accuracy of the assayed method was evaluated by performing replicate analyses against an analytical calibration curve and calculating the mean percentage differences between the theoretical values and the measured values. The accuracy values in the inter-day variation studies using LC at low, medium and high concentrations of quercetin, luteolin, paepalantine and 3,6-dimethoxy-1, 5,7-trihydroxyxanthone were evaluated in extracts A-D. Method precision, expressed as the percentage of the coefficient of variation (CV) of the replicate measurements, was tested for inter-day repeatability via LC. The inter-day variability of the method was determined from three different analyses (n = 5) of each sample with an addition of known amounts of analyte at low, medium and high concentrations.
2.9.5. Stability Study
The stability of the working standard solutions was tested at 22˚C (working temperature), 4˚C and –20˚C (storage temperatures). The stability of quercetin, luteolin, paepalantine and 3,6-dimethoxy-1,5,7-trihydroxyxanthone in the
 (a)
(a)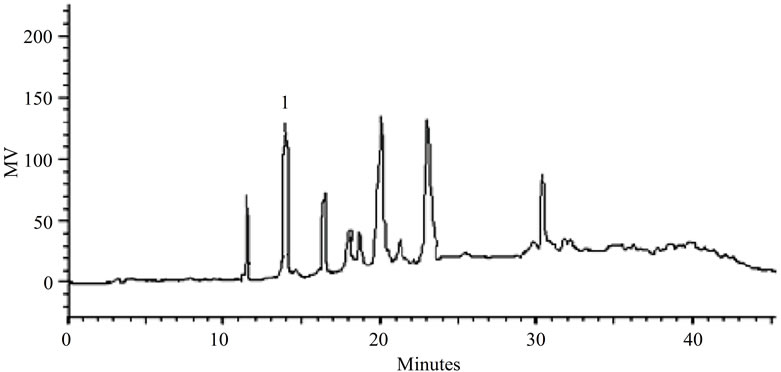 (b)
(b)
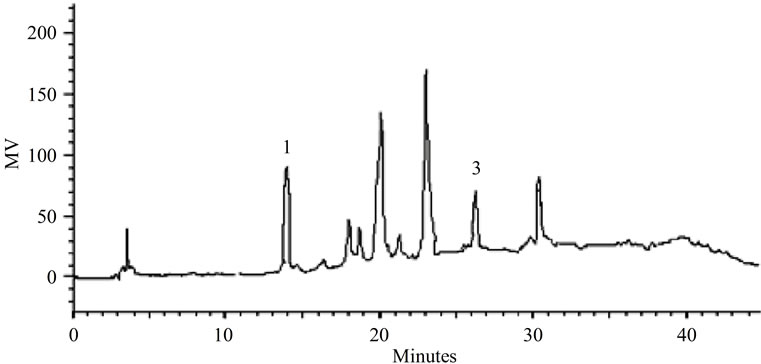
Figure 2. (a). LC-PDA of methanolic extract from S. nitens capitulae. For chromatographic conditions, see the experimental section; (b) LC-PDA of methanolic extract from S. nitens scapes. For chromatographic conditions, see the experimental section.
 (a)
(a)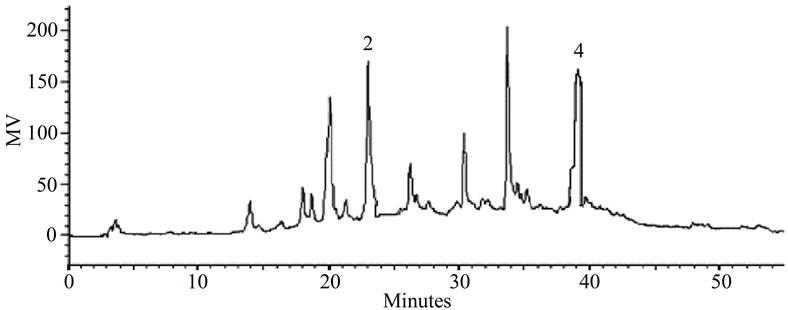 (b)
(b)
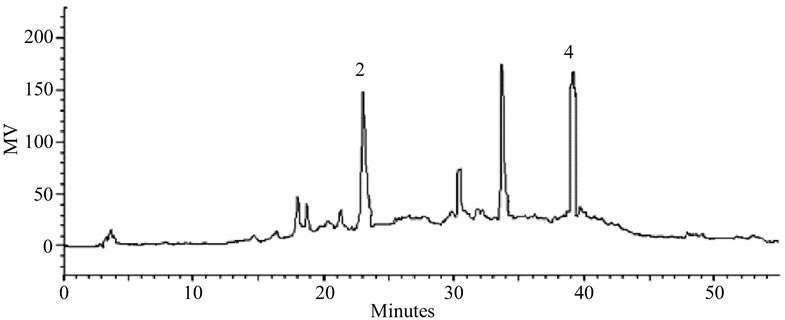
Figure 3. (a) LC-PDA of methanolic extract from P. giganteus capitulae. For chromatographic conditions, see the experimental section. (b) LC-PDA of methanolic extract from P. giganteus scapes. For chromatographic conditions, see the experimental section.
samples was evaluated during all the storage steps (i.e., at room temperature, at 4˚C and at –20˚C). Spiked samples were analyzed against the analytical calibration curves immediately after preparation (reference values) and after storage. Stability was defined as being less than 2% loss of the initial drug concentration in the stated time.
3. Results and Discussion
Earlier studies by our group have demonstrated promising antiulcer activity of S. nitens. Studies of capitulae and scapes of S. nitens [5] showed a high content of polar substances. The authors used the technique of high-speed countercurrent chromatography (HSCCC) for the study, thus yielding impure fractions in which some flavonoids and xanthones were identified. The complexity of the extracts comprised the phytochemical study of this material using traditional techniques of isolation, time and solvent consumption. The many other chromatographic techniques used to isolate the substances included gel permeation (GPC) using Sephadex LH-20 which was not efficient in the separation of secondary metabolites. Factors such as the strong adsorption of substances and the necessity of using large amounts of solvents for the elution of metabolites with subsequent steps for the regeneration of support led us to establish a new research strategy.
To assess P. giganteus, we are starting from its phytochemical studies; however, the paucity of material collected has also resulted in a lower amount of extract, which sometimes hinders the purification of substances for further identification. Thus, we opted to develop a methodology based on analysis by LC-PDA in an attempt to enable direct identification of major secondary metabolites using co-injection of authentic standards and /or substances previously isolated and characterized in species of the genus.
3.1. Identification of the Compounds with LC-PDA
Identification of the compounds with the aid of PDA detector scanning in the spectral range of 200 - 600 nm revealed two classes of compounds in the methanolic extracts of capitulae and scapes of P. giganteus (Figures 1(a) and (b)). Flavonols and naphthopyranones were easily identified based on their absorption spectra in the UV region. Flavonols exhibited bands around 260 nm and 346 nm [9]. The naphthopyranones showed maximum intensity of absorption bands in the region of 273 nm and a weak band at 325 nm [10], (Figures 1(a) and (b)).
Some of the secondary metabolites found in methanol extracts of capitulae and scapes from S. nitens (Figures 2(a) and (b)) and P. giganteus (Figures 3(a) and (b)) could be identified unambiguously by performing co-injection experiments in which aliquots of the extracts and standards were mixed and diluted to a known volume, and analyzed through LC by more than one elution method (gradient and/or isocratic). Thus, the peaks of the chromatograms in Figures 2(a), 2(b), 3(a), 3(b) with retention times tr (13.47; 24.22; 27.79 and 39. 04 min) were identified as luteolin, quercetin, 3,6-dimethoxy-1,5,7-trihydroxyxanthone and paepalantine, respectively (Tables 1 and 2, Figure 4). The relative standard deviation for the retention times of replicated injections (n = 5) were less than 5%, thereby demonstrating good repeatability.
3.2. Method Validation
3.2.1. Linearity
Calibration curves were determined by linear regression using LC (Table 3). The linearity for compounds 1-4 was assessed at 5 concentration ranges, respectively, for quercetin (0.20 - 40.00 µg·mL–1), luteolin (0.23 - 40.00 µg·mL–1), paepalantine (0.20 - 40.00 µg·mL–1) and 3,6- dimethoxy-1,5,7-trihydroxyxanthone (0.20 - 40.00 µg·mL–1) (Table 3). The average standard errors for the peak areas of replicated injections (n = 5) were less than 1%, thus showing good repeatability of the calibration curve. The respective coefficients of determination were r2 = 0.9998 for quercetin and paepalantine and r2 = 0.9999 for luteolin and 3,6-dimethoxy-1,5,7-trihydroxyxanthone.
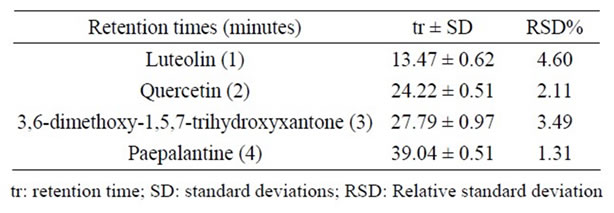
Table 2. Retention times of the standard samples.
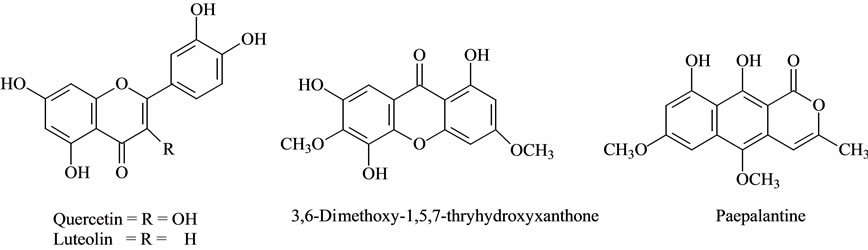
Figure 4. Structure of compounds identified.
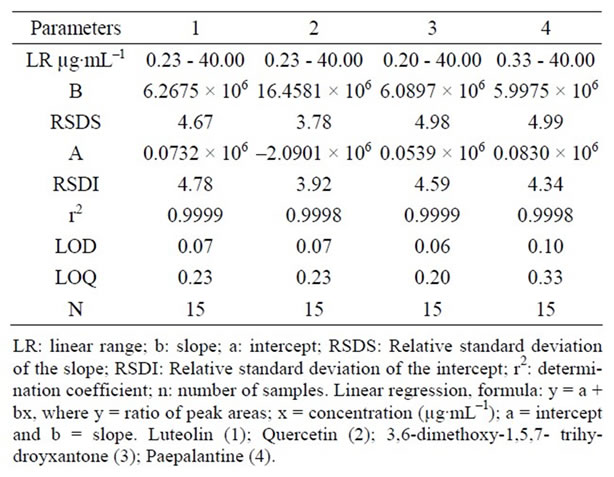
Table 3. Regression data of the analytical calibration curves for quantitative determination of the substances by LC.
The analytical procedure was verified from the linearity of the assayed method as expressed by the correlation coefficient obtained, which was evaluated by analyzing each sample (extracts A-D) spiked with a known amount of the analyte at low, medium and high concentrations, which showed that the response was proportional to the concentrations of the samples; the respective coefficients of determination were r2 = 0.9994 for quercetin and paepalantine and r2 = 0.9996 for luteolin and 3,6-dimethoxy- 1,5,7-trihydroxyxanthone.
3.2.2. Detection Limit
The detection limits were 0.07 µg·mL–1 for quercetin and luteolin, 0.06 µg·mL–1 for 3,6-dimethoxy-1,5,7-trihydroxyxanthone and 0.10 µg·mL–1 for paepalantine (Table 3) based on the signal-to-noise ratio (S\N) for 3.
3.2.3. Quantification Limit
The quantitation limit was defined as the lowest concentration level that provided a peak area with a signal-to-noise ratio higher than 10, a precision (% CV) within ±5% and accuracy (% recovery) between 98% and 100%. The quantification limits by LC were 0.23 µg·mL–1 for quercetin and luteolin and 0.20 µg·mL–1 for 3,6-dimethoxy-1,5,7-tri-and hydroxyxanthones 0.33 µg·mL–1 for paepalantine. The results suggest that substances in plants at these concentrations can be estimated accurately.
3.2.4. Extraction Recovery
The efficiency of each analytical procedure was evaluated by calculating the recovery values.
Recovery (98.01% - 99.99%) results show that the procedure utilized to extract the substances of interest was efficient with relative standard deviation lower than ±5% (Table 4), thus ensuring good recovery of these substances in plants.
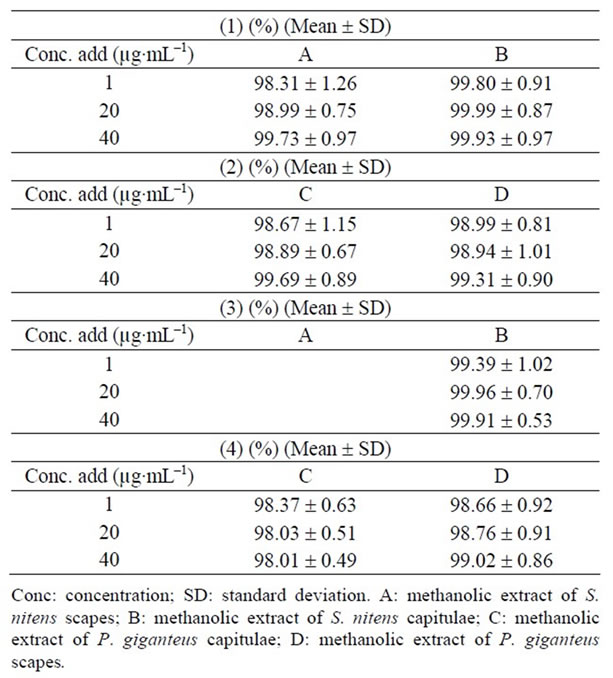
Table 4. Recovery of the (1) and (3) in methanolic extracts of S. nitens and (2) and (4) in methanolic extracts of P. giganteus (n = 5).
3.2.5. Stability
The identification of quercetin, luteolin, paepalantin and 3,6-dimethoxy-1,5,7-trihydroxyxanthone and the samples (methanolic extracts of capitulae and scapes of the species studied) was performed by comparing their retention times with authentic standards through adding standard solutions to the samples analyzed by LC and by comparing its UVVis spectrum [9,10]. There were no alterations detected in the working solutions prepared (quercetin, luteolin, 3,6- dimethoxy-1,5,7-trihydroxyxanthone and paepalantine) after 24 hours at 22˚C, two months at 4˚C and four months of storage at –20˚C. Therefore, this validated method for the determination of the flavonoids, xanthones and naphthopyranones can be considered adequate to indicate the stability of the solutions.
3.2.6. Accuracy and Precision
As to the precision of the assay, inter-day coefficients of variation were lower than ±5%. The international acceptance criteria (CV for precision better than 15% and recovery values) were met [11]. The inter-day variability of the method tested was determined at low, medium and high concentrations. The results are presented in Table 5. These data indicate that the method was reproducible on three different days.
3.3. Contents in S. nitens and P. giganteus
The contents of substances were described in Table 6.
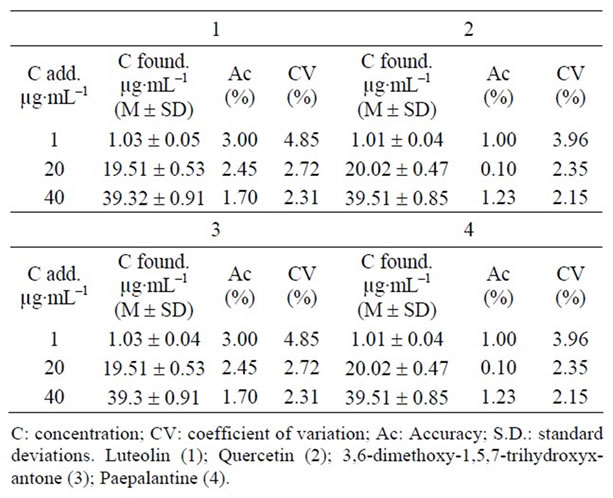
Table 5. Inter-day accuracy and precision of the LC method for determining the substances (1 - 4) in samples (n = 5 for each sample).
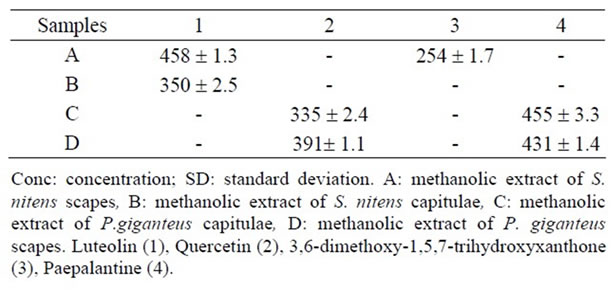
Table 6. Contents in μg/100 mg of extract A-D (mean ± SD) of the substances (1 - 4) employing the LC method.
Quercetin and paepalantine were quantified in methanolic extracts of capitulae and scapes of P. giganteus. Luteolin is present in methanolic extract of both the capitulae and scapes from S. nitens while 3,6-dimethoxy-1,5,7- trihydroxyxantone is present only in methanolic extract of the capitulae from S. nitens. The results from this method indicate that a variation coefficient of lower than 5% is adequate.
4. Conclusion
The implementation of the proposed work has demonstrated savings in time when compared to the classical phytochemical study. This factor avoids the performance of strenuous tasks that result in repeated substance isolation. The work also contributed to reducing the waste load generated by the use of organic solvents during the stages of preparation of extracts and chromatographic separation. This approach requires little organic solvent, which is easily discarded after the preparation steps. As an added benefit, small quantities of plant material are needed, which are indispensable in the study of Eriocaulaceae due to low mass preparation of the extracts presented. The recent literature does not report the presence of these substances in P. giganteus while for S. nitens the 3,6-dimethoxy-1,5,7-trihydroxyxanthone and luteolin have been identified by Pacifico [5]. On the other hand, flavonols and derivatives of paepalantine have been widely identified in the Paepalanthus genus [10,12]. Therefore, both species, when measured by LC-PDA, differ as to the existence of some flavonoids such as quercetin in extracts of P. giganteus and luteolin in S. nitens. Although the capitulae and scapes present similar profiles, the concentrations of these substances differ between the capitulae and scapes. Finally, we hope that our approach can contribute to the study of other plant species by providing a rapid and important strategy for the recognition of the chemical composition among Eriocaulaceae species. In addition, new parameters can be evaluated, especially when considering the occurrence of other classes of substances found in vegetable matrices, thus opening up new perspectives for future studies.
5. Acknowledgements
The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), for funding and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), for grants to L. C. Santos.
REFERENCES
- M. Giulietti, V. L. Scatena, P. T. Sano, L. R. Parra, L. P. Queiroz, R. M Harley, N. L. Menezes, A. M. B. Yseppon, A. Salatino, M. L. Salatino, W. Vilegas, L. C. Santos, C. V. Ricci, M. C. P. Bonfim and E. B. Miranda, “MultiDisciplinary Studies on Neotropical Eriocaulaceae,” In: K. L. Wilson and D. A. Morrison, Eds., Monocots: Systematics and Evolution, Melbourne, CSIRO Publishing, Vol. 1, 2000, pp. 580-588.
- P. T. Sano, “Actinocephalus (Körn.) Sano (Paepalanthus sect. Actinocephalus), a New Genus of Eriocaulaceae, and Other Taxonomic and Nomenclatural Changes Involving Paepalanthus Mart,” Taxon, Vol. 53, No. 1, 2004, pp. 99-107. doi:10.2307/4135493
- A. M. Giulietti, M. G. L. Wanderley, H. M. LonghiWagner, J. R. Pirani and L. R. Parra, “Estudos em sempre-vivas: Taxonomia com Ênfase nas Espécies de Minas Gerais, Brasil,” Acta Botanica Brasilica, Vol. 10, No. 2, 1996, pp. 329-377.
- B. Schmidt, I. B. Figueiredo, F. Borghetti and A. Scariot, “Produção e Germinação de Sementes de ‘capim dourado’, Syngonanthus nitens (Bong.) Ruhland (Eriocaulaceae): Implicações Para o Manejo,” Acta Botanica Brasilica, Vol. 22, No. 1, 2007, pp. 37-42. doi:10.1590/S0102-33062008000100005
- M. Pacifico, A. Napoletano, F. Hilario, C. Pizza, S. Piacente, W. Vilegas and L. C. Santos, “Metabolite Fingerprinting of Capim Dourado (Syngonanthus nitens), a Basis of Brazilian Handcrafts,” Industrial Crops and Products, Vol. 33, No. 2, 2011, pp. 488-496. doi:10.1016/j.indcrop.2010.10.023
- T. Stützel, “Monocotyledons: Alismatanae and Commelinanae (except Gramineae). Flowering Plants: The Families and Genera of Vascular Plants,” Springer-Verlag, K Kubitzki, Berlin, Vol. 4, 1998, pp. 197-207.
- V. L. Scatena and A. R. S. Moraes, “Anatomia de Raízes, Folhas e Escapos de Paepalanthus Subgênero Platycaulon (Eriocaulaceae) da Serra do Cipó-MG,” Arquivos de Biologia e Tecnologia, Vol. 39, 1996, pp. 1021-1035.
- W. Vilegas, C. A. L. Cardoso and A. E. Pires, “Química de Produtos Naturais, Novos Fármacos e a Moderna Farmacognosia,” Univali: Itajaí, SC, Brasil, 2009.
- T. J. Mabry, K. R. Markham and M. B. Thomas, “The Systematic Identification of Flavonoids,” Springer-Verlag, New York, 1970.
- W. Vilegas, A. L. Dokkedal, L. Rastrelli, S. Piacente and C. Pizza, “New Naphtopyranone Glycoside from Paepalanthus vellozioides and P. latipes,” Journal of Natural Products, Vol. 62, 1999, pp. 746-749. doi:10.1021/np980082t
- V. P. Shah, K. K. Midha, S. Dighe, I. McGilvery, J. P. Skelly, A. Yakobi, T. Layloff, C. T. Viswanathan, C. E. Cook, R. D. McDowall, K. A. Pittman and S. Spector, “Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies,” Pharmaceutical Research, Vol. 9, No. 4, 1992, pp. 588-592. doi:10.1023/A:1015829422034
- W. Vilegas, C. J. Nehme, A. L. DokkedaL, S. Piacente, L. Rastrelli and C. Pizza, “Quercetagetin 7-methyl Ether Glycosides from Paepalanthus vellozioides and P. latipes,” Phytochemistry, Vol. 51, No. 3, 1999, pp. 403-409. doi:10.1016/S0031-9422(99)00009-6
NOTES
*Corresponding author.