Journal of Biosciences and Medicines
Vol.04 No.07(2016), Article ID:68145,16 pages
10.4236/jbm.2016.47008
A Totally Absorbable Multilayer PLGA Implant Device Containing Doxorubicin Inhibited Tumor Growth and Metastasis without Systemic Toxicity in Murine Breast Cancer and an Ideal Pharmacological Paradigm for Regional Chemotherapy
Bennett Elzey1, Sandra Torregrosa-Allen2, Peterson Li3, Ben Ramsey2, Matthew Shaw3*
1Department of Urology, Indiana University School of Medicine, Indianapolis, USA
2Department of Comparative Pathobiology, Purdue University, West Lafayette, USA
3Shaw Scientific LLC, Carmel, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 24 May 2016; accepted 8 July 2016; published 11 July 2016
ABSTRACT
We hypothesize that a cylinder implant made of multilayer Poly-lactic-co-glycolic-acid (PLGA) mem- brane can be a method for controlled and extended drug release. We fashioned a multilayer cylindrical implant termed STID100 that released doxorubicin for 3 weeks in an orthotopic 4T1 breast cancer model in Balb/C mice. This implant starts as a thin doxorubicin-embedded PLGA membrane, and is then rolled into a cylinder containing an air gap between the membrane layers. Its controlled sustained release delivered 2× the amount of the intravenous (IV) equivalent of doxorubicin, inhibited the primary tumor, and prevented lung metastasis. Importantly it did not cause weight loss, splenomegaly, or cardiac toxicity vs systemically administrated doxorubicin. This favorable safety profile is further substantiated by the finding of no detectable plasma doxorubicin in multiple time points during the 3-week period, and low tumor doxorubicin concentration. The implant system delivered to the specification of an ideal pharmacological paradigm might offer a better coverage of the local tumor, significantly preventing metastatic spread with less drug toxicity to many vital organs, compared to the traditional pharmacology of IV route. The profile of STID made it an attractive therapeutic alternative in metastatic tumor prevention, pain management and many other diverse clinical scenarios.
Keywords:
PLGA, Doxorubicin, Implant, Multilayer Membrane, Metastasis, Controlled Release, Extended Release, Polymer, Tumor, Breast Cancer, Mice, Regional Chemotherapy, Pharmacology

1. Introduction
A therapeutic implant for localized disease should have many advantages over its IV or oral equivalent: minimal systemic side effects; elimination of daily dosing and maintenance of steady state drug concentration; better patient comfort and compliance, and only local drug delivery [1] [2] . This would also have high applicability for a host of chronic diseases in addition to cancer [3] .
Poly-lactic-co-glycolic-acid copolymer (PLGA) has been a favorite vehicle for such implants. It has been extensively used in orthopedic implants with a proven safety record. It is completely biodegradable and absorbable, generating minimal or no immune response, and is FDA approved for animal and human use [4] . Early on, it has been realized that drugs in PLGA tend to have a huge initial burst release (90% release in 1st 3 days), followed by a slow, often unpredictable core erosion release [5] - [7] . One way to overcome this challenge is through micro or nano particles [8]. The major problem is the elaborate process. The drug will go through several steps of dissolution in solvents, cross linking by chemical or thermal exposure (usually high temperature), followed by vacuum-mediated solvent removal with heat if necessary. For each drug, a unique manufacture process needs to be developed to each particular therapeutics [5] [9] .
Another way is using different geometrical solid forms [10] - [12] , but all require a complicated fabrication process. For example, Ortiz et al. made a cylindrical PLGA implant of 0.8 mm diameter × 8 mm long containing doxorubicin for placement in dog prostate [13] . The production process involved extrusion and required an elevated temperature. Fung et al. made a disc form of PLGA 1 mm thick × 10 mm diameter containing Paclitaxel and tested it in monkey brains. The process involved multiple steps of mixing drug and polymer, drying, and high pressure up to 8000 p.s.i. [14] .
Many innovative research efforts have tried different approaches to simplify the manufacturing process and to smooth out the drug release profile [9] [15] . These efforts are summarize in Table 1. Each technology has made strides towards the aim of smooth extended release to one degree or another. But the translation of those technologies into useful treatments has been slow to realize.
To address these deficiencies, we developed a simpler system to construct a solid therapeutic implant device with layered PLGA membranes. As shown in Figure 1, our prototype starts with a panel of thin PLGA membrane with the drug of interest evenly distributed inside the membrane. It is then rolled from one end into a multilayered cylinder in one simple step at or near room temperature. The multilayer structure theoretically can function as extended release concept because the drugs in the inner layer need to cross the outer layers to reach to the target tissue just outside the implant. The relationship of the number of layers vs time can be approximated by the equation below.
The therapeutics must diffuse across the membrane layers shown in Figure 2. There are 2 inter-material cross sections for each membrane. If the membrane is spaced by air, then the inter-material cross section will be air vs membrane. If the inter membrane was filled with tissue fluid, then the inter-material cross section will be tissue fluid vs membrane. There will be a specific coefficient for each therapeutic to the inter-material cross sections. The coefficient is determined by the physical character of the two interfacing material, and can be measured. The more layers a therapeutic has to cross, the longer it will take. Therefore therapeutic rate of release is inversely related to the number of the layers and correlated with the coefficients as shown in the equation.
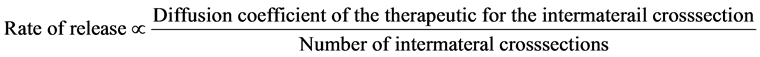
Another advantage of our multilayered solid therapeutic implant device (STID) is its simplicity of construction. The drugs do not have to be evenly distributed within the membrane, but can be painted or printed on just

Figure 1. The concept of multilayer membrane for STID construction. Upper left is a mem- brane panel with evenly distributed therapeutics on it. Upper right is the cross section of the cylinder formed, showing outer layers with more therapeutics. Middle left is a membrane panel with therapeutics concentrated more in one end. Middle right is the cross section of the cylinder formed, showing therapeutics evenly distributed in the cylinder. Lower is a 3 dimens- ional drawing of STID after the membrane is wrapped into the cylinder shape.

Figure 2. Illustration of mechanism of extended release for STID. Left is a 3 dimensional drawing of STID 100. Middle is a longitude section showing the multilayer membranes, and the diffusion direction (the arrow) of the therapeutics (represented by the dots). Also showing is the air layers between the membrane, and the capillary seal at the end of the membrane layers. Right is the count of the cross sections the therapeutics has to diffuse through, and rate of release of the therapeutics is affected by the cross sections.

Table 1. Comparison of STID with extended release platforms on the market.
one side of the membrane.
A uniformly solid cylinder implant will have an intrinsic problem in achieving linear extended release of embedded therapeutics. As shown in Figure 1, the outer region of the cylinder has a bigger diameter and the initial burst release will contain a larger portion of the drug than any subsequent release from the inner portion of the cylinder. Our multilayered membrane cylinder has the physical properties to overcome this intrinsic problem. Before rolling into final shape, a higher amount of drug can be place in the inner portion of the membrane relative to what will become the more outer portions to facilitate a more steady-state release.
An obvious application for such an implant is local, or regional chemotherapy for solid tumors. The current FDA-approved method for delivering drugs to solid tumors is still systemic administration, either by parenteral (intravenous) infusion, or oral intake. Many studies have indicated systemically administered drugs fail to penetrate solid tumor tissue evenly and are a major reason for therapy failure [16] - [18] . This uneven drug distribution occurs for several reasons but can be microscopically verified and may permit in the tumor tissue a seeding area for tumor relapse in the future [19] . On the temporal side, the effective concentration of chemotherapy needs to be sustained for an extended period of time. It has been estimated that around 10% to 15% of tumor cells are actively proliferating and susceptible to chemotherapeutics at a given time [20] . Drug exposure in this manner leads to higher resistance and is yet another way tumor cells to escape. Placement of a solid chemotherapy implant within or near the tumor mass might address this dilemma [21] .
To test our construct, we chose doxorubicin, a still widely used anthracycline antibiotic with potent chemotherapeutic properties. It is effective against several cancers including breast and prostate [22] . Unfortunately, it has many systemic side effects that limit its usage, notably the dose-accumulative cardiotoxicity [23] . To test our multilayered PLGA implant, we generated orthotopic breast tumors in Balb/c mice by injecting the aggressive murine mammary carcinoma 4T1 cells. We constructed the doxorubicin impregnated devices and placed them proximal to the 4T1 tumors. We found a construct that achieved these goals: effective suppression of tumor growth with minimal systemic side effect; and an initial pharmacological profile close to an ideal paradigm.
2. Materials and Methods
2.1. Chemical Reagents
Doxorubicin was purchased from Fisher Scientific, Pittsburg, PA. PLGA was purchased from Akina, Inc. West Lafayette, IN. Chloroform and acetyl nitrile were purchased from GFS chemical, Powell, OH.
2.2. STID100 and STID102 Construction
The biodegradable polymer is poly-lactic-glycolic copolymer (PLGA), with the ratio of lactic acid: glycolic of 85:15, and the average molecular weight of 55 K Dolton. 55 mg of the polymer and 5.5 mg of doxorubicin was dissolved in 3 ml of chloroform. The solution is then spread on a Taflon non-nonstick surface in a circle about 1 cm diameter, and air dried in a laminar flow hood for 6 hour at 0 celsius degree for STID100, or room temperature for STID102. The dried membrane is fold in half along a 1/32 inch (0.81 mm) diameter copper rod and rolled 3 times as showed in Figure 3. The highly volatile chloroform vaporized within 30 min. All the final membrane thus formed were collected and weighed to make sure their weight is less than the beginning material without the chloroform.
2.3. Cell Culture
4T1 cells were maintained in RPMI supplemented with 10% calf serum and 5% CO2. For tumor implantation, cells in log phase growth were trypsinized, counted, and stored on ice until fat pad injection. 2 × 104 or 1 × 105 cells in 50 - 100 µl were injected.
Animal care, mice breast fat pad implant procedure, blood and tissue collection.
10-week-old female Balb/C mice were purchased from The Jackson Laboratory and housed in Purdue’s AAALAC-accredited mouse facilities. All animal experimentation was performed under an Institutional Animal Care and Use Committee-approved protocol.
For each study, all mice are acclimated for one week before experimentation. They are then weighed and randomly divided into the three groups, with equal average starting weight. Mice were anesthetized and a 1 cm incision was made along the right pectoral group of nipples. A suitable fat pad was identified and injected with

Figure 3. Construction of STID.
4T1 cells, and the implants were placed at the same time near the fat pad. For the time lined test, the implants were placed 3 days later with re-incision by a 2nd anesthesia. The skin was closed with a 5 - 0 absorbable suture and reinforced with a surgical clip. The animals were then recovered from the surgery and observed for the next 21 days for tumor growth. The surgical clips were removed at 7 days post operation. The positive control groups received intravenous (IV) tail or intraperitoneal (IP) injection of doxorubicin every 2 to 3 days for 2 weeks for a total 320 µg, or otherwise indicated in the results section. The negative control group was given a placebo injection of phosphate buffered saline (PBS). Animals were observed daily for health per facility routine.
All the animals were weighed at beginning of the test and randomized to 3 groups so that all groups had about equal average weight at Day 0. Weight data then collected every 2 - 4 days for each animal, together with measurements of tumor size with a caliper.
Retro-orbital blood samples were collected by a heparin coated capillary tube at the indicated time, either post 2 or 24 hour of IP doxorubicin dosing for all animals. After centrifuge at 5000 rpm, about 25 microliter of plasma were collected and assayed for doxorubicin concentration. For Troponin test, terminal blood was collected by inferior vena cava, or rapid decapitation to avoid cardiac trauma.
2.4. Pathology
At about 21 days post implant, mice were euthanized and heart, lung, spleen, and tumor were dissected out and examined. Individual metastasis lung nodules were counted. Heart, lung, and tumor samples were formalin fixed, and then paraffin embedded. Standard 5 µm section slide were made and stained with hematoxylin and eosin for microscopic examination.
2.5. Doxorubicin and Troponin Measurement
The implants containing doxorubicin were completely dissolved in 1 ml of acetyl nitrile. Then 3 ml of water is added. The precipitates containing the PLGA polymers were spin down at 10,000 rpm for 20 min. 100 micro liter of the clear supernatant were then loaded to an Agilent Technology 6130 Quadrupole LC-MS with a Phenomenex Kinetex2 50 × 4.6 mm column. Mobile phase is 75:25 methanol: 0.1% formic acid in water, gradient adjusted to detect doxorubicin MW around 543 [24] . Internal control showed the extraction above 90%.
Retro orbital blood was collected from the specified time period with the heparin coated capillary tube. The blood was the spun for 5000 rpm for 5 min, the plasma collected were subjected for doxorubicin assay as described above.
For plasma doxorubicin measurement: 25 µl of retro orbital blood collected as above (diluted to 25 mcl total with standard serum if necessary, then later corrected for volume) were loaded to a AB Sciex API4000/5000/ 5500 mass spectroscopy with an auto-sampler from Shimadzu, Prominence. The column is Atlantis dC18 50 × 2.1 mm, 5 µm particle size, maintained at 35˚C. The mobile phase A (MPA): 0.1 formic acid in water. The mobile phase B (MPB): 0.1% formic acid in ACN: methanol (50:50), with flow rate of 0.7 mL/min, and 35% of MPB initially.
Tumor tissue doxorubicin extraction: tumor tissue was cut into small pieces and suspends in 10% wt/vol 2N HCl, and homogenized in a 2 ml Potter-S homogenizer. 200 µl of the homogenates was then add to 50 µl Triton X-100, and 870 µl of Isopropanol, extracted overnight in −20 degree [25] . The samples were then spun 13,700 rpm for 20 min at room temperature, the supernatant were assayed for doxorubicin concentration by fluorescence emission 590 nm with excitation 470 nm. A Spectro Max M5 96 well plate reader was used.
Mice tropon in levels were measured by an Ultra Sensitive Elisa kit from the Life Diagnostic, West Chester, PA 19380. A Promega plate reader was used.
2.6. Statistical Analysis
Anova analysis, student T test are performed with Microsoft Excel 2010.
3. Results
One of the major concerns using implants containing a high dose of chemotherapy agent is local tissue irritation, skin degradation, and inability to heal the surgical site. Implant constructs containing serial dosages of doxorubicin were placed subcutaneously in mice flank or breast. With the size of 1 mm diameter and 10 mm long, the implants containing about 750 µg are well tolerated. Various cylinder implants around these perimeters were then constructed and placed near orthotopic 4T1 cells in juvenile female fat pads for 21 days to suppress tumor growth. This served as a screen tool to identify the right construction. PLGA with molecular weight range of low of 7 KD to high of 100 KD, and varying LG ratio were tried. The initial tests were negative until we tried STID100 (Figure 4), underscore the difficulty of local cancer treatment. One of the typical versions of implants that tested negative is STID102, shown in Figure 4. Under the same grafting procedure, STID102 has no tumor suppression effect.
The one-time treatment of the STID100 implant suppressed the primary tumor 68%, vs the IV doxorubicin’s 44% (4 × 4 mg/kg dosing, a total of 320 mcg) in 5 independent test total 66 mice, shown in Figure 4 and Tables 2-4. In the experimental setting of Figure 4, 105 4T1 cells were injected into the right breast fat pad, the STID implants were placed at the same time. The animals were all subject to one anesthesia. It showed that the separation of tumor growth started early and was sustained throughout the 21 day period.
The STID 100 contained about 877 mg of doxorubicin, double the amount of the total doxorubicin given to the mice as a positive control group. The PLGA in STID100 has MW 55 KD, and a lactide vs glycolide ratio of 85:15. It is in the middle range of many PLGA choices. But the most unique aspect of STID100’s creation compared to all the failed implants is the temperature at the time of membrane formation. Instead of room temperature, the PLGA polymer and doxorubicin mixture was placed on a 0˚C plate and air-dried. The membrane thus formed is smooth, and not sticky to the touch. When it is rolled into the final cylinder shape, it has a bit more recoil, but is still able to stay in the cylinder shape. STID102 has the same membrane material, same dosage of doxorubicin, same dimensions, and same layered cylinder structure as STID100, but the membrane was dried at the room temperature.
The measurement of a typical batch of implant STID100 is summarized in Table 2. Most of the volume in the 10 mm long × 1 mm diameter cylinder is air, above 69%. The malleability of the membrane is such that it can stay in cylinder configuration once rolled up without being totally reduced to a flat sheet of the membrane. Given the fact of malleability and non-stickiness, we assume the air is evenly distributed within the multilayered structure. The specific measurement of STID102 is similar to that of STID100 in terms of length and diameter. Therefore, STID102 has comparable air volume. The main difference is the stickiness of the implant. The STID 102 inter membrane layers stick to each other more than in STID100. Once it is made, STID102 has less recoil than STD100, and it cannot be easily reduced to its original sheet without some major damage.
The length of the study is limited by the concern for animal’s wellbeing, when the 4T1 tumor grows too big and affects mice’s mobility. To evaluate how much longer the implant can work, and correlate the study of doxorubicin cardiac toxicity (see cardiac toxicity discussion below) with the plasma concentration, we changed the experimental setting to extend the test longer, as shown in Figure 5. 15 Balb/C mice were used in this time lined study, and 2 × 104 4T1 cells were injection at day 0. The STID 100 implants were placed 3 days later in the 5 mice implant group with a 2nd anesthesia procedure. The IP doxorubicin group included 5 mice. The dosing were 2 mg/kg IP each time starting Day 3; 3× per week for the first 2 weeks, then 1× per week thereafter, total

Figure 4. STID100 suppression of primary tumor. 4T1 tumor growth in Balb/C female mice breast fat pad, grafted at Day 1. Tumor size calculated as length × width × width/2. Data points represent the average tumor size measured at the corres- ponding days. (#) Number of mice in the group from 5 different tests. IV-PBS, groups that received phosphate buffered saline (PBS) injection in tail vein. IV-Dox, groups that received doxorubicin injection at tail vein. STID102 and STID100, groups that received STID102 and STID100 implants respectively.
Table 2. Measurements of STID 100. Membrane diameter and thickness is measured after the doxorubicin-PLGA is dried. The implant diameter and length is measured after the implant is made from the procedure in Figure 3. All number average from the measurements. (#) Number of measurements.
Table 3. Efficacy and side effect profile of STID100. 105 4T1 cells were injected and mice euthanized in 3 weeks. Lung metastasis is the average numbers of metastasis nodule by eye count. Both lung and spleen exam has 15 mice, 5 each in IV-PBS, IV-Dox and STID100. (#) Number of mice for the average. *2/19 mice in the STID 100 group have none healing wound at doxorubicin implant site, but otherwise behaved normally.
Table 4. Dose related tumor suppression and weight analysis. Total 6 tests were conducted repeating the 105 4T1 cells test condition. # of animals is the sum of animal in each test group in all the independent tests. Anova analysis of single factor for 21 day tumor size were conducted for the 4 test groups (exclude IV 2 mg) using Microsoft Excel 2010. Anova of two factors were conducted for weight gain for 5 points (Day 1, 5, 8, 18, 21): 9.67E−08 for two factors (IV-PBS vs IV-Dox); 3E−09 for two factors (IV-Dox vs STID100).

Figure 5. Time lined study. 4T1 tumor growth in Balb/C female mice breast fat pad, with 2 × 10 4T1 cells grafted at Day 0. STID100 were placed at day 3. Data points represent the average tumor size measured at the corresponding days ± standard deviation. (#) Number of mice in the group. IP-PBS: group that received intraperitoneal (IP) PBS injection. IP-Dox: groups that received IP doxorubicin injection. STID100: group that received STID100 implants at Day 3 with another general anesthesia procedure.
14 mg/kg, or 280 mcg per mouse. 3 mice in the IP-PBS group received the PBS injection with the same schedule. The tumor inhibition was by 21 days was 67% for the STID100 group, and 42% for the IP doxorubicin group. The tumor suppression by STID100 was significant into the middle of 4th week, but started showed signs of wearing down in the middle of the 4th week, shown in Figure 5.
Doxorubicin plasma concentration was on day 8, and day 15, 2 hour post IP dose of doxorubicin; then again 24 hour post of day 15 dose in the IP group. The blood was collected by the right retro-orbital puncture from one eye from all the 15 animals on Day 8. One Day 15, the contralateral eye was used for the blood collection. 24 hour later, the right eye was used once more. The bleeding was stopped by gentle pressure and all animals recovered well after the 3 blood collections. The control and implant groups showed no detectable doxorubicin (consistently below our instrumental limit of 1 ng/ml). In contrast, IP-doxorubicin group showed average plasma doxorubicin concentration of 15.9 and 16.2 ng/ml at 2 hour post IP dosing, on day 8 and Day 15 respectively. The numbers are consistent with most literatures. For example, Johanson et al reported a plasma doxorubicin concentration of 100 ng/ml 2 hour post IP doxorubicin dosing of 12 mg/kg. For the Day 15 time point, the average plasma doxorubicin concentration then drop to 4.8 ng/ml 24 hour later, consistent with the literatures that the 1/2 life of plasma doxorubicin is 8 hour [26] . The STID100 group mostly had no detectable plasma doxorubicin, except one mouse. That mouse had an abnormally high doxorubicin concentration at day 8, but no detectable plasma doxorubicin signal at day 15 both 2 hour and 24 hour post dosing.
The time lined study was ended around day 24, when the tumor in the each group reached to the mass that affects the animal’s mobility. Doxorubicin in tumor were extracted and measured by fluorescence. The STID100 group showed a highest concentration of 0.081 µg/g, no significant different than the IP-doxorubicin group’s 0.071 µg/g, but is significantly higher than the IP-PBS group. The IP-PBS group showed a signal of 0.057 µg/g, near lower limit of quantification LLOQ of 0.0375 µg/g (background signal of the tissue extract when measured with the fluorescence). The most important finding is that all the measured tumor concentration is 10 fold less than that of the target of systemic doxorubicin treatment. Most workers reported 1 - 10 µg/g doxorubicin concentration as adequate part of area under the curve AUC for cancer treatment, anything below 0.1 µg/g usually is treated as clinically irrelevant
At the time of 21 days, most of the STID100 was easily located around the tumor and easily removed. The examination of the dissected tissue around the implant found most animals had no tissue irritation as well. There were no signs of local tissue bleeding, swelling, or signs of hardening or fibrosis. Most of the implant actually grew into the growing tumor and was encased in visible tumor septa. Most of the time, it curved around the tumor, assuming the contour of the growing tumor, as if the implant coexisted with the tumor nicely (picture available upon request). In one test, the doxorubicin measurement of this batch of pre-implants was average about 877 mcg, at the time of day 21, the post-implants contain an average of 206 mcg of doxorubicin, 23% of the pre-implant. In the test in Figure 5, at day 24, only 2 out of 5 implants were retrieved intact, they were noticeably smaller. The rest of them either none exist or easily fragmented during dissection, suggesting the acceleration of the implant degradation after day 21. Assay of the doxorubicin contents of the 2 intact post-implants showed an average 22% of that of the pre-implants.
The overall shape and dimensions of the 21 days post-implant were not much smaller as shown in Table 2. The average weight is about 1.5 times that of the pre-implant, likely hydrated with tissue fluid. The volume is 3 times that of the pre-implant stage. There was some wearing down on the surface, but overall insignificant degradation was noticed. The implant then was cut longitudinally, and examined under microscopy. Compared to the implant that has not undergone 3 weeks of subcutaneous implantation, there were tears and degradation. Overall, the multilayered structure is still there. There are even chambered spaces intact corresponding to the air space (picture available upon request).
During the dissection for STID102, we had a hard time finding anything remotely resembling our initial implant. Examination of the implant placement site near the tumor after 3 weeks found that the implant is mostly degraded; some of it had totally disappeared. There were colored patches of tissue that might be a remnant of the implant. The cylinder implant was completely unrecognizable. (This is strong evidence that the layered structure is vital to the durability of the implant under physiological subcutaneous conditions, and contributes to the extended release profile and tumor suppression ability.)
One of the most frequent sites that 4T1 metastasize is the lung. After 21 days, the lung tissues of the 3 groups were dramatically different. The IV-PBS control group had the most diseased looking lungs, with numerous tumor nodules. The STID100 group had almost normal, healthy-looking lungs, and the IV-Dox group is somewhere in between. The gross eye count of metastatic lung nodules is summarized in Table 3. Pathological examination of lung tissue slides semi-quantitatively counted the tumor foci per view, and confirmed the overall gross tumor count. Although a more detailed microscopic quantification with software for internal control was not performed at this time. Microscopic examination also showed the PBS control group had the most frequent rate of tumor thrombosis, a much less frequent finding in the IV-Dox group. The tumor thrombosis was absent in the implant group. Judging from these observations, the STID100 implants suppressed the tumor metastasis significantly.
Weight is a measurement for an animal’s general well-being under chemotherapy conditions, and is recorded in our experiment. As shown in Table 3 and Table 4, the control group, STID100, and STID102 groups are all gaining more than 2 g of weight over the course of the 21 day treatment. At the end of experiments, the average weight in the IV-Dox group receiving 320 mcg of doxorubicin is significantly lower than the other 3 groups, and is actually the only group that lost weight. Note that an ANOVA analysis of the weight of 4 groups of 51 animals at the end of the 3 weeks yields a significance of P much less than 0.001. The time lined study in Figure 5 and Table 5 best illustrated this observation. As the tumor grew, the STID100 and IP-PBS gained weight steadily, while IP-Dox group gradually lost weight.
Another measurement of an animal’s general well-being is the size of its spleen. Generally, the bigger the spleen size and its weight, the more stress it might have. As shown in Table 3, the control group has the biggest spleen size, consistent with the fact that rapid tumor growth does have systemic stressful effects. IV Dox group reduced tumor growth, thus decrease the stress, consist with report from Desai et al and others. The STID100 group has the lowest spleen size and weight, closest to normal (without any treatment).
The heart tissues from the 3 groups were examined at day 21 and none had any signs of increased heart size or any abnormalities. Under microscopy, all heart muscle fibers and z-line organizations are similar. The microscopic examination is not sensitive or specific enough to detect any significant differences among the 3 groups for doxorubicin cardiac toxicity at day 21 with 16 mg/kg doxorubicin. At day 24 in the time lined study, the mouse cardiac troponin I level was measured in Table 5. 3/5 IP-Dox mice showed detectable troponin level. The 5 mice receiving 14 mg/kg doxorubicin, is averaging 0.19 ng/ml, vs 0/5 STID100 group receiving around 35 mg/kg doxorubicin, p < 0.05 T-test. The measurements are all around the LLOQ of 0.069 ng/ml. 1/3 IP-PBS control group showed a detectable signal near the LLOQ. The data is consistent with the finding of Desai et al., who have demonstrated low mice troponin level of 0.02 ng/ml. It was started to be detectable only after about 4 week of total 20 mg/kg of IV doxorubicin treatment. The limiting factor in further extending our study beyond 24 days and 14 mg/kg of IP doxorubicin is the aggressive growth of the 4T1 tumor, making the further test unattainable in our animal facility. (The implant contained about 870 mg doxorubicin, with 24% left in the retrievable ones. A conservative estimation is the implant group received at least 35 mg/kg doxorubicin within 24 days.) The overall data supports that the implant caused no cardiac toxicity.
Table 5. The time lined study. The doxorubicin plasma and tumor concentrations, and mice troponin level in 15 Balb/C mice, correlated with their body weight at the collecting day, and 4T1 tumor growth in Figure 5. Data following ± is the standard error whenever is available. *Student T-test of IP-Dox vs STID100, P < 0.05. ξStudent T-test IP-PBS vs STID 100, P < 0.05. For **Student T-test IP-Dox vs STID100, no significant difference. ***Anova two factor with replication of IP-Dox vs STID 100 from Day 0, 8, 15, to end Day of test, P = 2.44 × 10−5. LLOQ is the lower limit of quantification. For plasma doxo- rubicin determination, LLOQ is 1 ng/ml. For tumor doxorubicin content determination, LLOQ is 40 ng/g. LLOQ for plasma troponin level is 69 pg/ml.
4. Discussion
This is a first report of construction of a cylindrical extended release doxorubicin implant built on a PLGA based multilayer structure and deposited near a solid tumor. The implant suppressed its growth and metastasis without systemic side effects of weight loss or splenomegaly. Also, we demonstrated that the multilayer membrane solid implant stayed in a mammal body for 21 days, and was retrieved with much of the multilayer structure intact and released twice the amount of systemically administrated doxorubicin with no detectable drug in plasma, or cardiac toxicity. Its degradation accelerated after 21 days, and was totally absorbable shortly after its intended therapeutic window. It is important to point out that the favorable safety profile of no weight loss, splenomegaly and cardiac toxicity is time lined with the plasma study in the same animal. The evidences reinforced each other and increased the statistical significance. In doing so, we identified the critical technical factor, which might be its ability to maintain its structural integrity under 37 degrees C temperature for that period of time. We have reason to believe the unique design and construction of the STID contributed to its success.
4.1. Overcome Burst Release
To mix doxorubicin with PLGA and place it near a tumor as cancer treatment is not a novel idea [13] [27] [28] . Our premise of layered membrane sought to overcome the technical challenge of the initial burst release of doxorubicin impregnated PLGA. Many of our failed early tests, for example STID102, indicated that simple construction of a cylinder implant from layered membrane is not enough. The initial burst release often is exacerbated when the implant is placed in vivo. The in vitro tests often fail to predict the in vivo results [5] . That is why we use the in vivo test to screen the working device solely. The same early test also suggested that to effectively suppress tumor growth locally, delivery of doxorubicin has to be at sufficient amount, but sufficient time is needed and might be more important [19] . That probably is why, despite the theoretical attractiveness of local chemotherapy, and despite the many commercially available extended release vehicles, the number of implants that can successfully treat the solid tumor locally is very limited.
Interestingly, STID100 is an incidental finding. But as soon as we realized it, it quite reliably reproduced the tumor suppression. Also the around 77% doxorubicin release during the 3 week period is quite consistent in multiple preparations. This is a smooth extended release, good technical end point for any PLGA impregnated drug release device, likely due to the multilayer membrane construction. More tests are needed to further validate how closely the release is following the proposed equation.
4.2. Temperature is the Key
A comparison study of STID100 and STID102 gave hints that the temperature at which the dissolved polymer mixture consolidates and forms into the membrane might be the critical technical element. More tests are needed to answer the question of why the temperature of membrane formation is important. One speculation is that the temperature influences the orientation of the polymers when they solidify onto each other, and consequently made it more durable. Therefore, we have the hypothesis that the membrane thus made had a special mechanical character that maintained its air layer, and the maintained air layer played a role.
4.3. Air Pocket is Important
Theoretically, this might make sense as shown in Figure 2. Once placed inside the mouse tissue and in a closed wound, the tissue fluid will start to infiltrate the implanted structure. The oxygen in the air pocket between the layers will be absorbed quickly, leaving still 80% of the volume of nitrogen still trapped in an air pocket. Due to the capillary effect, the thin nitrogen layers will be truly trapped and will take time to be absorbed. This will slow down the tissue fluid’s infiltration of the inner layer of the structure, thus making the overall structure more durable. Thus, with STID102 providing more sticking points between the membrane layers, the tissue infiltration can have more access to the inner membrane of the construct with those contact points. And the air pocket in STID102 might be more divided and more easily be infiltrated and absorbed.
An added benefit of the layered structure is the protection of the drugs in the inner part of the STID implant. This is a key feature that differentiates STIDs from most of the extended release vehicles on the market. A uniformed solid implant, such as the wafer implant from Gliadel [12] , will be subject to tissue fluid infiltration as soon as it is deployed. With one inter-material cross section, the infiltration of the most inner part will be complete in a matter of minutes to hours. All the drugs will be subject to some kind of erosion by the 37 degrees Celsius tissue fluid for the entire extended release period. With multiple inter-material interfaces and air pockets, the speed of tissue fluid’s infiltration of the inner side of the implant will be greatly impeded. For most therapeutics, a day’s or weeks’ time of staying dry inside a totally biodegradable implant is a huge advantage in retaining its efficacy. We are in the process of quantifying this protection. At this time, we have no proof STIDs’ insides stayed dry, or how long they stayed dry. Further tests are planned to answer this question.
Table 1 is a comparison of STIDs with other major commercial vehicles for extended release on the market [15] . Detailed pro and con analysis for each technology is beyond the scope of this publication, but a brief examination of the physical perimeters of each technology shows that the STID is the only one that has the theoretical potential of protection inside the construct.
4.4. Low Plasma Doxorubicin of the STID100 Implant Correlated with Safety Profile
In the time lined study Figure 5 and Table 5, the weight loss in IP group is correlated with the rise and fall of plasma doxorubicin to each animal to each particular day. For example, Day 8 and 15, post 2 hour dosing, the plasma doxorubicin concentration rise to 15.9 and 16.2 ng/ml, a nice tight data range, suggesting the IP dosing and blood collection are well run. The figure is similar to many published data. It dropped to 4.8 ng/ml, in 24 hour, again consistent with the plasma 1/2 life of IP doxorubicin of 8 hour [29] . In designing the time lined study, we speculate that the STID100 system release is close to a zero order profile in releasing the drug locally, supported by the evidence that a significant 23% of doxorubicin left at the end of the 21 day test. And tumor suppression started early and is maintained well throughout the 21 day period from the tumor growth curve (Figure 4 and Figure 5). We were suspecting a low but detectable plasma doxorubicin concentration to substantiate the zero pharmacokinetic. The lack of detectable plasma doxorubicin during the height of the tumor suppression of Day 8 - 15, is a surprise, and is vigorously tested in a GLP laboratory setting. The LLOQ of HPLC- MS for plasma is 1 ng/ml, better or equivalent to most published work.
The IP-doxorubicin group registered a low level of doxorubicin in tumor 3 days after last IP dosing of 2 mg/kg. This is consistent with many published works. For example, Laginha et al. observed a tumor doxorubicin rose to around 2 µg/g after an IV dosing of 9 mg/kg, then quickly drop below detectable level in 48 hours [25] . The low tumor doxorubicin concentration in the implant group is yet another interesting finding. Maybe part of the reason is that the implants started to disintegrate rapidly after 21 days. We could find only 2 out of 5 implants at day 25, and they are all smaller. The tumor growth after 21 days was accelerating (Figure 5), suggesting less doxorubicin is being released. Incidentally, this might be an adequate technical perimeter for STID100. A rapid disintegration of the implant after the intended therapeutic window might be more preferable than a long slow release of its remaining chemotherapy agent, not enough to have chemotherapy effect but raise the risk of mutagenesis.
Maybe the tumor in the implant group never had the concentration of doxorubicin as high as the bench mark, when doxorubicin is administrated systemically. Or the implants established a sharp zone of drug gradient near the surrounding tissue, and drop very quickly further away from the surface of the implant [16] [18] . Or the pharmacokinetic measurements will be vastly different from the systemic administrated route in other possibilities [21] . Yet this leads to another striking question: during those 21 days, close to 700 mcg of doxorubicin was released from the implant, twice that of the IV group. This begs the question: where did this significant portion of doxorubicin go? What did it do to the mice? A well designed time course study might be needed find the answer and beyond this scope of this report.
The well correlated time lined measurement data suggests that the implant released doxorubicin mostly locally, and very few will find its way to the systemic circulation. Showed in Figure 5, at day 8, and 15, the implants are at its height of tumor suppression, the doxorubicin concentration is below detection level, consistent with the finding of lack of increased cardiotoxicity in the implant group. If we assume that doxorubicin’s cardiac toxicity is dose-dependent, the overwhelming evidence is that the implant mice’s heart tissue sees far less than the 700 mcg dose released.
Part of it will probably find its way to blood circulation, but not at high enough concentrations in the blood to cause cardiac toxicity or suppress weight gain. Part of it is likely to stay in the connective tissue around the implant, where the blood flow is low compared to the perfusion-rich organs (i.e. muscle). Given the fact that doxorubicin tends to migrate to cell nuclei after absorption; some kind of sink effect might exist as well. One thing is for sure though: the PK of doxorubicin with subcutaneous implants is quite different from that delivered through IVs.
The observation of splenomegaly, together with metastatic cancer nodules in the lung, is quite consistent in findings in all the independent tests. Better suppression of primary tumors, better lung condition, and smaller spleen sizes are consistently observed. Most likely, the released doxorubicin in the breast fat pad area are all piped through the same venous or lymph circulation system, as are those of the broken-off cancer cells. The implant can sustain a high enough doxorubicin locally in those tissue and vessel system that some of the cancer cells are killed while going to distal organs.
4.5. An Ideal Pharmacological Paradigm Achievable
The metastasis data is circumstantial evidence that the lymphatic region of the mouse body has high concentrations of doxorubicin after STID100 placement, and tissue extraction is needed to determine its concentration and provide concrete evidence. Weighing the all evidence at this point, an ideal pharmacological paradigm emerged. Figure 6 is the working hypotheses to explain all our experimental findings regarding the pharmacokinetic and pharmacodynamics of the STID100. The physiological connective tissue of a tumor and its surroundings is in the low blood flow organs. This is the target of our implant. The combination of low blood flow plus extended release together created a physiological space where the doxorubicin concentration is highest and suppresses tumor growth. Then the doxorubicin is piped into the lymph drainage system, where the drug concentration is lower than the tumor site but high enough to suppress the metastatic tumor cells. The systemic circulation is where the drug is continuously piped into next, with even lowers concentration. Then another decrease of doxorubicin concentration occurs when it enters the rich blood flow organs such as the brain and heart. The concentration of the drug is so low that we do not see weight loss or cardiotoxicity. Finally, it is secreted or eliminated through bile or the kidneys.
This model is quite different from the IV infusion model, which mandates a high front load of drug concentration in systemic circulation. It will quickly saturate the rich blood flow organs, also known as vessel rich tissue [30] , like the heart and brain, with high enough concentrations to cause damage. Then it saturates tumors in the poor blood flow organs, the target of the therapy, with lower concentration likely depend on the extend of the delay and drug affinity to the tissue. The drug concentration goes down further when it reaches the lymphatic system, where some tumor cells escape to metastasis. Finally, secretion or elimination occurs through bile or the kidneys.
The working model illustrates a unique attractiveness of the regional chemotherapy if an implant can be engineered to deliver to both spatial and temporal specification, as shown in this study. The implant puts the highest concentrations of drugs where they needed the most, which are poor blood flow organs (which includes most solid tumor) and the lymphatic drainage system (where metastasis quite often involves). The blood-rich organs, where most of the bad systemic side effects occur, are protected by dilution of the drug in the circulation system.
This working hypothesis is consistent with some recent regional cancer therapy research. Shikanov, et al., injected a semiliquid extended release polymer loaded with Paclitaxel to an orthotopic R3327 rat prostate tumor, which inhibited the prostate tumor and its metastasis to lung and lymph nodes for 25 days [28] . Liu, et al., placed a gelatin sponge with PLGA microspheres containing Paclitaxel near orthotopic rat lung cancer, and found that during a 14 - 32 day period, the mediastinum lymph nodes have higher area under the curve AUC drug and fewer metastatic tumors than IV treated tumors [31] . Both of the two studies bear similarities to our report. The polymer released the chemotherapy agent for about 3 weeks; a single placement; most drugs released locally; and suppressed both the primary tumor and its metastasis. There is also growing evidence from animal and human studies that doxorubicin formulated with liposomes or nanoparticles have a higher preference to target lymph nodes or lymphatics [32] .
4.6. Change the Benefit vs Risk Profile of Doxorubicin, Other Chemotherapy Agents, Rescue Pathway for Experimental Stage Chemotherapy Agents
Currently, surgery and IV or oral chemotherapy is still the mainstream delivery method for cancer treatment. But, if STIDs can deliver close to the pharmacological paradigm as outlined above in a human body, they might improve the benefit vs risk ratio of doxorubicin dramatically in a specific anatomical region. Just a rough estimation: since STID delivered 2 × IV dose with better primary and metastatic tumor suppression without systemic

Figure 6. An ideal pharmacological paradigm to demonstrate the different pharmacokinetics of doxorubicin delivered by IV infusion (upper), vs by an implant.
effect, the benefit vs risk ratio increased by a factor of 2. For an older chemotherapy agent such as doxorubicin with many side effects, a higher dose might be used in some breast cancer situation and prevent its metastasis than currently possible with IV infusion. A combination of STID100 with systemic chemotherapy agents is also an attractive breast conservation option for some aggressive cancers post-surgery resection [33] [34] . Given the simplicity of the STID construction, STIDs containing new agents such as gemcitabine, Taxol, or even immunotherapy agents are theoretically possible with even better benefits and risk differentiation. Even though there are exciting progresses made fighting solid cancers, it is still widely believed that we still need all the tools we can get in the near future. For example, with the wider and longer clinical usage of the newest immunotherapy cancer agents, more and more of their systemic side effect are reported [35] [36] . Any regional therapy version with improved benefit risk calculation might still be useful.
Intuitively, all implant delivery platforms, many are not listed in Table 1, thrive to the ideal pharmacological paradigm. What special about STID100 is its efficacy, side effect, and pharmacological profiles are closest to the ideal, as far as we know. With the simplicity of construction of the multilayer membrane, it is not a stretch to imagine future research might produce more STIDs, or regional chemotherapy implants in general, closer to the ideal pharmacological paradigm. We hope this report demonstrated to the absorbable implant community that the ideal pharmacological paradigm is achievable and the improve benefit and risk profile calculation might be a benchmark for implant devices in the future.
One potential of STID’s application in cancer field is metastasis prevention. Beside the breast cancer situation discussed before, it can be useful for solid cancer with predictable origin. For some slow growing prostate cancers, if a single deposit of an STID can retard the cancer growth or its metastasis, it will be welcome news for frail patients who are physically not fit for surgery or chemotherapy [37] . Even the most benign radiation therapy can exhaust an advanced-age patient, or one who has much co-morbidity. Younger patients who want to avoid the sexual side effects of hormone therapy also benefit. Combination of STID with systemic chemotherapy to achieve a better synergistic effect in a region of the body might be another area of wider application. A logical extension is the rescue of many experimental stage chemotherapy agents, many of which are terminated due to unacceptable systemic side effect. But a change of delivery system, or benefit and risk ratio regionally might offer new usage for those agents, if the ideal pharmacological paradigm is followed [38] .
Chronic and acute pain management also have unmet demand that regional therapy can be of great help. Steroids injection has been the choice of treatment for many lower back pains, but is mostly a single shot procedure. The spill over injected steroids to systemic circulation causes weight gain, transient hyperglycemia, and bone loss, thus limit how many shots a patient can get in a year. Local anesthetic injection for terminal cancer pain, phantom limb pain, complex regional pain syndromes, etc, lasts hours to days typically, with the risk of life threatening overdose [3] [39] . STIDs of steroid or local anesthetics lasting week with minimal systemic blood concentration might greatly improve the treatment for those patients. By introducing the multilayer membrane STID, we hope its potentials are evident, and warrant further interest in its broad application.
Acknowledgements
M.Q. Shaw thanks partners from the Shaw Scientific LLC for their financial support; Karl Koehler for many scientific discussions.
Cite this paper
Bennett Elzey,Sandra Torregrosa-Allen,Peterson Li,Ben Ramsey,Matthew Shaw, (2016) A Totally Absorbable Multilayer PLGA Implant Device Containing Doxorubicin Inhibited Tumor Growth and Metastasis without Systemic Toxicity in Murine Breast Cancer and an Ideal Pharmacological Paradigm for Regional Chemotherapy. Journal of Biosciences and Medicines,04,66-81. doi: 10.4236/jbm.2016.47008
References
- 1. Arias, J.L. (2008) Novel Strategies to Improve the Anticancer Action of 5-Fluorouracil by Using Drug Delivery Systems. Molecules, 13, 2340-2369.
http://dx.doi.org/10.3390/molecules13102340 - 2. Weinberg, B.D., Blanco, E. and Gao, J. (2008) Polymer Implants for Intratumoral Drug Delivery and Cancer Therapy. Journal of Pharmaceutical Sciences, 97, 1681-1702.
http://dx.doi.org/10.1002/jps.21038 - 3. Al Malyan, M., et al. (2006) Polymer-Based Biodegradable Drug Delivery Systems in Pain Management. Journal of Craniofacial Surgery, 17, 302-313.
http://dx.doi.org/10.1097/00001665-200603000-00018 - 4. Steve, M.H., Siegel Steve, P., Drug, C. and Carrier, D. (2012) PLGA as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel), 3, 1-19.
- 5. Hines, D.J. and Kaplan, D.L. (2013) Poly(lactic-co-glycolic) Acid-Controlled-Release Systems: Experimental and Modeling Insights. Critical Reviews in Therapeutic Drug Carrier Systems, 30, 257-276.
http://dx.doi.org/10.1615/CritRevTherDrugCarrierSyst.2013006475 - 6. Graf, M.A. (2001) Feasibility Study of the Modification of PLGA Thin Layers.
- 7. Shuwisitkul, D. (2011) Biodegradable Implants with Different Drug Release Profiles.
http://www.diss.fu-berlin.de/diss/receive/FUDISS_thesis_000000023117 - 8. Gao, Z.-G., Tian, L., Hu, J., Park, I.-S. and Bae, Y.H. (2012) NIH Public Access. 152, 84-89.
- 9. Wolinsky, J.B., Colson, Y.L. and Grinstaff, M.W. (2012) Local Drug Delivery Strategies for Cancer Treatment: Gels, Nanoparticles, Polymeric Films, Rods, and Wafers. Journal of Controlled Release, 159, 14-26.
http://dx.doi.org/10.1016/j.jconrel.2011.11.031 - 10. Qian, F., Stowe, N., Saidel, G.M. and Gao, J. (2004) Comparison of Doxorubicin Concentration Profiles in Radiofrequency-Ablated Rat Livers from Sustained- and Dual-Release PLGA Millirods. Pharmaceutical Research, 21, 394-399.
http://dx.doi.org/10.1023/B:PHAM.0000019290.70358.30 - 11. Grossman, S.A., et al. (1992) The Intracerebral Distribution of BCNU Delivered by Surgically Implanted Biodegradable Polymers. Journal of Neurosurgery, 76, 640-647.
http://dx.doi.org/10.3171/jns.1992.76.4.0640 - 12. Bota, D.A., Desjardins, A., Quinn, J.A., Affronti, M.L. and Friedman, H.S. (2007) Interstitial Chemotherapy with Biodegradable BCNU (Gliadel) Wafers in the Treatment of Malignant Gliomas. Therapeutics and Clinical Risk Management, 3, 707-715.
- 13. Ortiz, R., Au, J.L.-S., Lu, Z., Gan, Y. and Wientjes, M.G. (2007) Biodegradable Intraprostatic Doxorubicin Implants. AAPS Journal, 9, E241-E250.
http://dx.doi.org/10.1208/aapsj0902027 - 14. Fung, L.K., et al. (1998) Pharmacokinetics of Interstitial Delivery of Carmustine, 4-Hydroperoxycyclophosphamide, and Paclitaxel from a Biodegradable Polymer Implant in the Monkey Brain Pharmacokinetics of Interstitial Delivery of Carmustine, and Paclitaxel from Monkey Brain. Cancer Research, 672-684.
- 15. Solanki, H.K., et al. (2010) Recent Advances in Implantable Drug Delivery Tm. International Journal of Pharmaceutical Sciences Review and Research, 4, 28.
- 16. Jonas, O., et al. (2015) An Implantable Microdevice to Perform High-Throughput in Vivo Drug Sensitivity Testing in Tumors. Science Translational Medicine, 7, 284ra57.
- 17. Jaggi, A.S., Jain, V. and Singh, N. (2011) Animal Models of Neuropathic Pain. Fundamental & Clinical Pharmacology, 25, 1-28.
http://dx.doi.org/10.1111/j.1472-8206.2009.00801.x - 18. Klinghoffer, R.A., et al. (2015) A Technology Platform to Assess Multiple Cancer Agents Simultaneously within a Patient’s Tumor. Science Translational Medicine, 7, 284ra58.
- 19. Zheng, J.H., Chen, C.T., Au, J.L.-S. and Wientjes, M.G. (2001) Time and Concentration Dependent Penetration of Doxorubicin in Prostate Tumors. AAPS PharmSci, 3, 69-77.
http://dx.doi.org/10.1208/ps030215 - 20. Post, J. and Hoffman, J. (1964) The Replication Time and Pattern of Carcinogen-Induced Hepatoma Cells. The Journal of Cell Biology, 22, 341-350.
http://dx.doi.org/10.1083/jcb.22.2.341 - 21. Shemi, A., Khvalevsky, E.Z., Gabai, R.M. and Domb, A. (2015) Multistep, Effective Drug Distribution within Solid Tumors. Oncotarget, 6, 39564-39577.
- 22. Staltiel, E. and McGuire, W. (1983) Doxorubicin (Adriamycin) Cardiomyopathy—A Critical Review. The Western Journal of Medicine, 139, 332-341.
- 23. Desai, V.G., et al. (2013) Development of Doxorubicin-Induced Chronic Cardiotoxicity in the B6C3F1 Mouse Model. Toxicology and Applied Pharmacology, 266, 109-121.
http://dx.doi.org/10.1016/j.taap.2012.10.025 - 24. Israel, M., Pegg, W.J., Wilkinson, P.M. and Garnick, M.B. (1978) Liquid Chromatographic Analysis of Adriamycin and Metabolites in Biological Fluids. Journal of Liquid Chromatography, 1, 795-809.
http://dx.doi.org/10.1080/01483917808060035 - 25. Laginha, K.M. (2005) Determination of Doxorubicin Levels in Whole Tumor and Tumor Nuclei in Murine Breast Cancer Tumors. Clincal Cancer Research, 11, 6944-6949.
http://dx.doi.org/10.1158/1078-0432.CCR-05-0343 - 26. Charrois, G.J.R. and Allen, T.M. (2004) Drug Release Rate Influences the Pharmacokinetics, Biodistribution, Therapeutic Activity, and Toxicity of Pegylated Liposomal Doxorubicin Formulations in Murine Breast Cancer. Biochimica et Biophysica Acta (BBA)—Biomembranes, 1663, 167-177.
http://dx.doi.org/10.1016/j.bbamem.2004.03.006 - 27. Hsu, B.B., Park, M.-H., Hagerman, S.R. and Hammond, P.T. (2014) Multimonth Controlled Small Molecule Release from Biodegradable Thin Films. Proceedings of the National Academy of Sciences of the United States of America, 111, 12175-12180.
http://dx.doi.org/10.1073/pnas.1323829111 - 28. Shikanov, S., et al. (2009) Intratumoral Delivery of Paclitaxel for Treatment of Orthotopic Prostate Cancer. Journal of Pharmaceutical Sciences, 98, 1005-1014.
http://dx.doi.org/10.1002/jps.21492 - 29. Johansen, P.B. (1981) Doxorubicin Pharmacokinetics after Intravenous and Intraperitoneal Administration in the Nude Mouse. Cancer Chemotherapy and Pharmacology, 5, 267-270.
http://dx.doi.org/10.1007/BF00434396 - 30. Barash, P., Cullen, B. and Stoelting, R. (2000) In: Barash, P., Cullen, B. and Stoelting, R., Eds., Clinical Anesthesia, Lippincott Williams & Wilkins, Philadelphia, 380-381.
- 31. Liu, J., Meisner, D., Kwong, E., Wu, X.Y. and Johnston, M.R. (2009) Translymphatic Chemotherapy by Intrapleural Placement of Gelatin Sponge Containing Biodegradable Paclitaxel Colloids Controls Lymphatic Metastasis in Lung Cancer. Cancer Research, 69, 1174-1181.
http://dx.doi.org/10.1158/0008-5472.CAN-08-1753 - 32. Forrest, M.L. (2011) Cancer Diagnosis and Therapies. Expert Opinion on Drug Delivery, 6, 785-792.
- 33. Polsky, D. (2003) Economic Evaluation of Breast Cancer Treatment: Considering the Value of Patient Choice. Journal of Clinical Oncology, 21, 1139-1146.
http://dx.doi.org/10.1200/JCO.2003.03.126 - 34. Bandyopadhyay, A., et al. (2010) Doxorubicin in Combination with a Small TGFβ Inhibitor: A Potential Novel Therapy for Metastatic Breast Cancer in Mouse Models. PLoS ONE, 5, e10365.
- 35. Casale, T.B. and Stokes, J.R. (2014) Immunotherapy: What Lies beyond. Journal of Allergy and Clinical Immunology, 133, 612-619.
http://dx.doi.org/10.1016/j.jaci.2014.01.007 - 36. Calabrese, L. (2016) Checkpoint Therapy—The Next-Horizon in Cancer and Autoimmunity. Interdisciplinary Autoimmune Summit 2016, 78-85.
https://ias2016.sched.org/event/4VHa/checkpoint-therapythe-next-horizon-in-cancer-and-autoimmunity - 37. Perlroth, D.J., Bhattacharya, J., Goldman, D.P. and Garber, A.M. (2012) An Economic Analysis of Conservative Management versus Active Treatment for Men with Localized Prostate Cancer. Journal of the National Cancer Institute Monographs, 2012, 250-257.
http://dx.doi.org/10.1093/jncimonographs/lgs037 - 38. Kenmogne, L.C., Ayan, D., Roy, J., Maltais, R. and Poirier, D. (2015) The Aminosteroid Derivative RM-133 Shows in Vitro and in Vivo Antitumor Activity in Human Ovarian and Pancreatic Cancers. PLoS ONE, 10, e0144890.
http://dx.doi.org/10.1371/journal.pone.0144890 - 39. Richard, B.M., et al. (2012) The Safety of EXPAREL? (Bupivacaine Liposome Injectable Suspension) Administered by Peripheral Nerve Block in Rabbits and Dogs. Journal of Drug Delivery, 2012, Article ID: 962101.
http://dx.doi.org/10.1155/2012/962101
Abbreviations and Acronyms
PLGA: Poly-lactic-co-glycolic-acid copolymer;
STID: Solid therapeutic implant device.
NOTES

*Corresponding author.