International Journal of Clinical Medicine
Vol.4 No.4(2013), Article ID:30901,4 pages DOI:10.4236/ijcm.2013.44040
Severe Ectopic Cushing’s Syndrome Due to ACTH-Secreting Pheochromocytoma
![]()
1Second Division of Internal Medicine and Hypertension Unit, Arcispedale Santa Maria Nuova, Reggio Emilia, Italy; 2Diabetologic Unit, Arcispedale Santa Maria Nuova, Reggio Emilia, Italy; 3Radiology Unit, Arcispedale Santa Maria Nuova, Reggio Emilia, Italy; 4Pathology Unit, Arcispedale Santa Maria Nuova, Reggio Emilia, Italy.
Email: *negro.aurelio@asmn.re.it
Copyright © 2013 Aurelio Negro et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 16th, 2013, revised March 12th, 2013; accepted April 17th, 2013
Keywords: Cushing’s Syndrome; Pheochromocytoma; Apparent Excess of Mineralocorticoid Syndrome
ABSTRACT
We report a new case of ectopic Cushing’s syndrome caused by an ACTH-producing pheochromocytoma. A 55-yearold woman presented with a history of severe proximal muscle weakness, polyuria, progressive virilization, anxiety, dyspnea on exercise, difficult to treat hypertension, and type 2 diabetes mellitus since 4 months. The laboratory data demonstrated ACTH-dependent hypercortisolism. The abdominal computed tomography scan showed a 30 mm welldefined mass in the left adrenal gland suggestive for pheochromocytoma. The adrenal veins were sampled, with intraprocedural cortisol measurement, to dosing selective ACTH and cathecolamines. The results established clearly the left adrenal gland as the source of ACTH overproduction. A left sided adrenalectomy was performed with subsequent resolution of Cushing’s syndrome. The patient was discharged in good clinical condition.
1. Introduction
Cushing’s syndrome (CS) is characterized by overproduction of cortisol resulting in significant morbidity and, when untreated, increased mortality. As a consequence, it is crucial to establish an early and correct diagnosis [1]. Traditionally, CS is divided into ACTH-dependent CS and ACTH-independent CS. ACTH-dependent CS, about 80% of cases, can be caused by a corticotroph pituitary adenoma and, more rarely, by ectopic ACTH production [2]. Critical illness appears to be related to the severity of glucocorticoid excess and can occur in CS of every cause. However, ectopic ACTH production is a distinctive cause of CS with a particular high mortality because the hypercortisolism can be rapidly progressive. Moreover, severe hypertension and hypokalemia are the most common and prominent symptoms and mime the apparent mineralocorticoid excess syndrome [3]. Ectopic CS are usually due to secretion of ACTH from bronchial carcinoid, islet cell tumor, small cell lung carcinoma, medullary carcinoma of the thyroid, thymic carcinoid and disseminated neuroendocrine tumors. CS due to ACTHsecreting pheochromocytoma has sporadically been reported in the literature and accounts for 3% of ectopic ACTH-dependent CS [4,5].
We report a new case of severe CS because of ACTH secretion by a pheochromocytoma.
2. Case Report
A 55-year-old woman presented to us with a history of severe proximal muscle weakness, polyuria, progressive virilization, anxiety, dyspnea on exercise, difficult to treat hypertension, and type 2 diabetes mellitus since 4 months. Her past medical history was characterized by hyperthyroidism from multinodular goiter well treated with methimazole.
At presentation, physical examination revealed signs of severe CS, with centripetal obesity, a “buff hump”, moon face, mild hirsutism, abdominal striae, dermographism, and truncal ecchimotic patches. Her supine blood pressure was 178/102 while standing blood pressure was 166/98 mmHg; the heart rate was 82 bpm. She was on treatment with a calcium antagonist, a selective a-receptor blocker and an ACE-inhibitor. The main laboratory findings were as follows: glucose 12.39 mmol/l, serum creatinine 70.4 mmol/l, serum Na+ 142 mmol/l, serum K+ 1.6 mmol/l, Cl− 96 mmol/l, pH 7.57, 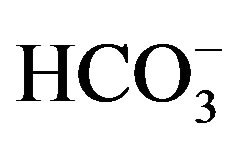 44 mmol/l, 24-hour urinary Na+ 226 mmol, 24-hour urinary K+ 144 mmol with a transtubular potassium gradient (TTKG) 23, no proteinuria at urinalysis. Plasma renin activity (PRA) and serum aldosterone levels were very low: 0.3 ng/ml/h and 19 pg/ml [normal range (n.r.) 35 - 160] respectively. Total 24-h urinary cortisol excretion was 6380 mg (n.r. 10 - 85). The plasma cortisol and ACTH concentrations in the morning were 1300 ng/ml (n.r. 45 - 250) and 273 pg/ml (n.r. 5 - 50) respectively; in the evening, 1182 ng/ml (n.r. 15 - 70) and 216 pg/ml (n.r. < 40), indicating an impaired circadian rhythm in the setting of ACTH-dependent Cushing’s syndrome. A high-dose dexametazone test did not show any suppression of cortisol and ACTH levels (960 ng/ml and 194 pg/ml respectively). Because of progressive deterioration of clinical condition, with marked hypokalemia despite intravenous potassium supplementation and spironolactone, we rapidly performed a magnetic resonance imaging of the pituitary, wich did not reveal any adenoma. An inferior petrosal venous sampling was not taken. A contemporary chest and abdominal computed tomography scan showed a 30 mm welldefined mass in the left adrenal gland with marked peripheral enhancement and high density (90 HU), altogether a central area of low attenuation due to necrosis with a thick enhancing septation (Figures 1(a) and (b)). The CT imaging was suggestive for pheochromocytoma. A 24-h urinary sample showed only a slightly increase of metanephrine excretion, 395.1 mg/24h (n.r. 74 - 297), while the normetanephrine resulted 76.2 mg/24h (n.r. 105 - 354); the methoxytyramine was within normal range. The plasma adrenaline and noradrenaline were 341.8 pg/ml (n.r. 10 - 50), and 296.7 pg/ml (n.r. 40 - 400), respectively. An 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) showed an area of high uptake in the left adrenal gland, with peripherally prevailing signal and central hypoactivity (Figure 1(c)); no other hyperactivity areas. Also, a 68GaDOTA-octreotate (DOTATATE) PET confirmed the peripheral high uptake in the left adrenal gland. Because of the lesion in the left adrenal gland, both adrenal veins were sampled, with intraprocedural cortisol measurement, to dosing selective ACTH and cathecolamines. The results established clearly the left adrenal gland as the source of ACTH overproduction (Table 1). Then, a left sided adrenalectomy was performed under treatment with a-blocker doxazosin and b-blocker atenolol. Histological examination revealed a 3 × 2.5 cm adrenal tumor consistent with a pheocromocytoma, without signs of malignancy and a PASS score 2. Additional immunostaining revealed positive immunostaining for chromogranin and s-100 protein (Figures 1(d) and (e)). Immunostaining for ACTH was not performed. As soon after surgery, the patient developed systemic inflammatory response syndrome by bilateral pneumonia with a 15-day stay in intensive care unit. However, clinical and biochemical signs of hypercortisolism rapidly improved; in fact, plasma ACTH and cortisol levels were 45 pg/ml and 207 ng/ml respectively. Also, serum glucose and potassium levels progressively returned within normal range. The successive postoperative course was uneventful and the patient was discharged in good clinical condition. At two months follow-up, plasma cortisol and ACTH were 72 ng/ml and 13 pg/ml respec-
44 mmol/l, 24-hour urinary Na+ 226 mmol, 24-hour urinary K+ 144 mmol with a transtubular potassium gradient (TTKG) 23, no proteinuria at urinalysis. Plasma renin activity (PRA) and serum aldosterone levels were very low: 0.3 ng/ml/h and 19 pg/ml [normal range (n.r.) 35 - 160] respectively. Total 24-h urinary cortisol excretion was 6380 mg (n.r. 10 - 85). The plasma cortisol and ACTH concentrations in the morning were 1300 ng/ml (n.r. 45 - 250) and 273 pg/ml (n.r. 5 - 50) respectively; in the evening, 1182 ng/ml (n.r. 15 - 70) and 216 pg/ml (n.r. < 40), indicating an impaired circadian rhythm in the setting of ACTH-dependent Cushing’s syndrome. A high-dose dexametazone test did not show any suppression of cortisol and ACTH levels (960 ng/ml and 194 pg/ml respectively). Because of progressive deterioration of clinical condition, with marked hypokalemia despite intravenous potassium supplementation and spironolactone, we rapidly performed a magnetic resonance imaging of the pituitary, wich did not reveal any adenoma. An inferior petrosal venous sampling was not taken. A contemporary chest and abdominal computed tomography scan showed a 30 mm welldefined mass in the left adrenal gland with marked peripheral enhancement and high density (90 HU), altogether a central area of low attenuation due to necrosis with a thick enhancing septation (Figures 1(a) and (b)). The CT imaging was suggestive for pheochromocytoma. A 24-h urinary sample showed only a slightly increase of metanephrine excretion, 395.1 mg/24h (n.r. 74 - 297), while the normetanephrine resulted 76.2 mg/24h (n.r. 105 - 354); the methoxytyramine was within normal range. The plasma adrenaline and noradrenaline were 341.8 pg/ml (n.r. 10 - 50), and 296.7 pg/ml (n.r. 40 - 400), respectively. An 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) showed an area of high uptake in the left adrenal gland, with peripherally prevailing signal and central hypoactivity (Figure 1(c)); no other hyperactivity areas. Also, a 68GaDOTA-octreotate (DOTATATE) PET confirmed the peripheral high uptake in the left adrenal gland. Because of the lesion in the left adrenal gland, both adrenal veins were sampled, with intraprocedural cortisol measurement, to dosing selective ACTH and cathecolamines. The results established clearly the left adrenal gland as the source of ACTH overproduction (Table 1). Then, a left sided adrenalectomy was performed under treatment with a-blocker doxazosin and b-blocker atenolol. Histological examination revealed a 3 × 2.5 cm adrenal tumor consistent with a pheocromocytoma, without signs of malignancy and a PASS score 2. Additional immunostaining revealed positive immunostaining for chromogranin and s-100 protein (Figures 1(d) and (e)). Immunostaining for ACTH was not performed. As soon after surgery, the patient developed systemic inflammatory response syndrome by bilateral pneumonia with a 15-day stay in intensive care unit. However, clinical and biochemical signs of hypercortisolism rapidly improved; in fact, plasma ACTH and cortisol levels were 45 pg/ml and 207 ng/ml respectively. Also, serum glucose and potassium levels progressively returned within normal range. The successive postoperative course was uneventful and the patient was discharged in good clinical condition. At two months follow-up, plasma cortisol and ACTH were 72 ng/ml and 13 pg/ml respec-
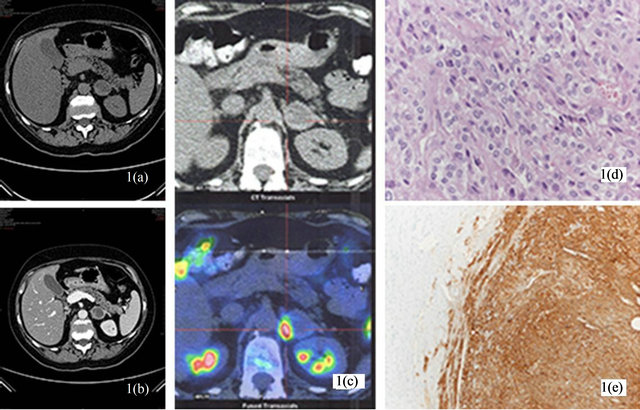
Figure 1. (a) Pheochromocytoma cells with finely granular basophilic cytoplasm and ovoid nuclei, bound by a delicate fibrovascular stroma; (b) Intense immunoreactivity for chromogranin in the tumor cells of pheochromocytoma (right).
tively, and blood pressure was 130/80 requiring only one antihypertensive drug (nebivolol).
3. Discussion
This patient represents a new case of ectopic CS caused by an ACTH-producing pheochromocytoma. ACTH-dependent CS is a rare endocrine disorder caused in most cases by a pituitary corticotroph adenoma. Ectopic ACTHdependent CS account for 10% - 15% of all cases with a reported incidence of 0.1 per million per year [2]. Adrenal pheochromocytoma is a rare cause of ectopic ACTH secretion [4,5]. To our knowledge, about thirty cases have been reported in the literature.
Pheochromocytoma is a challenging and life-threatening tumor, often associated with very difficult to interpret clinical manifestations. This neoplasm may secrete various substances such as somatostatin, enkephalins, calcitonin, vasoactive intestinal peptide, neuropeptide Y, renin, ACTH, PTH, erythropoietin, adrenomedullin, and DOPA in addition to catecholamines. The symptoms manifested by the patients depend on the prevailing secretory products secreted from the tumor [6]. ACTH eventually produced by the tumor stimulates the adrenal cortex to hypersecrete cortisol. Hence, as in our patient, the prevalent clinical manifestation is a Cushing’s syndrome. Our case is relevant because it presented as severe CS without manifest symptoms and signs of adrenergic hyperactivity. At first, the profound hypokalemia and the elevated TTKG point out an excess of mineralocorticoid activity. The relief of suppression of PRA and very low serum aldosterone levels indicated an apparent excess of mineralocorticoid activity. This potentially life-threatening condition was sustained by severe hypercortisolism in the setting of an ectopic ACTH-dependent CS. About 75% of patients with CS have hypertension, and this increases to over 95% in the subgroup of patients with ectopic ACTH-dependent CS. Moreover, the ectopic ACTH-dependent CS is characterized by excess of mineralocorticoid activity, with hypokalemic alkalosis in about 100% of cases, in contrast to 10% in other forms of CS. In severe hypercortisolism, all available cortisol cannot be converted to the inactive metabolite cortisone from the enzyme 11b-hydroxysteroid dehydrogenase type 2 (11bHSD2) and saturates the mineralocorticoid receptors to cause apparent hyperaldosteronism [7].
Surprisingly, in our patient CT scan and PET denied tumoral nodules in the most common sites of ectopic ACTH secrection [4], but showed a large adrenal mass suggestive for pheochromocytoma. As rewieved by Nijhoff et al. [5], only two of 24 patients with an ACTHsecreting pheochromocytoma did not manifest obvious clinical signs of catecholamines excess. This further emphasizes the diagnostic challenge in these cases. In our patient, only the selective adrenal vein sampling estabTable 1. Adrenal venous sampling with intraprocedural cortisol measurement: were dosed selective ACTH and cathecolamines. The results established the left adrenal gland as the source of ACTH overproduction.
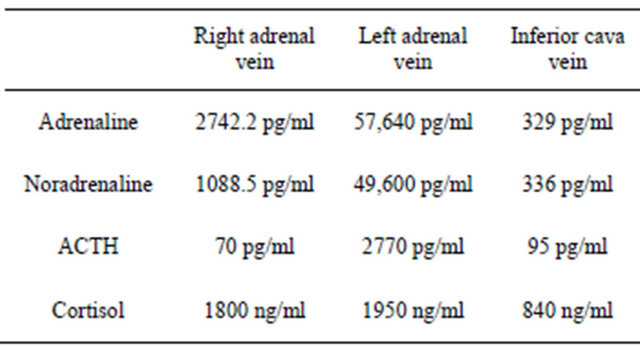
lished the left adrenal gland to be the source of ACTH overproduction. Moreover, as depicted in Table 1, the venous sampling demonstrated significant increase of catecholamines levels in the left vs the right adrenal vein too. Final diagnosis was made by immunohistological analysis. Therefore, the tumor was found to secrete ACTH predominantly, explaining the presentation of severe CS instead of a subclinical pheochromocytoma [6].
4. Conclusion
We report herein a new case of a very rare ectopic ACTH-dependent CS due to subclinical pheochromocytoma. Early diagnosis, adequate aand b-blockage before surgery and surgical removal of the adrenal tumor determined a good outcome for the patient.
REFERENCES
- J. Lindholm, S. Juul, J. O. Jorgensen, J. Astrup, P. Bjerre, U. Feldt-Rasmussen, C. Hagen, J. Jorgensen, M. Kosteljanetz, L. Kristensen, et al., “Incidence and Late Prognosis of Cushing’s Syndrome: A Population-Based Study,” The Journal of Clinical Endocrinology & Metabolism, Vol. 86, No. 1, 2001, pp. 117-123. doi:10.1210/jc.86.1.117
- J. Newell-Price, X. Bertagna, A. B. Grossman and L. K. Nieman, “Cushing’s Syndrome,” Lancet, Vol. 367, No. 9522, 2006, pp. 1605-1617. doi:10.1016/S0140-6736(06)68699-6
- D. J. Torpy, N. Mullen, I. Ilias, et al., “Association of Hypertension and Hypokalemia with Cushing’s Syndrome Caused by Ectopic ACTH Secretion: A Series of 58 Cases,” Annals of the New York Academy of Sciences, Vol. 970, No. 1, 2002, pp. 134-144. doi:10.1111/j.1749-6632.2002.tb04419.x
- J. P. Aniszewski, W. F. Young Jr., G. B. Thompson, C. S. Grant and J. A. van Heerden, “Cushing Syndrome Due to Ectopic Adrenocorticotropic Hormone Secretion,” World Journal of Surgery, Vol. 25, No. 7, 2001, pp. 934-940. doi:10.1007/s00268-001-0032-5
- M. F. Nijhoff, O. M. Derkers, L. J. Vleming, J. W. A. Smit, J. A. Romijn and A. M. Pereira, “ACTH-Producing Pheocromocytoma: Clinical Considerations and Concise Review of the Literature,” European Journal of Internal Medicine, Vol. 20, No. 7, 2009, pp. 682-685. doi:10.1016/j.ejim.2009.08.002
- M. Mannelli, J. W. M. Lenders, K. Pacak, G. Parenti and G. Eisenhofer, “Subclinical Phaeochromocytoma,” Best Practice and Researche Clinical Endocrinology and Metabolism, Vol. 26, No. 4, 2012, pp. 507-515.
- P. M. Stewart, “Mineralocorticoid Hypertension,” Lancet, Vol. 353, No. 9161, 1999, pp. 1341-1347. doi:10.1016/S0140-6736(98)06102-9
NOTES
*Corresponding author.