International Journal of Clinical Medicine
Vol. 3 No. 6 (2012) , Article ID: 24546 , 4 pages DOI:10.4236/ijcm.2012.36089
Novel p.C620L RET Mutation Detected in a Patient with Medullary Thyroid Carcinoma
![]()
1ARUP Institute for Clinical and Experimental Pathology, Salt Lake City, USA; 2Department of Pathology, University of Utah Medical School, Salt Lake City, USA.
Email: *rebecca.margraf@aruplab.com
Received July 24th, 2012; revised August 27th, 2012; accepted September 12th, 2012
Keywords: RET; MEN2; MTC; Medullary Thyroid Carcinoma; High Resolution Melting Analysis; Inherited Thyroid Cancer
ABSTRACT
A patient with an apparent sporadic medullary thyroid carcinoma was tested for RET germline mutations by Sanger sequencing of RET exons 10, 11, and 13 - 16. The patient was heterozygous for two known mutations causative of Multiple Endocrine Neoplasia type 2 disorder, and both mutations were within codon 620 of RET exon 10, c.1859G > T (p.C620F) and c.1860C > G (p.C620W). In order to determine if these adjacent mutations were in cis or in trans, an unlabeled probe method and high-resolution melting analysis were utilized. The mutations were confirmed to occur in cis, representing a novel mutation, c.1859_1860delinsTG (p.C620L). Sanger sequencing of parental samples did not identify any changes at codon 620, so the p.C620L mutation is also de novo. The early age of onset for medullary thyroid carcinoma and the presence of lymph node metastasis in this patient suggests individuals with the p.C620L mutation should be treated and screened (for pheochromocytomas and parathyroid hyperplasia) as Multiple Endocrine Neoplasia type 2 patients with other RET codon 620 mutations (American Thyroid Association risk level B).
1. Introduction
Multiple endocrine neoplasia type 2 (MEN2, OMIM 171400) is an autosomal dominant inherited disorder with the hallmark symptom of medullary thyroid carcinoma (MTC). MEN2 consists of three syndromes: familial medullary thyroid carcinoma (FMTC; have MTC only), MEN2A (MTC, Pheochromocytoma, and parathyroid hyperplasia) and MEN2B (MTC, Pheochromocytoma, and other characteristic findings) [1-3]. MEN2 is caused by pathogenic RET proto-oncogene germline mutations.
The transmembrane RET protein is involved in cell growth and differentiation. In the presence of ligand, the RET monomers dimerize, autophosphorylate, and activate downstream signaling. In RET exon 10 there are four conserved cysteine residues (at codons 609, 611, 618, and 620) that are hotspots for MEN2 causative, gain-of-function mutations [3,4]. These cysteine residues are involved in intramolecular disulfide bonding. It is hypothesized that cysteine residue substitution disrupts the intramolecular disulfide bond, allowing the unpaired partner cysteine to aberrantly create a disulfide bond between the mutant RET monomers, resulting in ligandindependent RET dimerization and activation [5-7]. Mutations at these four codons have been detected in patients with MEN2A or FMTC, and the age of disease onset is variable ranging from childhood to adulthood (see the MEN2 RET database: www.arup.utah.edu/database/MEN2/MEN2_welcome.php [4]).
This report describes the detection of two known pathogenic RET heterozygous changes at exon 10, both within codon 620, by Sanger sequencing in a patient with MTC. High-resolution melting analysis (HRM) and unlabeled probes were used to determine the adjacent mutations occur on the same allele and represent a novel RET mutation, c.1859_1860delinsTG (p.C620L).
2. Materials and Methods
2.1. Samples
This study was conducted under the University of Utah IRB board 00028725, and patient consent was obtained. Patient and parental peripheral blood specimens were collected and DNA was extracted from blood leukocytes using the MagNa Pure Compact (Roche) procedure following the manufacture recommended protocol.
2.2. Sequencing of the RET Protooncogene
Samples were amplified by PCR for the coding regions and intron/exon boundaries of RET exons 10 - 11, and 13 - 16, then bidirectional Sanger sequencing was performed. Mutation Surveyor® software (SoftGenetics, State College, PA) was used for data analysis.
2.3. Unlabeled Probe and High Resolution Melting Analysis
Sample DNA was amplified by asymmetric PCR, highresolution melting (HRM) analysis was performed on the HR-1TM (Idaho Technology, Salt Lake City, UT), and data were analyzed with custom software written in LabView (National Instruments, Austin TX) as described previously [8]. Briefly, after PCR, the unlabeled probe hybridizes to the amplicon template which is measured by LCgreen, a DNA intercalating dye. During HRM analysis, the amplicons with mismatches to the probe have a lower melting temperature (Tm) of dissociation than amplicons with complementary sequence to the probe [9].
The PCR primers used for RET exon 10, codon 620 interrogation were (forward 5’GGGCAGCATTGTTGGGGGAC3’ and reverse 5’TGGTGGTCCCGGCCGCCA3’). The unlabeled probes were 5’GGAGAAGTGCTTCTGCGAGCCCGAAGACATC3’ [wild type sequence at codon 620 (TGC)] and 5’GGAGAAGTGCTTCTTGGAGCCCGAAGACATC3’ [c.1859_1860delinsTG mutation sequence at codon 620 (TTG)]. Unlabeled probes have a blocking 3’amino modifier (IDT, Coralville, IA) to prevent extension during the PCR step.
3. Results
3.1. Clinical Report
In 2005, the patient presented at 19 years of age with a palpable thyroid nodule. The nodule was found to be 1.3 cm in the left thyroid lobe by ultrasound. The patient did not have dysphagia, voice changes, breathing issues, palpitations, previous radiation exposures, pain, or family history of MTC/MEN2. Fine needle aspiration showed neoplastic proliferation most consistent with medullary thyroid carcinoma (MTC). In concordance with the MTC diagnosis, calcitonin and carcinoembryonic antigen (CEA) levels were elevated: 1181 pg/mL and 53.3 ng/mL, respectively. The patient was screened for additional findings associated with MEN2, but had normal levels of parathyroid hormone, serum calcium, catecholamines, metanephrine and creatinine. Since the patient has no family history of endocrine problems or malignancy and the biochemical examination did not detect other findings of MEN2, sporadic MTC was suspected at the time of the surgery.
Total thyroidectomy with central compartment node dissection was preformed. A tumor of 1.2 × 1.1 × 1.0 cm was found in the left thyroid lobe. Multicentric or bilateral involvement was not identified. The tumor did not extend beyond the resection margins and capsular invasion was not identified. One of three lymph nodes tested positive for metastasis. The surgery confirmed the MTC diagnosis with small focus of perithyroidal connective tissue involvement and discovered metastasis to one of three perithyroidal lymph nodes. No unusual findings were found in the parathyroid tissue samples submitted for pathological examination. The left inferior parathyroid was autotransplanted to the left sternocleidomastoid. Calcitonin levels post-surgery remain borderline high at 13 - 15 pg/mL and were rising, suggesting the potential for recurrence.
3.2. RET Proto-Oncogene Mutation Identification
The patient was tested at ARUP laboratories for germline RET mutations in exons 10, 11, and 13 - 16 by clinical Sanger sequencing (Figure 1(a)). Two heterozygous mutations were found, adjacent to each other within codon 620, a known hotspot for MEN2 causative RET mutations. Sanger sequencing of the parental samples, targeting RET exon 10 yielded only wild type sequence at codon 620. So, were these two RET mutations in trans c.[1859G > T;1860C > G] (p.[C620F;C620W]) or in cis c.1859_1860delinsTG (p.C620L)?
Since these two mutations affect adjacent base pairs, phase was determined using HRM and the unlabeled probe method [9] as described previously for RET mutation identification [8]. A probe complementary to the wild type RET sequence for codon 620 (TGC) was used (Figure 1(b)). Two control DNA samples each heterozygous for one codon 620 mutation [c.1859G > T (p.C620F) or c.1860C > G (p.C620W)] demonstrated a decreased Tm (melting temperature) for the mutation alleles compared to their wild type alleles. The patient’s sample demonstrated an allele with an even lower Tm than either of the control alleles harboring single nucleotide mutations. The patient sample also had an allele with the same Tm as the wild type control sample (which had only wild type alleles). In addition, a probe that complemented the c.1859_1860delinsTG mutation sequence at codon 620 (TTG) was used (Figure 1(c)). The patient’s DNA sequence complemented the probe resulting in a variant
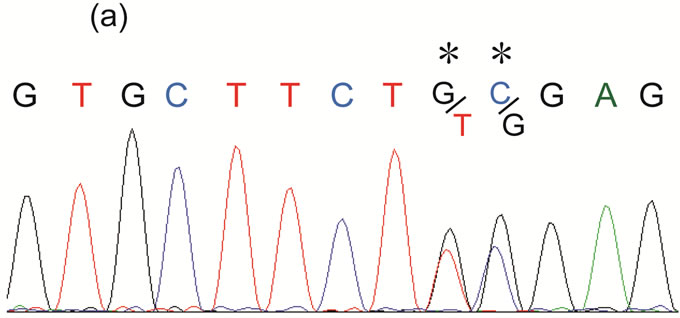
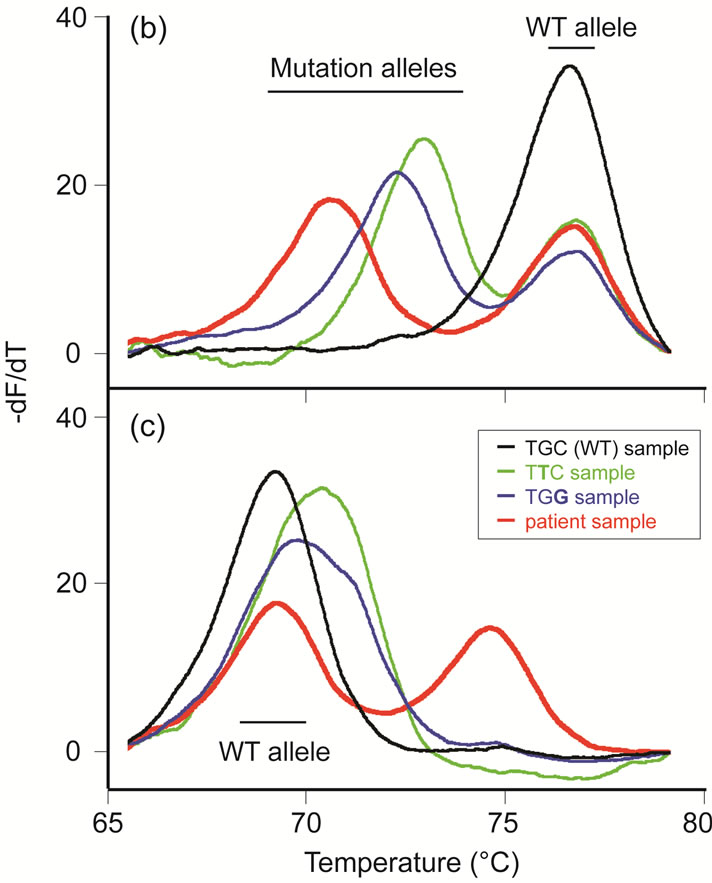
Figure 1. (a) Sanger sequencing data is shown covering codon 620, with the G/T and C/G heterozygous mutations starred (*); (b) and (c) unlabeled probe HRM data is shown as a derivative plot. Each sample line colors are displayed in the figure (on panel (c)). A control wild type (WT) sample with only wild type sequence at codon 620 (TGC) are the black traces. Green traces represent a sample heterozygous for the c.1859G > T (p.C620F) mutation (codon sequence TTC). Blue traces represent a sample heterozygous for the c.1860C > G (p.C620W) mutation (codon sequence TGG). The red traces represent the patient’s sample; (b) HRM data with unlabeled probe complementary to wild type codon 620 (TGC) sequence. The range of Tm for the wild type (WT) and mutation alleles are marked on the panel; (c) HRM data with unlabeled probe complementary to codon 620 (TTG) mutation sequence (c.1859_1860delinsTG). The range of Tm for the wild type (WT) allele is marked.
allele with a higher Tm than either the wild type allele or the mutation alleles from the controls harboring single nucleotide mutations at codon 620. And again, the patient also had a wild type allele. The patient’s parents were Sanger sequenced at RET codon 620 and both had wild type sequence.
4. Discussion
Since an apparently sporadic MTC (i.e., isolated, nonfamilial MTC) can be the presenting clinical feature for some MEN2 patients, RET genotyping is often performed for patients with sporadic MTC [10]. The most common areas of MEN2 causative germline RET mutations are exons 10, 11, 13 - 16 [3,11-13]. Molecular sequencing of the RET gene was performed to determine if this patient with apparently isolated MTC had a heritable cause of disease and may require further screening for other MEN2 manifestations. Sequencing detected two adjacent heterozygous RET mutations, each known to be causative of MEN2. It was important to define the chromosomal configuration of these two mutations to aid in risk assessment and genetic counseling for family members. Unlabeled probe high resolution melting analysis methods determined these two changes were on the same allele, and therefore identified the patient’s mutation as c.1859_1860delinsTG (p.C620L). This mutation appears to be novel, since it was not found in NCBI dbSNP135, 1000 Genomes, or other publically available databases. Also, the p.C620L change was not found in a self-reported MEN2/MTC unaffected cohort (136 people) tested for variations in RET introns and exons 9 through 16 [14]. The patient’s parents had wild type sequence at codon 620, so the mutation is de novo as well.
Any mutation changing the RET 620 cysteine residue to any other amino acid has been reported to result in MTC or MEN2A [4,15], supporting the presumed pathogenicity of the novel p.C620L mutation. As seen with the other RET codon 620 mutations [5-7], it is likely that the p.C620L mutation disrupts intra-cellular disulfide bonding, allowing intermolecular disulfide binding with other mutant RET monomers, leading to constitutive activity and signaling. The youngest age of MTC onset associated with RET codon 620 mutations is 5 years and prophylactic thyroidectomy is general recommended before or at 5 years of age [1,16]. The young age of onset in our patient as well as the detected lymph node metastasis, indicates this mutation should follow the American Thyroid Association’s recommendations for risk level B, which is the classification assigned to other codon 620 mutations [16].
In conclusion, the two adjacent Sanger sequencing detected mutations were determined to be in cis and therefore the patient had the novel and de novo RET mutation c.1859_1860delinsTG (p.C620L). The p.C620L mutation is very likely MEN2 causative due to loss of cysteine at codon 620 and the young age of MTC metastasis in the patient. Since this patient was the only affected family member and had MTC but no other features of MEN2, the specific MEN2 subtype association with this mutation cannot be predicted. Because other RET codon 620 mutations are associated with Pheochromocytomas (youngest reported age of onset in the literature was of 19 years of age) and the much rarer parathyroid hyperplasia; the patient should be followed for potential development of these additional MEN2 manifestations. The novel p.C620L mutation will be added to our online, publicly available MEN2 RET mutation database [4].
5. Acknowledgements
Thank you to Dr. Theodore H. Humble for the patient clinical information, and also to the patient and her parents for participation in our study.
REFERENCES
- J. W. de Groot, T. P. Links, J. T. Plukker, et al., “Ret as a Diagnostic and Therapeutic Target in Sporadic and Hereditary Endocrine Tumors,” Endocrine Reviews, Vol. 27, No. 5, 2006, pp. 535-560. doi:10.1210/er.2006-0017
- P. J. Morrison and N. C. Nevin, “Multiple Endocrine Neoplasia Type 2B (Mucosal Neuroma Syndrome, Wagenmann-Froboese Syndrome),” Journal of Medical Genetics, Vol. 33, No. 9, 1996, pp. 779-782. doi:10.1136/jmg.33.9.779
- C. Eng, D. Clayton, I. Schuffenecker, et al., “The Relationship between Specific Ret Proto-Oncogene Mutations and Disease Phenotype in Multiple Endocrine Neoplasia Type 2. International Ret Mutation Consortium Analysis,” Journal of the American Medical Association, Vol. 276, No. 19, 1996, pp. 1575-1579. doi:10.1001/jama.276.19.1575
- R. L. Margraf, D. K. Crockett, P. M. Krautscheid, et al., “Multiple Endocrine Neoplasia Type 2 Ret Protooncogene Database: Repository of Men2-Associated Ret Sequence Variation and Reference for Genotype/Phenotype Correlations,” Human Mutation, Vol. 30, No. 4, 2009, pp. 548-556. doi:10.1002/humu.20928
- M. Santoro, F. Carlomagno, A. Romano, et al., “Activation of Ret as a Dominant Transforming Gene by Germline Mutations of Men2a and Men2b,” Science, Vol. 267, No. 5196, 1995, pp. 381-383. doi:10.1126/science.7824936
- S. Chappuis-Flament, A. Pasini, G. De Vita, et al., “Dual Effect on the Ret Receptor of Men 2 Mutations Affecting Specific Extracytoplasmic Cysteines,” Oncogene, Vol. 17, No. 22, 1998, pp. 2851-2861. doi:10.1038/sj.onc.1202202
- A. Z. Lai, T. S. Gujral and L. M. Mulligan, “Ret Signaling in Endocrine Tumors: Delving Deeper into Molecular Mechanisms,” Endocrine Pathology, Vol. 18, No. 2, 2007, pp. 57-67. doi:10.1007/s12022-007-0009-5
- R. L. Margraf, R. Mao, W. E. Highsmith, et al., “Ret Proto-Oncogene Genotyping Using Unlabeled Probes, the Masking Technique, and Amplicon High-Resolution Melting Analysis,” Journal of Molecular Diagnostics, Vol. 9, No. 2, 2007, pp. 184-196. doi:10.2353/jmoldx.2007.060091
- L. Zhou, A. N. Myers, J. G. Vandersteen, et al., “ClosedTube Genotyping with Unlabeled Oligonucleotide Probes and a Saturating DNA Dye,” Clinical Chemistry, Vol. 50, No. 8, 2004, pp. 1328-1335. doi:10.1373/clinchem.2004.034322
- M. Wiench, Z. Wygoda, E. Gubala, et al., “Estimation of Risk of Inherited Medullary Thyroid Carcinoma in Apparent Sporadic Patients,” Journal of Clinical Oncology, Vol. 19, No. 5, 2001, pp. 1374-1380.
- D. P. Smith, C. Houghton and B. A. Ponder, “Germline Mutation of Ret Codon 883 in Two Cases of de Novo Men 2b,” Oncogene, Vol. 15, No. 10, 1997, pp. 1213-1217. doi:10.1038/sj.onc.1201481
- K. Frank-Raue, S. Rondot, E. Schulze, et al., “Change in the Spectrum of Ret Mutations Diagnosed between 1994 and 2006,” Clinical Laboratory, Vol. 53, No. 5-6, 2007, pp. 273-282.
- I. Berndt, M. Reuter, B. Saller, et al., “A New Hot Spot for Mutations in the Ret Protooncogene Causing Familial Medullary Thyroid Carcinoma and Multiple Endocrine Neoplasia Type 2a,” Journal of Clinical Endocrinology & Metabolism, Vol. 83, No. 3, 1998, pp. 770-774. doi:10.1210/jc.83.3.770
- R. L. Margraf, J. D. Durtschi, J. E. Stephens, et al., “Determination of Ret Sequence Variation in an Men2 Unaffected Cohort Using Multiple-Sample Pooling and NextGeneration Sequencing,” Journal of Thyroid Research, Vol. 2012, 2012, Article ID: 318232. doi:10.1155/2012/318232
- K. Frank-Raue, L. A. Rybicki, Z. Erlic, et al., “Risk Profiles and Penetrance Estimations in Multiple Endocrine neoplasia Type 2a Caused by Germline Ret Mutations Located in Exon 10,” Human Mutation, Vol. 32, No. 1, 2010, pp. 51-58. doi:10.1002/humu.21385
- R. T. Kloos, C. Eng, D. B. Evans, et al., “Medullary Thyroid Cancer: Management Guidelines of the American Thyroid Association,” Thyroid, Vol. 19, No. 6, 2009, pp. 565-612. doi:10.1089/thy.2008.0403
Abbreviations:
HRM: High-Resolution Melting;
MEN2: Multiple Endocrine Neoplasia Type 2;
MTC: Medullary Thyroid Carcinoma;
NOTES
*Corresponding author.