Open Journal of Blood Diseases
Vol.3 No.1(2013), Article ID:29307,7 pages DOI:10.4236/ojbd.2013.31008
Successful Management of Sickle Cell Intrahepatic Cholestasis with Combined Use of Exchange Transfusion and Single-Pass Albumin Dialysis: A Case Report
![]()
1Hematology Department, Papageorgiou General Hospital, Thessaloniki, Greece; 2Second Department of Internal Medicine, Medical School, Aristotle University, Thessaloniki, Greece; 3Nephrology Department, Papageorgiou General Hospital, Thessaloniki, Greece; 4Blood Center and Blood Transfusion Service, AHEPA University Hospital, Thessaloniki, Greece; 5Thalassemia and Hemoglobinopathies Unit, First Department of Internal Medicine, Medical School, Aristotle University, Thessaloniki, Greece.
Email: *vassiliospap@gmail.com
Received December 31st, 2012; revised February 1st, 2013; accepted February 12th, 2013
Keywords: Sickle-Cell Intrahepatic Cholestasis; Exchange Transfusion; Single-Pass Albumin Dialysis (SPAD); Hydroxyurea
ABSTRACT
Sickle-cell intrahepatic cholestasis (SCIC) is an uncommon complication of sickle-cell disease (SCD), which can be life-threating if left untreated. We present the case of a 28-year-old man with SCD, who presented with jaundice and abdominal pain, one month after hydroxyurea discontinuation. Laboratory investigation revealed solely increased serum bilirubin, mainly conjugated, and imaging studies revealed choledocholithiasis. The patient unterwent an endoscopic sphincterectomy, but his jaundice deteriorated. Sickle-cell intrahepatic cholestasis was suspected and he received aggressive exchange transfusion therapy in combination with hydroxyurea. The jaundice had no signs of improvement; in fact total bilirubin raised up to 1053.7 μmol/L (61.62 mg/dl). Subsequently, the patient was treated with single-pass albumin dialysis (SPAD), in order to remove the excess bilirubin and protect mainly the brain and liver cells from its toxic effects. His laboratory values started to improve after one-and-a-half months of treatment. For the next two months, exchange transfusions were continued and bilirubin gradually returned to baseline values. The successful response appeared after the combined use of exchange transfusion and SPAD, which is being reported for the first time.
1. Introduction
Sickle-cell intrahepatic cholestasis (SCIC) is a difficult clinical problem. It is a quite rare entity, which could become a fatal complication of sickle cell disease (SCD). Higher death rate may be observed in older patients, with a prior hepatic disease and with homozygous sickle cell anemia [1]. The pathophysiology of SCIC remains uncertain, but extensive sickling within liver sinusoids is thought to cause vascular stasis and subsequent ballooning of hepatocytes leading to intracanalicular cholestasis. The only effective treatment in most cases is the early onset of exchange transfusion aiming at lowering the hemoglobin S (HbS) fraction, in an attempt to minimize the intrahepatic sickling.
In SCD patients, gall-stones is a common medical condition, therefore cholestasis can easily be attributed to cholelithiasis and treated with endoscopic retrograde cholangiopancreatography (ERCP).
In this report, we present a case of a young male with SCIC, treated with multiple manual exchange transfusions and supportive care, who achieved complete recovery of hepatic function after nearly two months of treatment.
2. Case Presentation
A 28-year-old Caucasian male with SCD, homozygous for the gene of HbS, was admitted to the emergency ward of the regional hospital, due to deep jaundice and choluria, started three days ago, and abdominal pain reflecting on flank. Physical exam demonstrated deep jaundice and right upper quadrant tenderness. The patient did not present fever, diarrhea, or bleeding. There were no signs of hepatic encephalopathy. Past medical history included a self-limited episode of painless jaundice at the age of 16 years and cholecystectomy five years ago. The last painful crisis, for which he was hospitalized, occurred three years ago. Since then he has been taking hydroxyurea on a regular basis, until a month ago, when he could no longer obtain the drug. He was referred to our hospital for further investigations and treatment.
The laboratory data on admission were: total bilirubin 624.66 μmol/L (36.53 mg/dL), direct (conjugated) bilirubin 378.6 μmol/L (22.14 mg/dL), hemoglobin concentration (Hb) 119 g/L, hematocrit (Hct) 0.346, white blood cell (WBC) count 7.61 ´ 109/L, platelet count 564 ´ 109/L, alanine aminotransferase (ALT) 79 U/L, aspartate aminotransferase (AST) 165 U/L, alkaline phosphatase (ALP) 171 U/L, gamma-glutamyltransferase (γ-GT) 165 U/L, albumin 39 g/L, international normalized ratio (INR) 1.05 and activated partial thromboplastin time (aPTT) 33.3 sec. The serology for hepatitis B and C viruses and for human immunodeficiency virus was negative, and the tests for hepatitis A virus, cytomegalovirus and EbsteinBarr virus were positive for IgG-antibodies. The immunological tests for antinuclear antibodies (ANA), antimitochondrial antibodies (AMA) and anti-liver/kidney microsomal antibodies (anti-LKM), were negative, too.
Imaging studies included a magnetic resonance cholangiopancreatography (MRCP), which demonstrated the presence of gall-stones into the common bile duct and distention of both intraand extra-hepatic bile ducts. Due to the markedly elevated direct bilirubin, and the imaging findings, the patient was treated for choledocholithiasis with ERCP and sphincterotomy. Despite the intervention, the jaundice deteriorated‑total bilirubin 830 μmol/L (48.54 mg/dL), direct/conjugated bilirubin 521.5 μmol/L (30.5 mg/dL)—while the clinical performance status remained excellent and liver biochemistry remained normal or only slightly influenced. At that time, hematologist consultation was requested.
The possibility of hepatic crisis or intrahepatic cholestasis due to sickling of red blood cells into the liver was taken into consideration. The patient was submitted to aggressive exchange transfusion of packed red blood cells (pRBCs) in order to achieve HbS fraction less than 30% and avoid further sickling. Due to the scarceness of patient’s red blood cells phenotype, that is group B, Rhesus CCD.ee, Duffy b negative, Kidd a negative, several consecutive manual exchange transfusions were performed, instead of a single session of large volume exchange transfusion. A few days later, hydroxyurea (HU) therapy was restarted.
On the 8th day of treatment, although HbS declined gradually to less than 40%, the bilirubin raised up to more than 1000 μmol/L (60 mg/dL). Thus, it was decided to commence a bilirubin-removing therapy using single-pass albumin dialysis (SPAD) along with exchange transfusions.
During the first twenty days of such aggressive treatment, the value of bilirubin showed a rapid fall of approximately 170 μmol/L (10 mg/dL) just after the SPAD session, and then started to increase slowly to the presession values on 1 - 2 days. The patient underwent a second MRCP, which did not produce additional findings, as well as a comprehensive laboratory investigation, which produced no evidence of primary biliary cirrhosis, autoimmune hepatitis and liver hemosiderosis. After the 4th session of SPAD, the bilirubin started to decline. In addition, ursodeoxycholic acid capsules were administered to increase bile flow and facilitate bilirubin excretion. According to the literature and the international guidelines, the patient continued exchange transfusions, in order to keep the hemoglobin S fraction below 30%.
Throughout his hospitalization, the patient remained clinically stable without any complications and with no significant changes in other laboratory values. He was discharged after 43 days when the total bilirubin dropped to 586.87 μmol/L (34.32 mg/dl). Exchange transfusion sessions were continued for another two months period, at a schedule of one session per week and bilirubin levels continued to drop steadily. They returned to patient’s baseline levels 73 days after the first exchange transfusion. The patient’s course is illustrated in Figure 1. The actual values of laboratory results and dates of procedures are included in Table 1 in the Appendix.
3. Discussion
3.1. Liver Complications of SCD
Hepatobiliary complications of SCD include viral hepatitis, hepatic crisis, cirrhosis, cholelithiasis/cholecystitis, intrahepatic cholestasis [2]. The clinical features at presentation of the complications range from benign hyperbilirubinemia to fulminant hepatitis [3]. Sickle cell intrahepatic cholestasis is an uncommon complication with a possible fatal outcome that may be observed in patients with SCD [4]. SCIC may have a high death rate; as negative prognostic factors are considered older age, homozygosity for HbS sickle cell anemia and an underlying hepatic disease [1]. SCIC may appear both in patients homozygous for HbS, as well as in those who are heterozygous for HbS and β-thalassemia [3,5]. The incidence of SCIC is greater in the pediatric than the adult population. In children, the clinical course is mild and often self-limiting, although the mortality rate may sometimes reach up to 30% [3].
The etiology and pathophysiology of SCIC is poorly understood, but it is believed that extensive deformed red blood cells adhesion to the hepatic vascular endothelium results in slugging and congestion of vascular beds leading to tissue ischemia, infarction and generalized liver dysfunction.
The diagnosis of SCIC is one of exclusion and is established be the characteristic clinical and laboratory
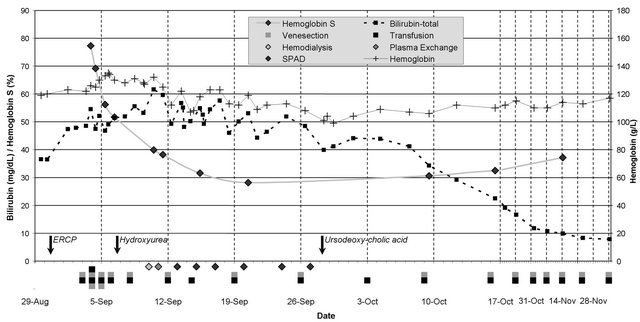
Figure 1. The patient’s course. Values of total bilirubin and HbS fraction are shown on left axis, the hemoglobin concentration on right axis. Arrows show the date of intervention (ERCP) and start dates of medications (hydroxyurea, ursodeoxycholic acid). Grey and black boxes indicate the units of blood removed by venesection and transfused respectively. Based on hemoglobin, patient underwent venesection or transfusion alone. Diamonds indicate the dates of the various dialysis procedures.
findings. The clinician should first exclude: viral hepatictis, hepatic sequestration and cholecystitis/cholelithiasis which are entities which may share clinical features. Positive serology helps to diagnose viral hepatitis. Hepatic sequestration shows tender hepatomegaly, sudden decrease in Hb concentration, reticulocytosis, mild to moderate elevation of aminotransferases, but not extreme hyperbilirubinemia. In SCIC, liver biopsy and autopsy show extreme intrahepatocellular and intracanalicular cholestasis with dilated intrahepatic canaliculus [2]. Sinusoidal engorgement, sickle cell thrombi, Kupffer cell hypertrophy, scattered bile stained micro-infarcts may also be found [2,6-10]. Since percutaneous liver biopsy seems to carry quite a lot of risks in sickle cell hepatopathy, it should be contraindicated in the diagnostic approach of SCIC [11].
Clinical features of SCIC may include: upper quadrant pain, progressive hepatomegaly, extreme total hyperbilirubinemia levels may reach up to 3420 μmol/L (200 mg/dL)—and acholic or light stools [2-3,6]. In most cases, mild to moderate elevation of aminotransferases is observed (ALT and AST <3 times of normal levels). In contrast, both total and direct (conjugated) bilirubin are markedly increased [3]. In more severe cases, renal dysfunction, encephalopathy and coagulopathy may be observed [4].
Ahn and colleagues [12] classified the patients with SCIC into two groups according to the severity of the clinical presentation. In Group I, there were patients with a mean maximum total bilirubin level of 472 μmol/L (27.6 mg/dl) and no other signs of hepatic dysfunction. Group II patients had a mean bilirubin level of 1313.3 μmol/L (76.8 mg/dl) and severe hepatic failure (coagulopathy and/or encephalopathy). Mortality rate was 4% and 64% respectively.
Our patient had the mild clinical and laboratory findings of the Group I except for the much higher bilirubin levels; even then, he had no other signs of functional liver failure, such as encephalopathy, due to failing detoxifying function, or coagulopathy, due to poor synthetic function. In the literature there are case reports mainly of children with benign extreme hyperbilirubinemia, which was self-limited [13]. There have also been reported cases of patients with SCIC, presenting with multi-organ failure, which responded to exchange transfusions [5,8].
Treatment patterns of SCIC are not well established, most probably due to the rarity of the disease. After a systematic review of the published literature, it has been found that the treatment ranges from supportive care to exchange transfusions [1]. Exchange transfusion attempts to lower the HbS fraction, with a target of <20% - 30%, trying to minimize intrahepatic sickling.
3.2. The Effects of Hydroxyurea
The use of hydroxyurea has been associated with reduced incidence of painful crises and hospitalization, increased HbF production through yet undetermined mechanisms, decreased neutrophil and reticulocyte counts and reduced hemolysis through improved erythrocyte hydration. It also releases nitric oxide with potential vasodilation and improved vascular permeability [14]. This way, HU prevents vaso-occlusive crises and seems to improve survival in SCD patients. In vitro studies suggest that HU may be beneficial in the acute vaso-occlusion as well, by means of reducing neutrophil adhesion to the endothelium [15]. These effects of HU seem to revert after discontinuation of the drug [16]. The fact that our patient had stopped taking HU may have been a triggering factor for the crisis, through waning of the above mentioned beneficial effects.
3.3. Increased Bilirubin: When to Worry? What to Do?
After about a week on exchange transfusions, our main concern was whether we should proceed to a bilirubinlowering procedure, although no other signs of liver failure were present, besides jaundice. It is known that bilirubin and other toxins that accumulate in hepatic failure have toxic effects in brain cells and hepatocytes, leading to encephalopathy and coagulopathy. To the best of our knowledge, there is not a consensus on the threshold value of bilirubin, above which cellular toxicity is more probable to occur. Moreover, high levels of bilirubin predict a poor outcome in a number of prognostic models used for assessing the severity of acute (King’s College criteria [17]) or chronic (Child-Pugh score [18], MELD formula [19]) liver disease. In order to prevent cellular toxicity, when total bilirubin levels reached 1053.7 μmol/L (61.62 mg/dl), conjugated 639.9 μmol/L (37.42 mg/dl), we decided to remove bilirubin by means of an extracorporeal liver support system, such as those used in acute and acute-on-chronic liver failure.
Albumin dialysis, that is, dialyzing blood against an albumin-containing solution across a high-flux membrane, is the most commonly used system for the effecttive elimination of albumin-bound toxins. The molecular adsorbent recirculating system (MARS) is a commercially available system using this principle. However, it was not available locally, it had technical difficulties and high cost. Therefore, the use of SPAD was decided after nephrologist’s suggestion. Single-pass albumin dialysis in principal works like MARS, but can be performed with any standard continuous venovenous hemodialyzer. SPAD might be therefore a cheaper and more convenient alternative to MARS, yet equally effective in retrospecttive evaluation [20,21].
4. Conclusions
The case we report underlines the effectiveness of exchange transfusions in the treatment of SCIC in combination to bilirubin removal with SPAD technique [22] which to the best of our knowledge has not been reported in earlier published literature. Early onset of exchange transfusion should be introduced and the clinician should bear in mind that SCIC may be fatal if not treated. The duration of treatment may be long term. In the cases of SCIC where the patient’s age was >30 years old, treatment with exchange transfusion had even reached up to a month [4,8].
In general, exchange transfusion seems to be the cornerstone of SCIC’s treatment, with simultaneous use of supportive care (RBCs, FFP) aiming at the correction of coagulopathy.
In conclusion, SCIC is a rare complication of SCD that may be fatal if not diagnosed and treated early. Despite the generally favorable outcomes, unsuccessful results may be observed. Our case reinforces the need for early onset of exchange transfusion and underlines the need for collaboration between different medical specialists (hematologists, gastroenterologists, nephrologists, radiologists), in order to achieve the best outcome for the patient. Finally, it is worth mentioning that the successful response appeared after the combined use of exchange transfusion and SPAD, which is being reported for the first time in the published literature.
REFERENCES
- D. M. Brunetta, A. C. Silva-Pinto, M. do Carmo Favarin de Macedo, S. C. Bassi, J. V. Piccolo Feliciano, F. B. Ribeiro, et al., “Intrahepatic Cholestasis in Sickle Cell Disease: A Case Report,” Anemia, Vol. 2011, 2011, Article ID: 975731. doi:10.1155/2011/975731
- T. W. Sheehy, “Sickle Cell Hepatopathy,” Southern Medical Journal, Vol. 70, No. 5, 1977, pp. 533-538. doi:10.1097/00007611-197705000-00008
- S. Banerjee, C. Owen and S. Chopra, “Sickle Cell Hepatopathy,” Hepatology, Vol. 33, No. 5, 2001, pp. 1021-1028. doi:10.1053/jhep.2001.24114
- D. B. Costa, R. A. Miksad, M. S. Buff, Y. Wang and B. J. Dezube, “Case of Fatal Sickle Cell Intrahepatic Cholestasis Despite Use of Exchange Transfusion in an AfricanAmerican Patient,” Journal of the National Medical Association, Vol. 98, No. 7, 2006, pp. 1183-1187.
- A. Betrosian, M. Balla, G. Kafiri, C. Palamarou and N. Sevastos, “Reversal of Liver Failure in Sickle Cell VasoOcclusive Crisis,” American Journal of Medical Sciences, Vol. 311, No. 6, 1996, pp. 292-295. doi:10.1097/00000441-199606000-00012
- S. H. Shao and E. P. Orringer, “Sickle Cell Intrahepatic Cholestasis: Approach to a Difficult Problem,” American Journal of Gastroenterology, Vol. 90, No. 11, 1995, pp. 2048-2050.
- S. C. Gilli, I. F. Boin, L. Sergio Leonardi, A. C. Luzo, F. F. Costa and S. T. Saad, “Liver Transplantation in a Patient with S(Beta)O-Thalassemia,” Transplantation, Vol. 74, No. 6, 2002, pp. 896-898. doi:10.1097/00007890-200209270-00030
- I. Khurshid, L. Anderson, G. H. Downie and G. S. Pape, “Sickle Cell Disease, Extreme Hyperbilirubinemia, and Pericardial Tamponade: Case Report and Review of the Literature,” Critical Care Medicine, Vol. 30, No. 10, 2002, pp. 2363-2367. doi:10.1097/00003246-200210000-00029
- F. M. Klion, M. J. Weiner and F. Schaffner, “Cholestasis in Sickle Cell Anemia,” The American Journal of Medicine, Vol. 37, No. 5, 1964, pp. 829-832. doi:10.1016/0002-9343(64)90031-2
- D. M. Owen, J. E. Aldridge and R. B. Thompson, “An Unusual Hepatic Sequela of Sickle Cell Anemia: A Report of Five Cases,” American Journal of Medical Sciences, Vol. 249, No. 2, 1965, pp. 175-185. doi:10.1097/00000441-196502000-00006
- N. Zakaria, A. Knisely, B. Portmann, G. Mieli-Vergani, J. Wendon, R. Arya, et al., “Acute Sickle Cell Hepatopathy Represents a Potential Contraindication for Percutaneous Liver Biopsy,” Blood, Vol. 101, No. 1, 2003, pp. 101-103. doi:10.1182/blood-2002-06-1823
- H. Ahn, C. S. Li and W. Wang, “Sickle Cell Hepatopathy: Clinical Presentation, Treatment, and Outcome in Pediatric and Adult Patients,” Pediatric Blood & Cancer, Vol. 45, No. 2, 2005, pp. 184-190. doi:10.1002/pbc.20317
- G. R. Buchanan and B. E. Glader, “Benign Course of Extreme Hyperbilirubinemia in Sickle Cell Anemia: Analysis of Six Cases,” Journal of Pediatrics, Vol. 91, No. 1, 1977, pp. 21-24. doi:10.1016/S0022-3476(77)80436-8
- R. E. Ware, “How I Use Hydroxyurea to Treat Young Patients with Sickle Cell Anemia,” Blood, Vol. 115, No. 26, 2010, pp. 5300-5311. doi:10.1182/blood-2009-04-146852
- C. B. Almeida, C. Scheiermann, J. E. Jang, C. Prophete, F. F. Costa, N. Conran, et al., “Hydroxyurea and a cGMPAmplifying Agent Have Immediate Benefits on Acute Vaso-Occlusive Events in Sickle Cell Disease Mice,” Blood, Vol. 120, No. 14, 2012, pp. 2879-2888. doi:10.1182/blood-2012-02-409524
- M. H. Steinberg, F. Barton, O. Castro, C. H. Pegelow, S. K. Ballas, A. Kutlar, et al., “Effect of Hydroxyurea on Mortality and Morbidity in Adult Sickle Cell Anemia: Risks and Benefits up to 9 Years of Treatment,” The Journal of the American Medical Association, Vol. 289, No. 13, 2003, pp. 1645-1651. doi:10.1001/jama.289.13.1645
- J. Polson and W. M. Lee, “Aasld Position Paper: The Management of Acute Liver Failure,” Hepatology, Vol. 41, No. 5, 2005, pp. 1179-1197. doi:10.1002/hep.20703
- C. G. Child and J. G. Turcotte, “Surgery and Portal Hypretension,” In: C. G. Child, Ed., The Liver and Portal Hypertension, Saunders, Philadelphia, 1964, pp. 50-64.
- M. Malinchoc, P. S. Kamath, F. D. Gordon, C. J. Peine, J. Rank and P. C. ter Borg, “A Model to Predict Poor Survival in Patients Undergoing Transjugular Intrahepatic Portosystemic Shunts,” Hepatology, Vol. 31, No. 4, 2000, pp. 864-871. doi:10.1053/he.2000.5852
- U. Boonsrirat, K. Tiranathanagul, N. Srisawat, P. Susantitaphong, P. Komolmit, K. Praditpornsilpa, et al., “Effective Bilirubin Reduction by Single-Pass Albumin Dialysis in Liver Failure,” Artificial Organs, Vol. 33, No. 8, 2009, pp. 648-653. doi:10.1111/j.1525-1594.2009.00758.x
- A. Kortgen, F. Rauchfuss, M. Gotz, U. Settmacher, M. Bauer and C. Sponholz, “Albumin Dialysis in Liver Failure: Comparison of Molecular Adsorbent Recirculating System and Single Pass Albumin Dialysis—A Retrospective Analysis,” Therapeutic Apheresis and Dialysis, Vol. 13, No. 5, 2009, pp. 419-425. doi:10.1111/j.1744-9987.2009.00760.x
- M. Wisniewski, M. Barwina, M. Zajac and J. Sein Anand, “[SPAD—New Possibilities?],” Przeglad Lekarski, Vol. 68, No. 8, 2011, pp. 483-485.
Abbreviations
ALP: Alkaline Phosphatase;
ALT: ALanine aminoTransferase;
AMA: Anti-Mitochondrial Antibodies;
ANA: Anti-Nuclear Antibodies;
anti-LKM: anti-Liver/Kidney Microsomal antibodies;
aPTT: activated Partial Thromboplastin Time;
AST: ASpartate aminoTransferase;
ERCP: Endoscopic Retrograde Cholangio-Pancreatography;
FFP: Fresh Frozen Plasma;
g/L: grams per Liter;
Hb: Hemoglobin Concentration;
HbS: Hemoglobin S;
Hct: Hematocrit;
HU: Hydroxyurea;
IgG: Immunoglobulin G;
INR: International Normalized Ratio;
MARS: Molecular Adsorbent Recirculating System;
MELD: Model for End-Stage Liver Disease;
mg/dL: milligrams per deciliter;
MRCP: Magnetic Resonance Cholangio Pancreatography;
pRBCs: Packed Red Blood Cells;
SCD: Sickle-Cell Disease;
SCIC: Sickle-Cell Intrahepatic Cholestasis;
SPAD: Single-Pass Albumin Dialysis;
U/L: Units per Liter;
WBC: White Blood Cells;
γ-GT: gamma-GlutamylTransferase;
μmol/L: micromolars per Liter.
Appendix
Table 1. Patient’s course in numbers.
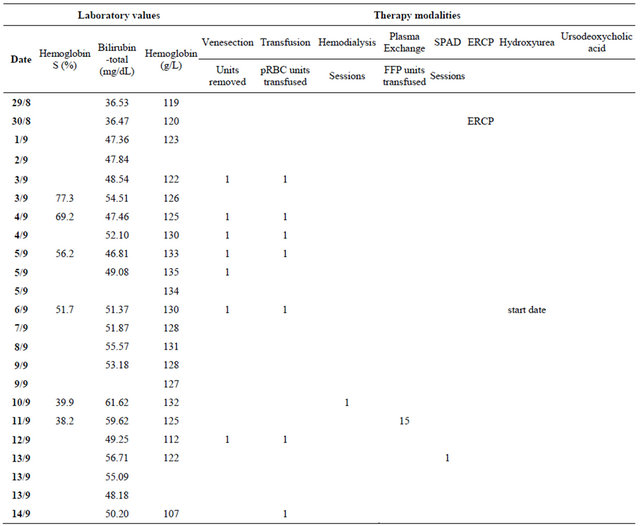
Continued
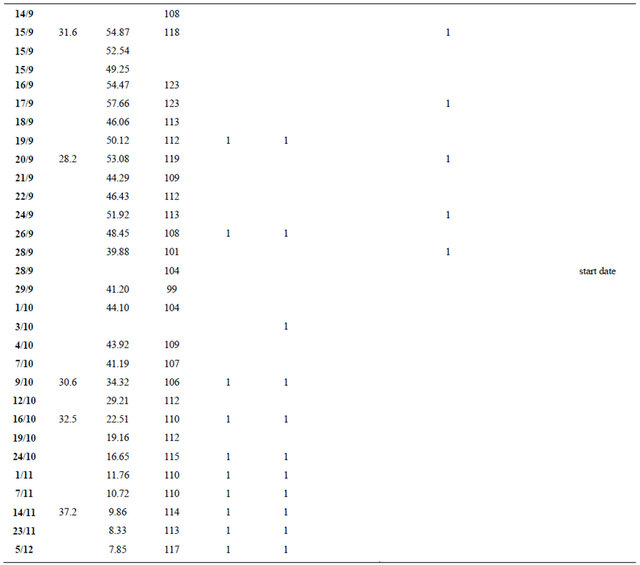
Abbreviations: pRBC: packed red blood cells; FFP: fresh frozen plasma; SPAD: single-pass albumin dialysis; ERCP: endoscopic retrograde cholangiopancreatography.
NOTES
*Corresponding author.