Open Journal of Pathology
Vol.3 No.2(2013), Article ID:30269,3 pages DOI:10.4236/ojpathology.2013.32017
A Case Report of a Sarcomatoid Carcinoma Arising in the Renal Pelvis with Exuberant Osteosarcomatous Element
![]()
Department of Pathology, College of Medicine, Hanyang University, Seoul, South Korea.
Email: *sspaik@hanyang.ac.kr
Copyright © 2013 Hye In Ahn et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 23rd, 2013; revised February 23rd, 2013; accepted March 20th, 2013
Keywords: Sarcomatoid Carcinoma; Osteosarcoma; Pelvis; Kidney
ABSTRACT
Sarcomatoid carcinoma is a rare malignant tumor that has both malignant epithelial and mesenchymal components. We describe a sarcomatoid carcinoma arising in the right renal pelvis of a 68-year-old man. The dominant component of the tumor was osteosarcomatous, but there were also focal carcinomatous areas. The sarcomatous tumor cells produced abundant osteoid matrix surrounded by osteoblastic cells. The carcinomatous tumor cells consisted of papillary urothelial carcinoma. Immunohistochemical assay showed that the sarcomatous tumor cells were positive for vimentin and negative for cytokeratin. The papillary urothelial carcinoma was positive for cytokeratin and negative for vimentin. After surgery, the patient underwent adjuvant chemotherapy. Four months later, he presented with recurrence in the right subphrenic area and metastasis in the right middle lobe of the lung.
1. Introduction
Sarcomatoid carcinoma is a rare high-grade malignant neoplasm that shows morphologic and immunohistochemical evidence of both epithelial and mesenchymal differentiation [1]. This tumor can occur in various organs including the bladder, breast, larynx, esophagus, kidney and female genital tract [2,3]. In the urinary tract, sarcomatoid carcinoma occurs predominantly in the urinary bladder [1]. Sarcomatoid carcinoma of the kidney is usually a variant of renal cell carcinoma [3]. The renal pelvis is an extremely rare site of origin for sarcomatoid carcinoma. To the best of our knowledge, fewer than 20 cases of sarcomatoid carcinoma of the renal pelvis have been reported [2]. Here, we report an additional case of renal pelvic sarcomatoid carcinoma showing exuberant osteosarcomatous differentiation.
2. Case Report
A 68-year-old male non-smoker presented with a monthlong history of right flank pain and hematuria. He had a previous history of kidney stone disease. He was given extracorporeal shock wave lithotripsy 10 times. Urine cytology revealed many red blood cells and neutrophils, but no malignant or atypical cells. Blood biochemistry showed low hemoglobin levels (9.0 g/dL). Physical examination revealed no palpable mass in the abdomen. Abdominal computed tomography revealed a large heterogeneous solid mass in the upper pole of the right kidney (Figure 1). There were several enlarged para-aortic lymph nodes. The possibility of renal pelvic malignancy was suggested and a radical nephroureterectomy was performed.
The resected kidney had a bulging outer contour in the upper pole. The cut surface revealed a large, soft, necrotic tumor that filled and dilated the pelvicalyceal system (Figure 2). The renal cortical tissue was compressed with uniform thinning with fibrotic change. Microscopically, the tumor showed exuberant sarcomatous differenttiation: 95% of the tumor consisted of an osteosarcomatous component (Figure 3), and less than 5% was a focal residual papillary urothelial carcinomatous component (Figure 4). In the sarcomatous areas, there were anaplastic osteoblastic tumor cells with scattered osteoclast-like giant cells. The sarcomatous tumor cells displayed edosteosarcomatous heterologous differentiation with abundant production of osteoid matrix surrounded by osteoblastic cells. The carcinomatous tumor was composed of papil-
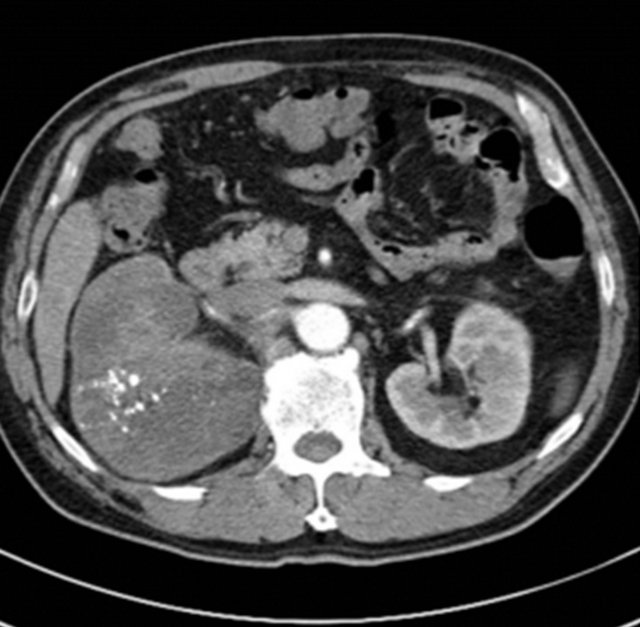
Figure 1. Computed tomography from the abdomen showing a large heterogeneous tumor with calcification in the right kidney.
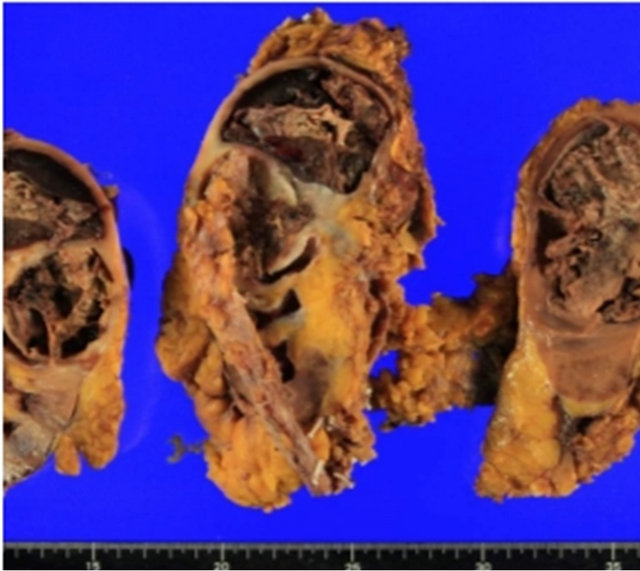
Figure 2. The resected kidney showing a dark brown tumor with hemorrhage and necrosis in the upper pole.
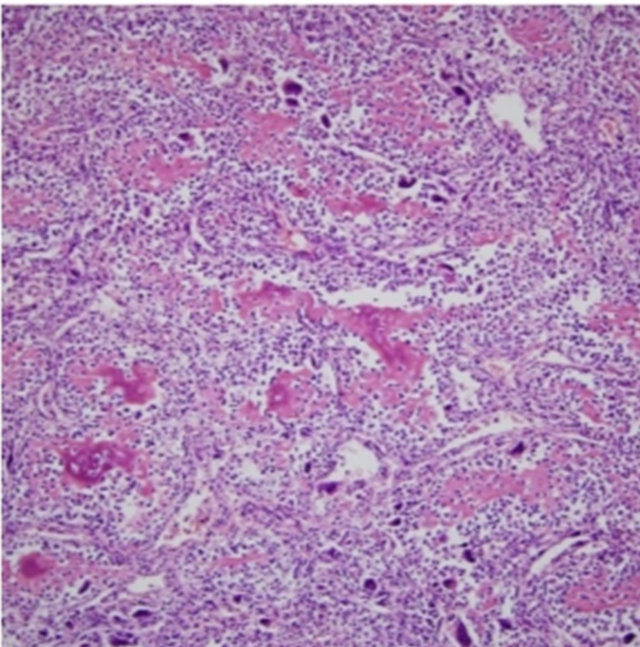
Figure 3. Section showing exuberant osteosarcomatous differentiation with abundant osteoid production (H&E 100×).
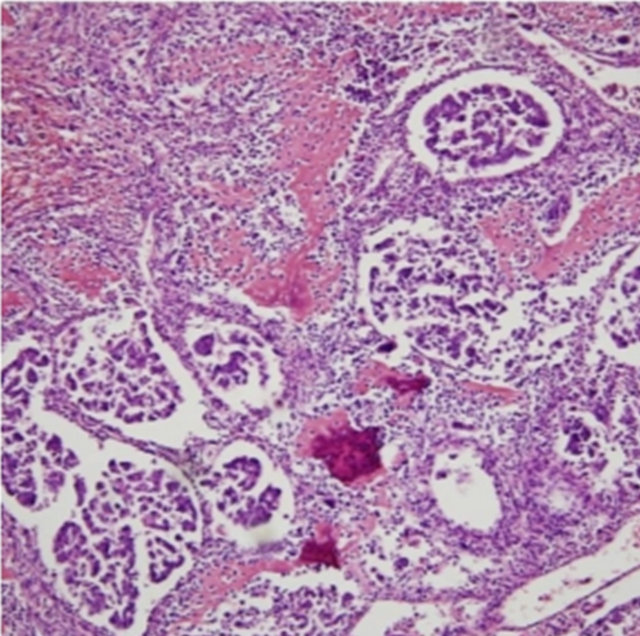
Figure 4. Section showing focal papillary urothelial carcinoma (H&E 100×).
lary urothelial carcinoma cells. Areas of hemorrhage and necrosis were present, and lymphatic tumor invasion had occurred. Immunohistochemically, the sarcomatous tumor cells were positive for vimentin and negative for cytokeratin. The papillary urothelial carcinoma cells were positive for cytokeratin and negative for vimentin. The postoperative period was uneventful and the patient was discharged 10 days after operation. He underwent adjuvant chemotherapy. Four months later, he had recurrence in the right subphrenic area and metastasis in the right middle lobe of the lung.
3. Discussion
In this report we have described an exceedingly rare case of renal pelvic sarcomatoid carcinoma consisting mainly of malignant mesenchymal tissue with osteosarcomatous differentiation and small areas of malignant epithelial tissue forming papillary urothelial carcinoma. The first case of sarcomatoid carcinoma in the renal pelvis was reported by Fauci et al. [4]. To our knowledge, fewer than 20 cases of sarcomatoid carcinoma of the renal pelvis have been reported [2,3]. In most of these cases, the sarcomatous element consisted of poorly formed fascicles of undifferentiated spindle cells with or without osteoclasttype giant cells. However our case showed exuberant osteosarcomatous differentiation in most sarcomatous element. Osteosarcomatous differentiation is extremely rare histopathologic finding in renal pelvic sarcomatoid carcinoma [2].
Sarcomatoid carcinoma is a high-grade malignant neoplasm with a biphasic microscopic appearance caused by the presence of both epithelial and mesenchymal components [3]. The terms “carcinosarcoma” and “sarcomatoid carcinoma” have both been used in reports. In 2004, the World Health Organization Classification of Tumors simplified the approach to these tumors [5]. The term “sarcomatoid carcinoma” should be used for all biphasic malignant neoplasms that exhibit morphological and/or immunohistochemical evidence of epithelial and mesenchymal differentiation. Immunohistochemically, epithelial elements react with cytokeratins and sarcomatous elements react with vimentin or other mesenchymal specific markers. In our case, most areas of the tumor consisted of an osteosarcomatous mesenchymal element with limited focal areas of a papillary urothelial carcinomatous element. Immunohistochemical staining results revealed vimentin positivity in sarcomatous areas and cytokeratin positivity in carcinomatous areas.
The exact pathogenesis of sarcomatoid carcinoma is not known. Two opposing theories have been proposed. The monoclonal theory states that the carcinomatous and sarcomatous tumor cells are both derived from a single pluripotent stem cell that undergoes divergent epithelial and mesenchymal differentiation [6]. The multiclonal theory states that the sarcomatoid carcinoma is a collision tumor composed of the derivatives of two or more stem cells of separate epithelial and mesenchymal origin [7]. In their study of loss of heterozygosity and X-chromosome inactivation in 30 sarcomatoid urothelial carcinomas, Sung et al. [8] supported the idea of a monoclonal origin from a primitive pluripotent stem cell [8]. They found identical non-random X-chromosome inactivation and significant overlap of loss of heterozygosity in the carcinomatous and sarcomatous components.
The most common presenting symptoms of sarcomatoid carcinoma are hematuria, dysuria, nocturia, acute urinary retention and lower abdominal pain [2]. Our patient had right flank pain and hematuria, and blood biochemistry revealed decreased hemoglobin levels. The main differential diagnosis includes true sarcoma and pseudosarcomatous mesenchymal proliferations. Mixed malignant epithelial and mesenchymal components are diagnostic, and immunohistochemical staining for epithelial and mesenchymal markers is helpful. In our case, there were undifferentiated sarcomatoid cells with osteosarcomatous differentiation and abundant osteoid production, and focal areas of papillary urothelial carcinoma. Immunohistochemical staining clearly revealed two different mesenchymal and epithelial tumor components. This tumor has a worse prognosis than the more common urinary tract carcinomas, and recurrence and metastasis are frequent [9]. Our patient suffered recurrence at the operation site and lung metastasis four months after surgery.
In summary, we described an extremely rare case of sarcomatoid carcinoma arising in the renal pelvis with predominantly osteosarcomatous differentiation and focal papillary urothelial carcinoma. Although this tumor is very rare, sarcomatoid carcinoma should be included in the differential diagnosis of the renal pelvis tumors.
REFERENCES
- L. Cheng, S. Zhang, R. Alexander, G. T. MacLennan, K. B. Hodges, B. T. Harrison, A. Lopez-Beltran and R. Montironi, “Sarcomatoid Carcinoma of the Urinary Bladder: The Final Common Pathway of Urothelial Carcinoma Dedifferentiation,” American Journal of Surgical Pathology, Vol. 35, No. 5, 2011, pp. 34-46. doi:10.1097/PAS.0b013e3182159dec
- S. Gill-Samra, A. Brooks and C. H. P’ng, “Sarcomatoid Carcinoma Involving the Renal Pelvis And Ureter with Heterologous Osteosarcomatous Differentiation,” Pathology, Vol. 44, No. 4, 2012, pp. 367-369. doi:10.1097/PAT.0b013e328353be4a
- M. F. Acikalin, S. Kabukcuoglu and C. Can, “Sarcomatoid Carcinoma of the Renal Pelvis with Giant Cell TumorLike Features: Case Report with Immunohistochemical Findings,” International Journal of Urology, Vol. 12, No. 2, 2005, pp. 199-203. doi:10.1111/j.1442-2042.2005.01002.x
- P. A. Fauci Jr., H. G. Therhag and J. E. Davis, “Carcinosarcoma of the Renal Pelvis,” Journal of Urology, Vol. 85, 1961, pp. 897-902.
- J. N. Eble, G. Sauter, J. I. Epstein and I. A. Sesterhenn, “World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs,” IARC Press, Lyon, 2004, pp. 102-103.
- C. A. Bloxham, M. K. Bennett and M. C. Robinson, “Bladder Carcinosarcomas: Three Cases with Diverse Histogenesis,” Histopathology, Vol. 16, No. 1, 1990, pp. 63-67. doi:10.1111/j.1365-2559.1990.tb01062.x
- M. R. Wick and P. E. Swanson, “Carcinosarcomas: Current Perspectives and a Historical Review of Nosological Concepts,” Seminars in Diagnostic Pathology, Vol. 10, No. 2, 1993, pp. 118-127.
- M. T. Sung, M. Wang, G. T. MacLennan, J. N. Eble, P. H. Tan, A. Lopez-Beltran, R. Montironi, J. J. Harris, M. Kuhar and L. Cheng, “Histogenesis of Sarcomatoid Urothelial Carcinoma of the Urinary Bladder: Evidence for a Common Clonal Origin with Divergent Differentiation,” Journal of Pathology, Vol. 211, No. 4, 2007, pp. 420-430. doi:10.1002/path.2129
- H. Ikegawa, H. Iwasaki, Y. Ohjimi, T. Takeuchi, A. Ariyoshi and M. Kikuchi, “Sarcomatoid Carcinoma of the Urinary Bladder: A Clinicopathologic and Immunohistochemical Analysis of 14 Patients,” Human Pathology, Vol. 31, No. 3, 2000, pp. 332-340. doi:10.1016/S0046-8177(00)80247-6
NOTES
*Corresponding author.