Open Journal of Obstetrics and Gynecology
Vol. 2 No. 4 (2012) , Article ID: 24441 , 6 pages DOI:10.4236/ojog.2012.24076
Antenatal and postnatal management of congenital cystic adenomatoid lung malformation diagnosed by ultrasound and Magnetic Resonance Imaging (MRI)
![]()
1Department of Obstetrics, Gynaecology and Reproductive Medicine, Hannover Medical School, Hannover, Germany
2Department of Obstetrics and Gynaecology, Justus-Liebig-University of Giessen, Giessen, Germany
3Institute of Diagnostic and Interventional Neuroradiology, Hannover Medical School, Hannover, Germany
4Department of Biochemistry, University of Nairobi, Nairobi, Kenya
5Department of Pediatric Radiology, University of Giessen, Giessen, Germany
6Department of Pediatric Pulmonology, Allergology and Neonatology, Hannover Medical School, Hannover, Germany
Email: *gruessner.susanne@mh-hannover.de, *susanne.gruessner@gmx.de
Received 4 September 2012; revised 3 October 2012; accepted 10 October 2012
Keywords: Congenital Cystic Adenomatoid Lung Malformation; Antenatal Serial Ultrasound Studies; Antenatal MRI; Imaging Surveillance
ABSTRACT
Purpose: Antenatal diagnosis of congenital cystic adenomatoid lung malformation (CCAM) is vital for disease surveillance and postnatal care. Ultrasonography (US) has been the imaging gold standard for antenatal CCAM assessment. However, one of the limitations of US is the “vanishing phenomenon” caused by isoechogenicity of CCAM tissue and adjacent normal lung parenchyma. Methods: Antenatal serial US were concurrently used with magnetic resonance imaging (MRI) to monitor macroand microcystic lesions. Results: In both pregnant women, antenatal US and MRI confirmed the presence, in the fetus, of cystic lesions and predicted disease regression/progression as well as the need for postnatal surgical intervention. Several advantages were detected by using both—serial US and MRI (over serial US alone)—including improved signal intensity, exact volume size measurements, precise CCAM location in particular for patients with adverse ultrasound conditions. Both neonates underwent surgical resection and had an uneventful post-operative course. Conclusions: Antenatal use of MRI as well as serial US improved information regarding tissue resolution and delineation of CCAM. The information from two imaging modalities was complementary. Our literature review confirmed the emerging role of prenatal MRI for postnatal monitoring and management of CCAM.
1. INTRODUCTION
Congenital cystic adenomatoid lung malformation (CCAM) is characterized by various cystic intrathoracic masses caused by proliferative terminal respiratory bronchioles and by a reduction in the number of normal alveoli. It usually affects a single pulmonary lobe; multi-lobular or bilateral involvement is rare [1]. CCAM was first reported in 1949 by Ch’in and Tang [2]. The estimated incidence of CCAM ranges from 1:10.000 to 1:35.000 live births [3-5], with male and female neonates equally affected [4]. The possible influences of genes, such as HOXB5, Fdf7, and PDGF-BB, have been described, but, as of now, are not thought to be of any diagnostic or prognostic value [5,6]. The histopathologic classification by Stocker et al. (1977) describes 3 forms of CCAM (type I-III). Type I is the most common form, detected in up to 65% of patients with CCAM: the single or multiple cysts are more than 20 mm in diameter and are lined by ciliated columnar or pseudostratified epithetlium, mucussecreting cells, and, perhaps prominently, cartilage. Type II is the second most common form, detected in up to 20% to 35% of patients with CCAM: the multiple cysts are less than 20 mm in diameter and are lined by cylindrical or cuboidal epithelium, with prominent smooth or striated muscle. Type III is the least common form, detected in up to 10% of patients with CCAM: the solid microcysts are rarely larger than 2 mm in diameter and are lined by cuboidal epithelium, often intricately folded [1]. CCAM ranges in severity from intrauterine regression to progressive nonimmune hydrops and/or mediastinal shifting. Treatment options include prenatal intrauterine laser therapy, postnatal thoracoscopic local excision, or thoracotomy with appropriate surgical resection. Without treatment, macrocystic lesions, in particular, tend to increase the risk of pulmonary infections, of mediastinal shifting with lung compression, and of malignnancy late in life [7-10].
The earliest reported diagnosis of CCAM with ultrasonography (US) was made in 1975 [11]. The sonographic appearance of CCAM was classified by Adzick et al., who distinguished between subgroups of macrocystic (≥5 mm in diameter) and microcystic (<5 mm in diameter) CCAM [12]. Serial antenatal US has been considered the imaging gold standard for antenatal monitoring and management. The disadvantages of US in particular regarding unfavorable ultrasound conditions (i.e. elevated body mass [BMI] indices), however, include imprecise measurement of exact volume size, reduction in signal intensity, and need for a high number of serial imaging studies [13-16].
In contrast, the advantages of magnetic resonance imaging (MRI) especially in cases of attenuate ultrasound signals include exact measurement of the lesion(s)-tolung volume ratio, superior tissue resolution, with determination of disease progression or regression and precise determination of CCAM location, and need for a low number of studies [17-19].
US and MRI are complementary. Therefore, we used both modalities, in combination, in our two patients; then, we compared our observations with the current literature. Ethical clearence was obtained from the relevant office and experiments conducted in accordance with the Helsinki declaration of 1982 regarding use of human subjects.
2. METHODS AND RESULTS
2.1. Case 1
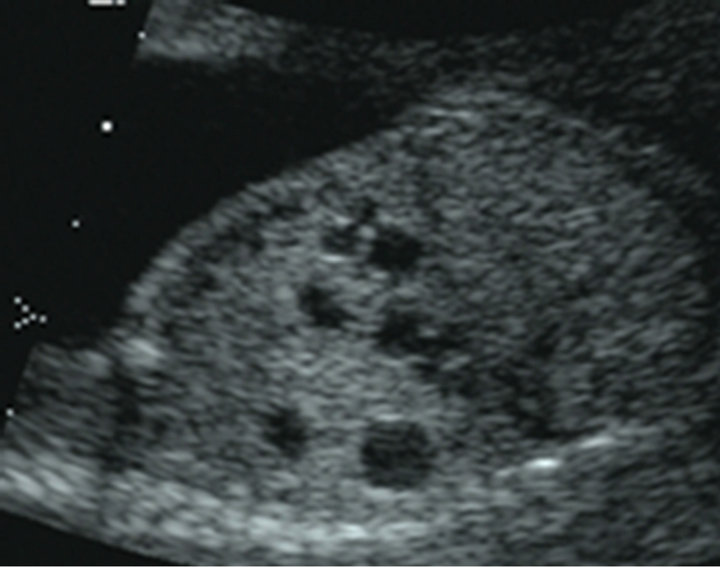
A 29-year-old woman (primigravida) was referred, at 21 weeks of gestation, to our tertiary center for a comprehensive US study to assess fetal chest masses. Earlier in her pregnancy, echocardiography excluded any associated anomalies; amniocentesis excluded chromosomal pathology (46 XX). We diagnosed CCAM in the right lung of the fetus with transabdominal sonography, using Acuson 128 XP/10 (Acuson, Mountain View, CA, USA) and Sonoline G 60 (Siemens Medical Systems, Inc., Issaquah, WA, USA) systems, both equipped with a probe of 3.5 to 5 MHz. Our initial US study revealed multiple macroand micro-cystic malformations, 5 to 12 mm in diameter, in the right lung (Figure 1(a)). Adverse ultrasound conditions caused by maternal increased BMI let to fast T2-weighted MRI (acquisition time, about 10 seconds, HASTE sequence) at 21 weeks of gestation; we used a Multiarray GyroScan 1.5-T MRI unit (Philips, Germany). Our MRI study not only confirmed the initial US diagnosis but also provided superior tissue resolution and revealed the exact location of the lesions (lower lobe of the right lung; Figures 1(b) and (c)).
Our US follow-up studies also confirmed the primary location (lower lobe of the right lung) and subsequent CCAM regression. During the course of gestation, our serial prenatal US studies excluded the presence of hydrops, of mediastinal shifting, of polyhydramnios, and of other anomalies. Spontaneous premature delivery occurred at 35 weeks of gestation (the neonate’s birth weight, 3160 gm; Apgar score, 9/10/10; pH value, 7.39). Initially, the neonate had no respiratory symptoms of concern. For postnatal monitoring, the neonate was transferred to the neonatal intensive care unit, given its capability for extracorporeal membrane oxygenation (ECMO) and intensive pediatric surgical care.
 (a)
(a)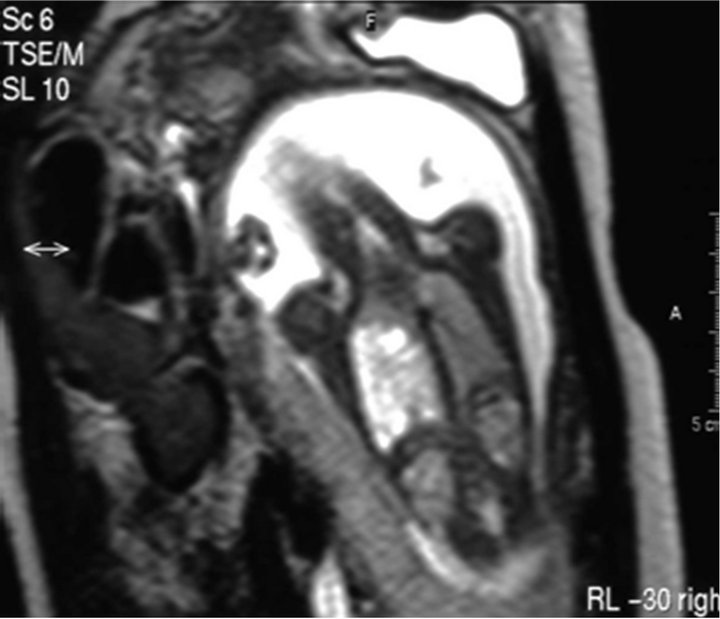 (b)
(b)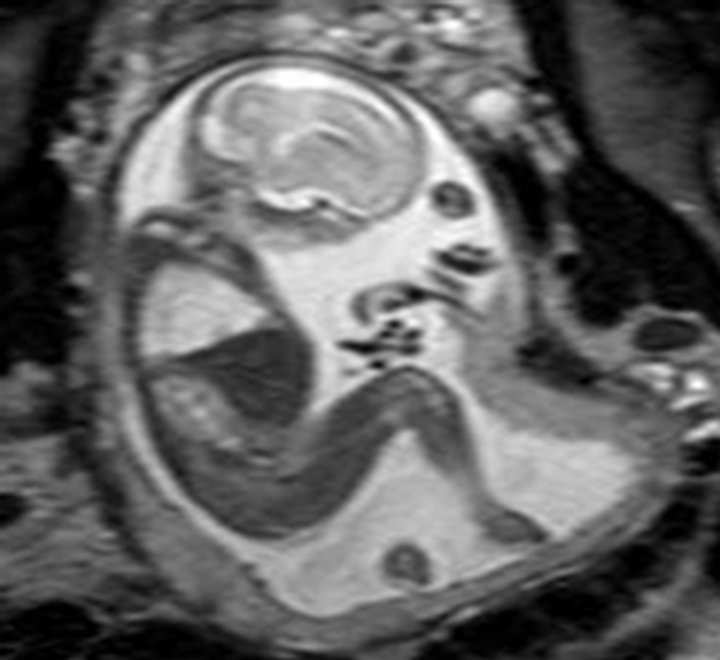 (c)
(c)
Figure 1. (a) Sonographic feature of the chest 21 weeks of gestation; (b)-(c) MR images of the macroand microcystic chest masses.
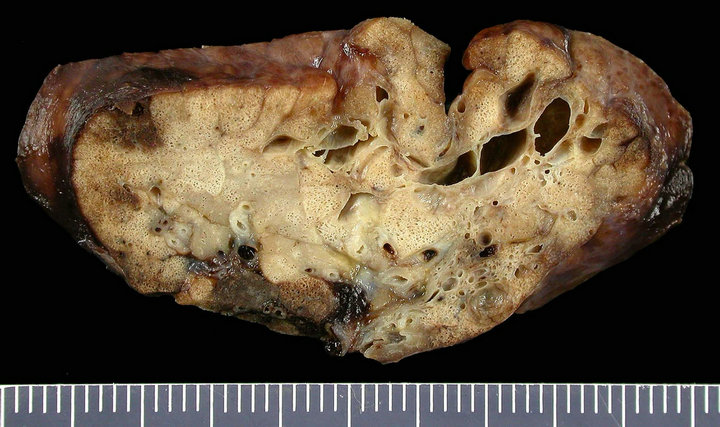
The postnatal chest computed tomography (CT) scan confirmed the diagnosis of macroand micro-cystic lung lesions of the lower lobe of the right lung. The initially asymptomatic neonate developed clinical signs of CCAM during the first week of life, including tachypnoe and dyspnea. To prevent the risk of recurrent infection and late malignant transformation, we performed an elective posterolateral thoracotomy, with resection of the lower lobe of the right lung, when the neonate was 11 days old (Figure 2(a)).
Histopathologic examination confirmed the diagnosis of CCAM type II (Figure 2(b)). The neonate’s postoperative course was uneventful.
2.2. Case 2
A 24-year-old woman (gravida II, para I) was referred, at 30 weeks of gestation, to our tertiary center for comprehensive US screening. Earlier in her pregnancy, elective amniocentesis showed no chromosomal pathology (46 XX). Our initial US study showed a single macrocystic lesion, 20 mm in diameter, in the left lung of the fetus (Figure 3(a)).
After detection of CCAM, we performed an extensive sonographic examination, including fetal echocardiography, which excluded fetal cardiac failure and other associated anomalies. Inappropriate terms of ultrasound caused by reduced amniotic fluid volume let to fast T2-weighted MRI at 30 weeks of gestation; we used a
 (a)
(a)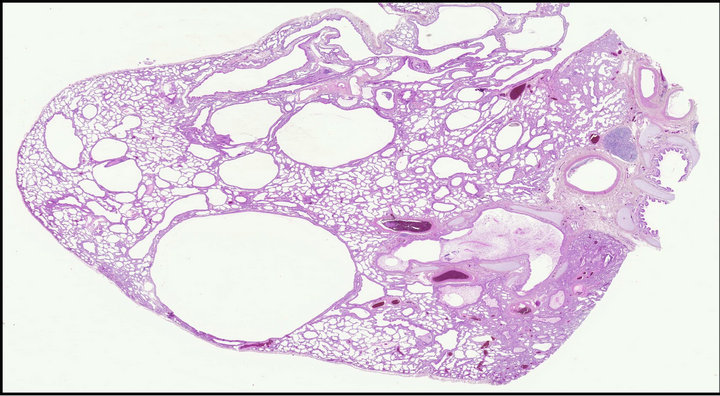 (b)
(b)
Figure 2. (a) Fixed gross pathologic specimen of the CCAM; (b) Histopathology of the CCAM.
Multiarray GyroScan 1.5-T MRI unit (Philips). Again, our MRI study not only confirmed the initial US diagnosis but also provided superior tissue resolution and revealed the exact location of the lesion (lower lobe of the left lung; Figure 3(b)); concurrent microcystic lesions in other parts of the lungs were not present. During the course of gestation, our serial prenatal US studies revealed a decrease in the size of the single cystic malformation; at term, it was 16 mm in diameter. Our follow-up US studies confirmed the location of the macrocystic lesion (lower lobe of the left lung) and excluded the presence of hydrops, of mediastinal shifting, and of polyhydramnios. At 40 weeks of gestation, the woman underwent a cesarean section (the neonate’s birth weight, 3510 gm; Apgar score, 8/8/10; pH value 7.31). A postnatal chest CT scan confirmed the diagnosis of a single macrocystic lesion in the lower lobe of the left lung.
The primary asymptomatic condition of the neonate changed within the first three days of life, when clinical signs of infection developed. Five days after delivery, before serious respiratory infection and/or dysfunction
 (a)
(a) (b)
(b)
Figure 3. (a) Sonographic feature of the chest 30 weeks of gestation; (b) MR image of the macrocystic lung lesion (CCAM).
could occur, the neonate underwent elective segmentectomy of the lower lobe of the left lung. Histopathologic examination confirmed the diagnosis of CCAM type I. The neonate’s postoperative course was uneventful.
3. DISCUSSION
CCAM is the most common (77%) of congenital cystic lung diseases, followed by pulmonary sequestration, bronchogenic cysts, and bronchial atresia [7-9,20]. The disease spectrum of CCAM is quite variable. Lesions with spontaneous regression and, ultimately, complete disappearance during the third trimester have been reported; so have rapidly growing lung masses with nonimmune hydrops and in utero demise [7,16,20]. Close perinatal monitoring is essential to optimize outcome [7-9,16,20-22]. Serial antenatal US studies have been the imaging gold standard to characterize the features of CCAM, including the size of lesions, their appearance (macrovs micro-cystic), and their location (lobe involvement) [13-16,23]. However, US studies, even when done serially, have disadvantages, including less than ideal CCAM resolution and imprecise initial CCAM location, particularly in obese pregnant women [13-16].
Other imaging modalities have hardly been used in the prenatal period. Antenatal CT studies are not indicated, because of the amount of irradiation.
In contrast, MRI, particularly in the third trimester, appears to be relatively safe and provides excellent tissue resolution [17,18]. Of note, MRI was used in one of our cases as early as 21 weeks into the pregnancy, i.e. at an early stage of gestation. MRI also distinguishes CCAM from other chest lesions, clearly identifies the exact lung location, and demonstrates the presence of compressed normal lung [10,16]. Thus, antenatal MRI enables clear identification of high-risk fetuses and may help optimize postnatal intensive care. In addition, quantification and assessment of pulmonary hypoplasia by antenatal MRI can offer insights about such options as termination of the pregnancy, elective delivery, and planned postnatal surgical procedures [16,23].
In our experience, especially in cases of unfavorable terms of US, information provided by the initial MRI study was superior to the initial US study with regard to tissue resolution and precise CCAM location. The advantage of serial US studies lies in the documentation of morphologic changes (e.g., size, appearance) over time. Therefore, we believe that in cases of attenuate ultrasound conditions the combination of an initial MRI study and serial US studies provides the best information regarding changes in CCAM appearance.
To date, despite the use of serial prenatal US studies to monitor the lesions of CCAM, no standard predictors for disease progression or regression have been pinpointed. It remains to be determined whether or not a second MRI study, during or close to the end of the third trimester, will help pinpoint any such predictors, which are essential to optimize outcome [17,18]. As mentioned above, the clinical symptoms of neonates with CCAM vary in severity. Neonates with severe and symptomatic cases, including in utero development of nonimmune hydrops or polyhydramnios require ventilatory support, and sometimes high-frequency ventilation or even ECMO shortly after birth because of hypoplasia of the lungs [24,25]. Initially asymptomatic neonates with isolated CCAM, in the absence of hydrops, may develop, shortly after birth, recurrent respiratory tract infections (10%) [15,23]. In addition, pleuropulmonary blastoma is a risk in childhood, and bronchoalveolar carcinoma is a risk in adolescence [20,21]. In contrast, nonhydropic fetuses with isolated CCAM have little or no hypoplasia of the lungs and, therefore, a much better prognosis [26-29]. Irrespective of the absence or presence of symptoms, close prenatal surveillance, carefully planned delivery, and rigorous postnatal evaluation at a facility with ECMO capability and pediatric surgical care all contribute to favorable outcome [21-25]. Aggressive in utero management, using both thoracoamniotic shunting (for macrocystic malformations) and intrauterine laser therapy or fetal surgery (for microcystic solid malformations), has been proposed in cases with a poor prognosis [7-10]. However, compared to noninterventional obstetric care, open fetal surgery has failed to improve survival rates [7,13]. Antenatal high-risk CCAM fetuses (with either mediastinal shifting or progressive non-immune hydrops) and symptomatic CCAM neonates (with respiratory insufficiency) require immediate postnatal emergency surgery [7-10, 25].
In contrast, treatment of asymptomatic lesions is controversial. Simple monitoring of asymptomatic CCAM has been suggested, but monitoring carries the risk of ongoing and recurrent pulmonary infections [26]. Although early surgical resection (within the first 4 weeks of life) is associated with an increased anesthetic risk, it maximizes compensatory lung growth at this early stage [9,26,28]. A consensus seems to be growing against the simple monitoring “wait and see” approach in cases of asymptomatic CCAM, because postnatal spontaneous resolution may not occur. Elective resection within the first 3 to 6 months of life has also been suggested, given the lifelong risk of recurrent lung infections and occult malignant lung cell proliferation [3,10,25-27]. The youngest described patient with malignant malformation due to CCAM was 13 months old [30]. Elective surgery is usually well tolerated, the risk of infection is lower, and compensatory lung growth with normal long-term respiratory function has been observed [13,20]. Despite current limitations, prognostic information is necessary to provide parental counseling throughout the pregnancy and appropriate perinatal management [7,8,27,31]. In hydropic CCAM fetuses, the presence of additional lifethreatening anomalies might lead to the decision to terminate the pregnancy. Prognosis is very favorable in fetuses without hydrops and with isolated, unilateral CCAM [13,14,20,25]. After extensive parental counseling, a noninvasive obstetric strategy should be adopted, including serial US studies and an initial antenatal MRI study. The value of a subsequent MRI study in the third trimester needs to be determined [17,18].
4. CONCLUSION
In summary, CCAM of the fetal lung remains a challenge for obstetricians, neonatologists, and pediatric surgeons. Improvements in antenatal monitoring are necessary to improve and better predict outcomes. The combination of prenatal MRI and serial US studies appears to optimize fetal surveillance and postnatal care.
REFERENCES
- Stocker, J.T., Madewell, J.E. and Drake, R.M. (1977) Congenital cystic adenomatoid malformation of the lung: Classification and morphologic spectrum. Human Pathology, 8, 155-171. doi:10.1016/S0046-8177(77)80078-6
- Ch’in, K.Y. and Tang, M.Y. (1949) Congenital cystic adenomatoid malformation of one lobe of a lung with general anasarca. Archives of Pathology, 48, 221-229.
- Calvert, J.K. and Lakhoo, K. (2007) Antenatally suspected congenital cystic adenomatoid malformation of the lung: Postnatal investigation and timing of surgery. Journal of Pediatric Surgery, 42, 411-414. doi:10.1016/j.jpedsurg.2006.10.015
- Gornall, A.S., Budd, J.L., Draper, E.S., et al. (2003) Congenital cystic adenomatoid malformation: Accuracy of prenatal diagnosis, prevalence and outcome in ageneral population. Prenatal Diagnosis, 23, 997-1002. doi:10.1002/pd.739
- Wilson, R.D., et al. (2006) Cystic adenomatoid malformation of the lung: Review of genetics, prenatal diagnosis, and in utero treatment. American Journal of Medical Genetics, A140, 151-155. doi:10.1002/ajmg.a.31031
- Volpe, M.V., Pham, L., Lessin, M., et al. (2003) Expression of Hoxb-5 during human lung development and in congenital lung malformations. Birth Defects Research. Part A, Clinical and Molecular Teratology, 67, 550-556. doi:10.1002/bdra.10086
- Azizkhan, R.G. and Crombleholme, T.M. (2008) Congenital cystic lung disease: Contemporary antenatal and postnatal management. Pediatric Surgery International, 24, 643-657. doi:10.1007/s00383-008-2139-3
- Davenport, M., Warne, S.A., Cacciaguerra, S., Patel, S., Greenough, A. and Nicolaides, K. (2004) Current outcome of antenatally diagnosed cystic lung disease. Journal of Pediatric Surgery, 39, 549-556. doi:10.1016/j.jpedsurg.2003.12.021
- Eber, E. (2007) Antenatal diagnosis of congenital thoracic malformations: Early surgery, late surgery, or no surgery? Seminars in Respiratory and Critical Care Medicine, 28, 355-366. doi:10.1055/s-2007-981656
- Mann, S., Wilson, R.D., Bebbington, M.W., Adzick, N.S. and Johnson, M.P. (2007) Antenatal diagnosis and management of congenital cystic adenomatoid malformation. Seminars in Fetal and Neonatal Medicine, 12, 477-481. doi:10.1016/j.siny.2007.06.009,
- Garrett, W.J., Kossoff, G. and Lawrence, R. (1975) Grey scale echocardiography in the diagnosis of hydrops due to fetal lung tumor. Journal of Clinical Ultrasound, 3, 45- 50. doi:10.1002/jcu.1870030109
- Adzick, N.S., Harrison, M.R., Glick, P.L., Golbus, M.S., Anderson, R.L., Mahoney, B.S., Callen, P.W., Hirsch, J.H., Luthy, D.A. and Filly, R.A. (1985) Fetal cystic adenomatoid malformation: Prenatal diagnosis and natural history. Journal of Pediatric Surgery, 20, 483-488. doi:10.1016/S0022-3468(85)80470-X,
- Cavoretto, P., Molina, F., Poggi, S., Davenport, M. and Nicolaides, K.H. (2008) Prenatal diagnosis and outcome of echogenic fetal lung lesions. Ultrasound in Obstetrics & Gynecology, 32, 769-783. doi:10.1002/uog.6218
- Chen, W.S., Yeh, G.P., Tsai, H.D. and Hsieh, C.T. (2009) Prenatal diagnosis of congenital cystic adenomatoid malformations: Evolution and outcome. Taiwanese Journal of Obstetrics and Gynecology, 48, 278-281. doi:10.1016/S1028-4559(09)60304-1
- Illanes, S., Hunter, A., Evans, M., Cusick, E. and Soothill, P. (2005) Prenatal diagnosis of echogenic lung: Evolution and outcome. Ultrasound in Obstetrics & Gynecology, 26, 145-149. doi:10.1002/uog.1921
- Kunisaki, S.M., Barnewolt, C.E., Estroff, J.A., Ward, V.L., Nemes, L.P., Fauza, D.O. and Jennings, R.W. (2007) Large fetal congenital cystic adenomatoid malformations: Growth trends and patient survival. Journal of Pediatric Surgery, 42, 404-410. doi:10.1016/j.jpedsurg.2006.10.014
- Epelman, M., Kreiger, P.A., Servaes, S., Victoria, T. and Hellinger, J.C. (2010) Current imaging of prenatally diagnosed congenital lesions. Seminars in Ultrasound, CT, and MRI, 31, 141-145.
- Liu, Y.-P., Chen, C.-P., Shih, S.-L., Chen, Y.-F., Yang, F.-S. and Chen, S.-C. (2010) Fetal cystic lung lesions: Evaluation with magnetic resonance imaging. Pediatr Pulmonol, 45, 592-600.
- Matsushita, M., Ishii, K., Tamura, M., Takahashi, Y., Kamura, T., Takakuwa, K. and Tanaka, K. (2008) Perinatal magnetic resonance fetal lung volumetry and fetal lung-to-liver signal intensity ratio for predicting short outcome in isolated congenital diaphragmatic hernia and cystic adenomatoid malformation of the lung. Journal of Obstetrics and Gynaecology Research, 34, 162-167. doi:10.1111/j.1447-0756.2008.00754.x
- Rocha, G., Fernandes, P.C., Proenca, E., Quintas, C., Martins, T., Azevedo, I. and Guimaraes, H. (2007) Congenital cystic adenomatoid malformation of the lung— The experience of five medical centres. Revista Portuguesa de Pneumologia, 13, 511-523.
- Calvert, J.K., Boyd, P.A., Chamberlain, P.C., Syed, S. and Lakhoo, K. (2006) Outcome of antenatally suspected congenital cystic adenomatoid malformation of the lung: 10 years’ experience 1991-2001. Archives of Disease in Chilhood, Fetal and Neonatal Edition, 91, F26-28. doi:10.1136/adc.2004.068866
- Chow, P.C., Lee, S.L., Tang, M.H., Chan, K.L., Lee, C.P., Lam, B.C. and Tsoi, N.S. (2007) Management and outcome of antenatally diagnosed congenital cystic adenomatoid malformation of the lung. Hong Kong Medical Journal, 13, 31-39.
- Stocker, J.T. (2001) The respiratory tract. In: Stocker, J.T. and Dehner, L.P., Eds., Pediatric Pathology, 2nd Edition, Lippincott/Williams & Wilkins, Philadelphia, pp. 445- 517.
- Farrugia, M.K., Raza, S.A., Gould, S. and Lakhoo, K. (2008) Congenital lung lesions: Classification and concordance of radiological appearance and surgical pathology. Pediatric Surgery International, 24, 987-991. doi:10.1007/s00383-008-2201-1
- Stanton, M., Njere, I., Ade-Ajayi, N., Patel, S. and Davenport, M. (2009) Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. Journal of Pediatric Surgery, 44, 1027-1033. doi:10.1016/j.jpedsurg.2008.10.118
- Hammond, P.J., Devdas, J.M., Ray, B., Ward-Platt, M., Barrett, A.M. and McKean, M. (2010) The outcome of expectant management of congenital cystic adenomatoid malformations (CCAM) of the lung. European Journal of Pediatric Surgery, 20, 145-149. doi:10.1055/s-0030-1249047
- Pelizzo, G., Barbi, E., Codrich, D., Lembo, M.A., Zennaro, F., Bussani, R. and Schleef, J. (2009) Chronic inflammation in congenital cystic adenomatoid malformations, An underestimated risk factor? Journal of Pediatric Surgery, 44, 616-619. doi:10.1016/j.jpedsurg.2008.10.064
- Tran, H., Fink, M.A., Crameri, J. and Culminane, F. (2008) Congenital cystic adenomatoid malformation: Moitoring the antenatal and short-term neonatal outcome. Australian and New Zealand Journal of Obstetrics and Gynaecology, 48, 462-466. doi:10.1111/j.1479-828X.2008.00887.x
- Diniz, E.M.A., Viera, R.A., Silvia, F.L.V.F., Ceccon, M.E.J., Krebs, V.L.J. and Vaz, F.A.C. (2006) Diffuse congenital cystic lung disease with spontaneous regression. Journal of Maternal-Fetal and Neonatal Medicine, 19, 745-748. doi:10.1080/14767050600964174
- Ozcan, C., Celik, A., Ural, Z., Veral, A., et al. (2001) Primary pulmonary rhabdomyosarcoma arising within cystic adenomatoid malformation: A case report and review of literature. Journal of Pediatric Surgery, 36, 1062- 1065. doi:10.1053/jpsu.2001.24747
- Bush, A. (2009) Prenatal presentation and postnatal management of congenital thoracic malformations. Early Human Development, 85, 679-684. doi:10.1016/j.earlhumdev.2009.08.056
NOTES
*Corresponding author.