World Journal of Neuroscience
Vol.3 No.2(2013), Article ID:31642,26 pages DOI:10.4236/wjns.2013.32014
Non-peptide ligands in the characterization of peptide receptors at the interface between neuroendocrine and mental diseases
![]()
1INM-5 Nuclear Chemistry, Institute of Neuroscience and Medicine, Jülich, Germany
2IBG-3, Institute of Bioand Geosciences, Research Centre Jülich, Jülich, Germany
Email: m.pissarek@fz-juelich.de, u.disko@fz-juelich.de
Copyright © 2013 Margit Pissarek, Ulrich Disko. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 20 February 2013; revised 22 March 2013; accepted 17 April 2013
Keywords: Feeding Receptor; Hypothalamus; Neuropeptide; Non-Peptide Antagonist; PET; SPECT
ABSTRACT
Hypothalamic receptors for neuropeptide Y, melaninconcentrating hormone, melanocortins and orexins/ hypocretins as well as for the downstream signaling corticotrophic factor have been discussed broadly for their influence on food intake and reward but also on several psychiatric disorders. For the development of non-peptide ligands for the in vivo detection of alterations in density and affinity of such G-protein coupled (GPCRs) peptide receptors the requirements to affinity and pharmacokinetics have been shifted to thresholds markedly distict from classical GPCRs to dissociation constants < 0.5 nM, partition coefficients log P < 3.5 and transcellular transport ratios, e.g. for the permeability glycoprotein transporter, below 3. Nevertheless, a multitude of compounds has been reported originally as potential therapeutics in the treatment of obesity among which some are suitable candidates for labeling as PET or SPECT-tracers providing receptor affinities even below 0.1 nM. These could be unique tools not only for better understanding of the mechanism of obesity but also for investigations of extrahypothalamic role of “feeding receptors” at the interface between neuroendocrine and mental diseases.
1. INTRODUCTION
1.1. Hypothalamus as a Potential Target for Drug Development in the Treatment of Obesity and Related Mental Diseases
Among the huge number of central neuropeptide receptors the hypothalamic G-protein coupled receptors
(GPCR) attracted early efforts of ligand development. Many regulatory peptides found in hypothalamic nuclei fulfill a dual function in the brain at the intercept between neuroendocrine metabolic and behavioral regulation [1-5]. One of the central motives for development of ligands of hypothalamic peptide receptors and hypothalamic-pituitary-axis (HPA) is the search for effective treatment strategies for endocrinological disorders and hormone-dependent tumor diseases [6]. Furthermore, effort is directed to potential therapeutics, prevention and diagnostics of obesity-related disorders. Many of feeding-related receptor agonists act not only in the hypothalamus but also in extrahypothalamic, telencephalic, mesencephalic and metencephalic areas thereby contributing to the regulation of behavior, as has been confirmed by numerous studies [5,7-17]. Consequently, rising attention is paid to ligands of feeding-related GPCRs as potential tools for the investigation of their role in behavioral disturbances.
The hypothalamic structural triad of the arcuate nucleus (Arc), paraventricular nucleus (PVH), and the ventromedial hypothalamus (VMH; referred to as “satiety” centre) as well as the neuronal relay formed by lateral hypothalamic area (LHA; in contrast referred to as “hunger” centre) all together with the perifornicular area (PFA) represent a regulatory core unit of food intake [18] (see Figure 1).
The Arc, surrounding the third ventricle and adjacent to the eminentia mediana as the blood to brain gate to the hypothalamus for circulating hormones contains two discrete neuronal populations which act as first order neurons for insulin, leptin, ghrelin and other circulating metabolic signals. One population contains pro-opiomelanocortin (POMC) as precursor of melanocyte stimulating hormones (α-MSH, ß-MSH, γ-MSH), β-endorphin as well as the cocaine-amphetamine-regulated transcript
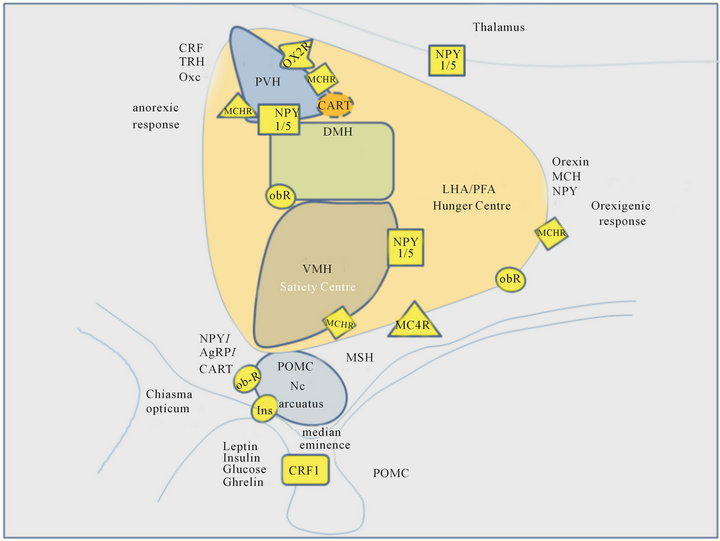
Figure 1. Scheme of expression of hypothalamic neuropeptide receptors involved in the regulation of food intake. Abbreviations: DMH dorsomedial hypothalamus. LHA lateral hypothalamic area, PFA perifornicular area, VMH ventromedial hypothalamus, PVH paraventricular nucleus, Ob-R leptin receptor, NPY1/5 neuropeptide Y1 and Y5 receptors, MCR melanocortin receptor, MCHR melanin-concentrating hormone receptor, CART cocaineamphetamine regulated transcript receptor, AgRP Agouti regulated peptide, MSH melanocyte stimulating hormone, CRF1 corticotrophin releasing factor receptor 1. Scheme modified according to [18,101,251-254].
(CART) and is stimulated by leptin which inhibits in turn the other cell population containing neuropeptide Y (NPY) and agouti-related peptide (AgRP) [19]. MSH, β- endorphin and CART (endogenous ligands at melanocortin receptors, µ-opioid receptor and still non-identified CART receptors, respectively) [20] inhibit food intake per se similarly to other melanocortins e.g. via corticotrophin releasing factor (CRF1). NPY (via NPY1- and NPY5 receptors) and AgRP acting as endogenous melanocortin antagonist at MC4-R may enhance food intake [9,21]. The balance of neuronal activity in both cell populations is therefore a major regulating factor in feeding behavior.
Both neuronal populations project to the second order regions PVH (where oxytocin, CRF and, thyrotropin releasing hormone (TRH) are released) as well as to the LHA/PFA (releasing orexin and melanin concentrating hormone; (MCH) increasing food intake) [18]. This system serves as the long-term regulator of food intake. Activation of the neurons in PVH results in a catabolic state.
Short-term regulation of food intake is realized by secondary neurons in the nucleus tractus solitarii (NTS) and nucleus dorsalis nervi vagi. The secondary neurons of the NTS project via Nc. parabrachilis and the thalamic Nc. ventroposterior parvocellulares to the insular cortex.
For some of the GPCRs involved in this system receptor densities have been reported as shown in Table 1. The data suggest that especially in non-diencephalic regions of the brain the requirements to affinity, specificity and pharmacokinetics are high if in vivo molecular imaging of those receptors shall be made possible.
1.2. Peptidic Versus Non-Peptidic Receptor Ligand
For the characterization of neuropeptide receptors in vivo two profound observations have to consider. First, almost all of these receptors exhibit relatively low receptor densities at their cerebral expression sites and second the uptake of peptidic tracers suitable for receptor labeling is possible per se but frequently limited due to inappropriate pharmacokinetic properties. Even small regulatory peptides with a molecular weight < 500 Da often show large differences in their potency to influence brain functions depending on the mode of application [22-27] and metabolic vulnerability [28,29].
Plasma half-life of peptide transmitters varies in a broad time range between several seconds and hours [23,24,30,31]. In general, the stability of endogenous peptides is difficult to predict and depends also from the presence and activity of cleavage enzymes in the blood or target regions.
Fifteen years ago it was postulated that neuropeptides released in the periphery either cannot cross the blood brain barrier (BBB) or can enter the brain only by very limited extent [32,33]. Although this hypothesis had been revised [27,34-36]), even today weak responses to exogenously applied peptidic receptor ligands are attributed to aggregation, poor enzymatic stability and various other limiting factors of BBB passage [23-25,27]. Further drawbacks to the suitability of peptidic compounds for therapeutic use are poor oral bioavailibility and high costs of production [37]. Alternative routes of drug delivery for instance transnasal administration can improve the accumulation of such drugs in the brain as has been shown for the somatostatin analogue octreotide [38]. However, this application route appears not suitable for nuclear medical purposes because of dosimetric limitations.
Although the affinity of peptidic tracers is frequently higher than that of non-peptidic ligands only few peptidic ligands developed to label brain receptors specifically could be introduced successfully into in vivo trials [15] due to limitations by the BBB. In consequence such tracers or drugs have been recommended and applied predominantly for therapy and diagnostics of peripheral tumors [6,15].
However, also a multitude of non-peptidic drugs and pharmacological probes [39] has been developed in parallel [39-41]. Some of those have been radio-labeled for in vivo imaging. Clinical trials have been performed for ligands at CRF1 receptors [39,42-47], at orexin receptors [48,49], at the NPY receptors [50,51], the opioid receptors [52], melanocortin -4-receptors (MC4-R) [53-55], melanin concentrating hormone receptors (MCH1-R) [56] and growth hormone secretagogue receptor 1 (ghrelin; GHS1-R) [57,58].
2. CRF-RECEPTORS
2.1. Endogenous Peptidic CRF Receptor Ligands
CRF (41 a.a.r.) is an early discovered prime coordinator of the synthesis of corticotrophin (ACTH; 39 a.a.r.) in the pituitary gland [59,60]. It is released from the neurons of the paraventricular region of the hypothalamus into the median eminence region which is one of the seven known circumventricular regions of the brain with interruption of the blood brain barrier [8,19,44,61,62]. Here, the paraventricular region is in close contact to the capillary plexus drained by long portal veins into the anterior pituitary a main target region of CRF [22].
Further endogenous peptides interacting with corticotrophin releasing factor receptors (CRF-R) are the urocortins 1 (Ucn1), (acting at CRF1-R and CRF2-R) Ucn2 and 3 (at CRF2-R) [63]. Two G-protein coupled receptor subtypes CRF1 and CRF2, which are members of the B-receptor family [64-69], as well as the CRF-binding protein [60] mediate the action of the transmitter peptides. CRF1-R as well as CRF2-R mainly couple to stimulatory Gs proteins [70,71] but trigger Gi and Gq dependent signal transduction mechanisms additionally [39,71].
CRFs-R are found wide spread throughout the brain but especially in stress responsive regions. They are expressed in relatively high densities in pituitary anterior (605 fmol/mg; human [72]) and intermediate lobe (200 fmol/mg; marmosets) [73]), but in rather moderate densities within prefrontal and cingulate cortex as well as in the subcortical part of the amygdala (centromedial nucleus) (60 - 150 fmol/mg; cynomolgus monkey) [62, 63,74] corresponding to the expression levels of the CRF1-R (see also Table 1). Hypersecretion of CRF has been postulated to be involved in the pathogenesis of several mood and anxiety disorders [5,15,75,76]. The CRF2-R has been found predominantly in the periphery [76] and is involved in the stress response, cardiovascular function and gastric motility [77]. In the rat the CRF2α variant of the CRF-R is expressed predominantly in the hypothalamus, while CRF2ß-R has been observed in the heart and skeletal muscle [78,79].The splice variant in the human brain is the CRF2γ-R [79, 80].
An urotensin-like immunreactivity has been found in the region of the Edinger Westphal nucleus and colocalized with CRF2-R [81,82].
2.2. Non-Peptide CRF Receptor Ligands
A comprehensive overview on endogenous and synthetic peptides and on non-peptide ligands at CRF-Rs as well as pharmacophore development has been provided by Zorilla and Koob [15] together with a review of phase II/III clinical trials for CRF1-R antagonists in depression, anxiety and irritable bowel syndrome.CRF2-R antagonism has been described for some peptide analogues of antisauvagine-30 suitable for detection of gastrointestinal CRF2-R in mice but obviously not able to cross the BBB
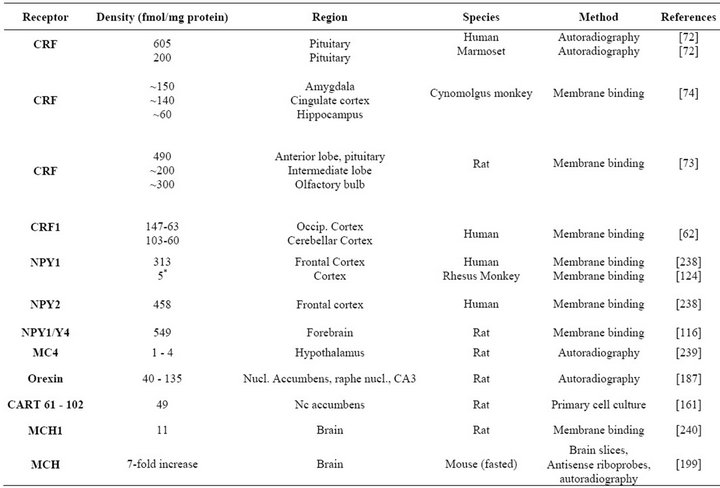
Table 1. Densities of feeding receptors in brain tissues of different species obtained by autoradiographic and membrane binding studies (rodent, monkey, human) available in the literature (in fmol/mg protein and in one exception * in nM).
[83].
Whereas Zorilla and Koob [15] summarize CRF-R antagonists discovered since the late 1990s, Kehne & Cain [84] review experiences from animal models of mental disorders.
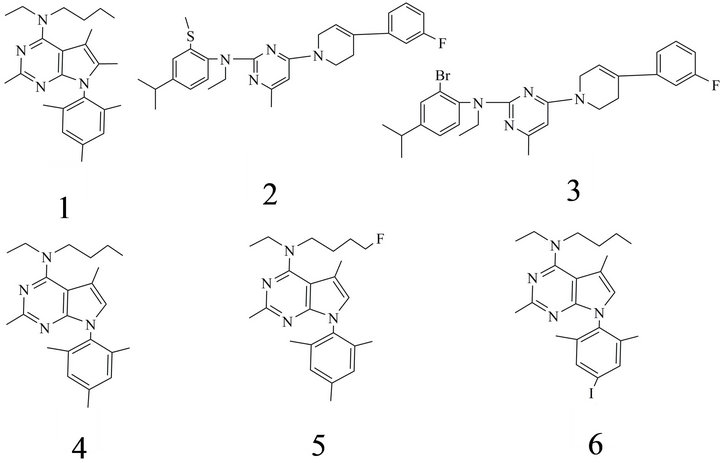
The first non-peptide CRF1-R antagonist was described in 1996 by Schulz et al. [85]. However, together with later reported ligands including antalarmin (1), NBI 27914, [86] DMP 904, CRA 1000 (2), CRA 1001 (3) SRA 12 5543A it shared the problem of high lipophilicity (Table 2) [87]. Chen et al. [88] reports for instance that CP 154, 526 (4) shows a volume distribution of 105 l/kg as well as a clog P of 8.43 and a half-life of 51 h [89] (Figure 2(a)). The lead compound NGD-98-2 presented in 2011 by Hodgetts et al. showed a log P of 6.3 [90].
Four non-peptide PET CRF1-ligands with affinities in the subnanomolar range have been described [47].
The radiosynthesis of a first CRF1-R PET ligand (5) was published for [18F]FBPPA [91,92]. Together with FBPPA also the SPECT tracer [123I]IBPPA (6) it was tested in vivo in Fischer rats (Figure 2(a)). Pharmacokinetic studies showed that the PET tracer was accumulated by 5.59% ID/g in the anterior pituitary. Fast wash out and poor specific binding in hypothalamus, amygdala and cerebellum were mainly accounted to its high log P > 6 [93]. The ratios pituitary/blood were for FBPPA 16.9:1 (5 min p.i.), 6.1:1 (60 min p.i.) and 5.6:1 (120 min p.i.).
Five minutes p.i. 4.28% ID/g of the SPECT tracer is presented in the pituitary declining to 1.98% ID/g until 120 min p.i. For [123I]IBPPA the respective ratios pituitary/blood were 7.4:1 (5 min), 13:1(60 min) and 9.4:1 (120 min) [93] reflecting a much slower wash out than for FBPPA.
The group of Zuev et al. [93] reported that in analogues of FBPPA the introduction of polar groups into the 8-(6-methoxy-2-methylpyridin-3yl) ring resulted in a dramatic loss of potency. The observation that too strong hydrophilicity impaires ligand binding to CRF1- Rs brought up the N-fluoroalkyl-8-(6-methoxy-2-methylpyridin-3-yl) 2,7-dimethyl-N-alkylpyrazolo [1,5-α]1,3,5- triazin-4-amine analogue (7).The compound was found to combine useful binding characteristics with appropriate lipophilicity (IC50 6.5 nM, log D = 3.5 (clog P 2.3). and was announced as a candidate for testing as a potential PET tracer [93].
Further efforts to get compounds of lower lipophilicity resulted in 3-phenylpyrazolo [1,5-a]pyrimidines like NBI 30545, with a log P of 3.18 and an appropriate Ki value,
 (a)
(a)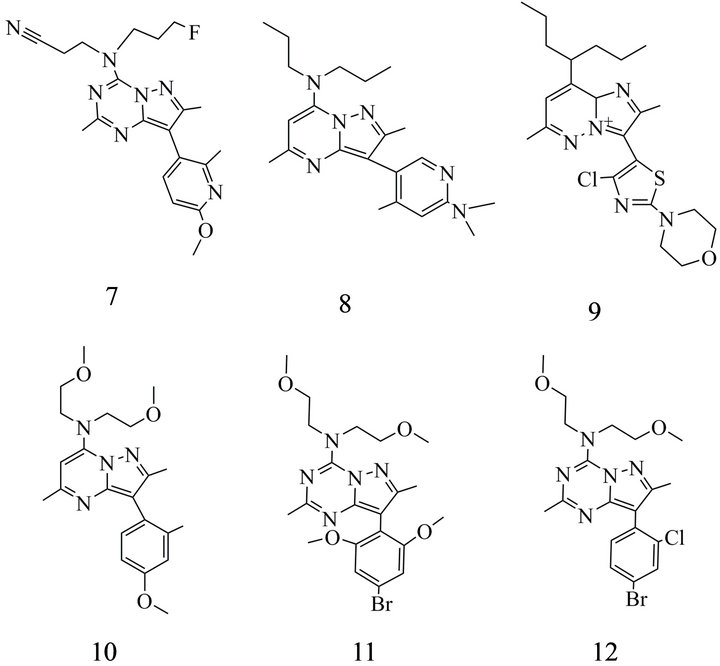 (b)
(b)
Figure 2. CRF1 receptor antagonists: (a) (1) Butyl-ethyl-[2,5,6- trimethyl-7-(2,4,6-trimethyl-phenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]-amine; (2) Ethyl-{4-[4-(3-fluoro-phenyl)-3,6-dihydro-2Hpyridin-1-yl]-6-methyl-pyrimidin-2-yl}-(4-isopropyl-2-methylsulfanyl-phenyl)amine; (3) (2-Bromo-4-isopropyl-phenyl)-ethyl- {4-[4-(3-fluoro-phenyl)-3,6-dihydro-2H-pyridin-1-yl]-6-methyl-pyrimidin 2-yl}amine; (4) Butyl-[2,5-dimethyl-7-(2,4,6-trimethylphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]-ethyl-amine; (5) [2,5- Di-methyl-7-(2,4,6-trimethyl-phenyl)-7H-pyrrolo[2,3-d] pyrimidin-4-yl] ethyl-(4-fluoro-butyl)-amine; (6) Butyl-ethyl-[7-(4-iodo2,6-dimethyl-phenyl)-2,5-dimethyl-7H-pyrrolo[2,3-d] pyrimidin-4-yl]- amine; (b) CRF1 receptor antagonists: (7) 3-{(3-Fluoro-propyl)- [8-(6-methoxy-2-methylpyridin-3-yl)-2,7-di-methyl-pyrazolo
[1,5-a][1,3,5] triazin-4-yl]-amino}-propionitrile. (8) [3-(6-Dimethylamino-4-methyl-pyridin-3-yl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidin-7-yl]-dipropyl-amine; (9) 3-(4-Chloro-2- morpholin-4-yl-thiazol-5-yl)-2,6-dimethyl-8-(1-propyl-butyl)8aH-imidazo-[1,2-b]pyridazin-4-ylium; (10) Bis-(2-methoxy-ethyl)- [3-(4-methoxy-2-methyl-phenyl)-2,5-dimethyl-pyrazolo[1,5-a]
pyrimidin-7-yl]-amine; (11) [8-(4-Bromo-2,6-dimethoxy-phenyl)-2, 7-dimethyl-pyrazolo [1,5-a][1,3,5] triazin-4-yl]-bis-(2-methoxyethyl)-amine; (12) [8-(4-Bromo-2-chloro-phenyl)-2,7-dimethylpyrazolo [1,5-a][1,3,5] triazin-4-yl]-bis-2-methoxy ethyl)-amine. ChemDraw (Cambridgesoft.com) was used for verification of the IUPAC names.
as well similar compounds obtained by incorporation of polar alkoxy groups [88]. Modifications of this lead structures allowed the identification of NBI 307775 (R121919) (8) (Figure 2(b)) and of a fluorinated analogue (3-(4 methyl-6-dimethylaminopyridin-3-yl)2,5-dimethyl-6-fluoro-7-dipropylaminopyrazolo [1,5-a] pyrimidine) as a potential PET tracer. However, first in vivo investigations in the baboon brain with the high affinity [11C]R121919 were not successful, probably due to low receptor density of CRF1 subtype in these animals [62].
Promising drugs with appropriate physicochemical properties are also the imidazol pyridazines presented by Gehlert et al. [87] ( MTIP (9) as well as by Richardson et al. (MPZP (10) (Figure 2(b)) [94]. MTIP has been described with a Kd of 0.22 nM, a log P of 3.55 as well as a bioavailability of 90% [15] (Table 2) and a distribution volume of 1.7 l/kg markedly improved in comparison to former candidates of in vivo imaging probes (e.g. the well known R121919 with 75.7 l/kg). Moreover, the intermediate plasma-half life of 3.3 h after p. o. administration is regarded as an advantage for potential therapeutic use.
MPZP showed a lower affinity (4.7 nM) but further improved lipophilic properties with log P of 2.95 [94].
More recently a Br-76 labelled non-peptidic CRF antagonist, (4-[76Br]BMK-152) (11) (Figure 2(b)), has been described [95] which exhibited high affinity to rat and monkey frontal cortex (0.23 nM and 0.31 nM). Moreover, 4-[76Br]BMK-152 showed an acceptable log P of 2.6 and is not a substrate of the P-glycoprotein (P-gp). During in vivo tests in monkey and rat it revealed binding of the tracer in brain regions consistent with the distribution of CRF -R.
3. NPY RECEPTORS
3.1. Endogenous Ligands of NPY Receptors
The 36 a.a.r. compound NPY is one of the most abundant neuropeptides in the brain. NPY-receptors (NPY1-R, NPY2-R, NPY4-R and NPY5-R) belong to the Class A (rhodopsin-like) GPCRs [96-100].
Insight into the network of NPY-Rs is given by many reviews e.g. by Kamiji & Inui in 2007 [101]. NPY has a dual effect on food intake. On one hand it is regarded as the most potent endogenous orexigenic agent [102] acting via NPY1-R and NPY5-R [103]. On the other hand it was found to mediate anorexia at the NPY2-R and NPY4-R [101]. In the Arc leptin depolarizes POMC neurons but hyperpolarizes NPY/GABA/AgRP neurons reducing the transmitter release from these terminals. The decrease of GABA is responsible additionally for the disinhibition of POMC neurons. Projections from NPY/AgRP and POMC/CART (rodents) neurons innervate neuron populations in the VMH, DMH, which also synthetize NPY, and to PVH which expresses NPY1- and NPY5-Rs [104]. The lateral hypothalamic area
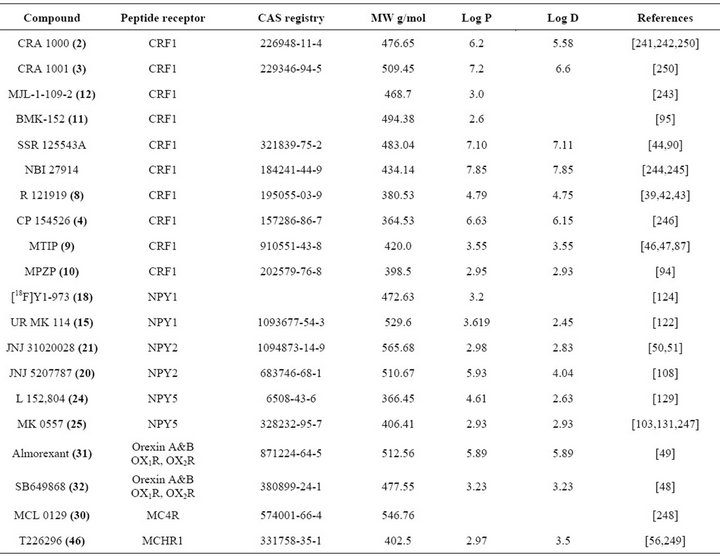
Table 2. Partition coefficients log P and log D as well as molecular weights for selected neuropeptide receptor ligands. Bold and parenthesized numbers correspond to the number of the respective formula in the figures (ACD/ChemSketch and Scifinder software was the source of some of the data).
(LHA) releases melanin-concentrating hormone, orexin and NPY [105]. Its neurons communicate with the cerebral cortex where also afferent signals from the vagus nerve and the sympathetic nerve system arrive particularly via nucleus of tractus solitarii (expressing NPY2-R and NPY4-R) what allows the cortical coordination of feeding behavior [104].
Because of their potential role in obesity, NPY1-R and NPY5-R are the main targets for ligands in anti-obesity drug development. A multitude of antagonists for their identification has been reported [106]. With view to anxiety disturbances most attention has been paid to NPY1-R and NPY2-Rs [36]. mRNA of the four functional receptors has been found also in the amygdala supporting a role of NPY-Rs as prime candidates for the regulation of emotion, learning and fear memory [107]. This forces the interest in NPY-R ligands suitable for treatment of anxiety disorders. The NPY1-R receptor is also localized in cerebral cortex, caudate-putamen and thalamus [108], while NPY2-R is found primarily in hypothalamus, hippocampus, substantia nigra and cerebellum. Moreover, the NPY2-R has been found as preand postsynaptic receptor [109-111]. The distribution of NPY5- R has been verified by Weinberg et al. [112] by means of hybridization studies in mouse brain to be discretely limited to regions within the hypothalamus including nucleus suprachiasmaticus, anterior hypothalamus, bed nucleus striae terminalis and ventromedial nucleus. NPY-R signaling is mediated by G-proteins especially via pertussis toxin dependent Gi/o proteins and decrease of cAMP formation. Hitherto among the six known NPYRs four (NPY1-R, NPY2-R, NPY4-R and NPY5-R) have been confirmed to couple to Gi-protein [113], whereas the existence of NPY3-R has been postulated but not demonstrated and the NPY6-R gene is not found functional in primates.
Moreover, several downstream signaling pathways mediate biological actions of NPY-Rs in the brain via Ca2+ channels or G-protein coupled inwardly rectifying potassium channels (GIRK) [36,113,114] as well as mitogen activated protein kinase (MAPK). Different phospholipase C subtypes and IP3 have been shown to be involved in signal transduction by NPY in different tissues and many questions on special regulatory chains also in brain cells remained unanswered even today.
3.2. Non-Peptide Ligands of NPY Receptors
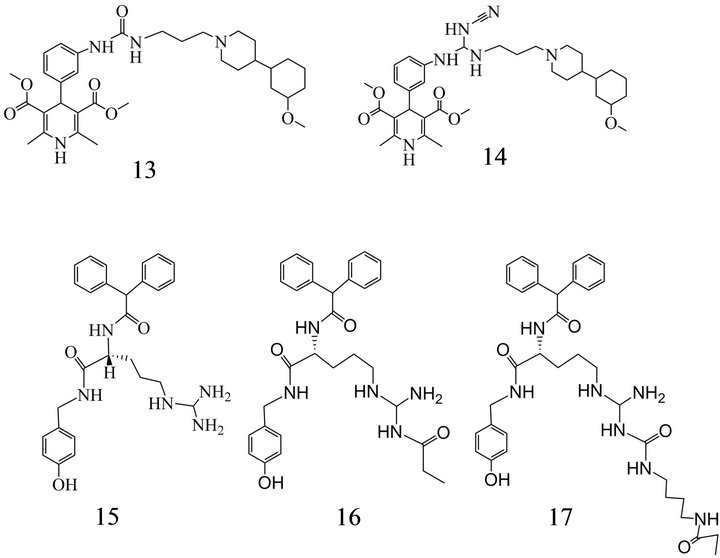
The NPY1-R subtype was the major target of efforts in research groups of several companies which developed antagonists with relatively high affinity and selectivity as J-104870, J-115814, BMS-193885 (13), BMS205749 (14) (Figure 3(a)), SAR-135966 ,1229U91 (GW 1229 or GR 231118) [115-120].
 (a)
(a) (b)
(b)
Figure 3. NPY1 receptor antagonists: (a) (13) 4-[3-(3-{3-[4-(3- Methoxy-cyclohexyl)-piperidin-1-yl]-propyl}-ureido)-phenyl]- 2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid dimethyl ester; (14) 4-{3-[(Cyanoamino-{3-[4-(3-methoxy-cyclohexyl)- piperidin-1-yl]-propyl-amino}-methyl)-amino]-phenyl}-2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid dimethyl ester; (15) 5-(Diaminomethyl-amino)-2-diphenylacetylamino-pentanoic acid 4-hydroxy-benzylamide; (16) 5-[(Amino-propionylaminomethyl) amino]-2-diphenylacety-lamino pentanoic acid-4-hydroxy-benzyl-amide; (17) 5-({Amino-[3-(4-propionylamino-butyl)- ureido]-methyl}-amino)-2-diphenylacetyl-amino-pentanoic acid 4-hydroxy-benzyl-amide. (b) NPY1 receptor antagonists: (18) [6-(5-Ethyl-4-fluoromethyl-thiazol-2-ylsulfanylmethyl)-4- mopholin-4-yl-pyridin-2-yl]-(6-methyl-pyridin-2-yl-methyl)- amine; (19) 6’-(81,5-Dimethyl-1H[1,2,4]triazol-3-ylsulfanylmethyl)-4,4-difluoro-3,4,5,6-tetrahydro-2H-[1,4’] bipyridinyl- 2’-yl]-(2,2,2-trifluoro-ethyl)-amine.
The first orally active Y1 antagonist was the pyrrolidine derivative SR 120819A [121]. Up to date, however, no ligands for the in vivo imaging of NPY1 receptors in clinical use for humans have been presented [51,103].
Keller et al. [122,123] and Weiss et al. [124] recommended the synthesis of ligands using substitution of argininamide type compounds with an acylguanidine entity at the guanidine as in BIBP 3226 (15) or the acylation of the guanidine group with propionic acid as in UR-MK114 (16) (Figure 3(a)) leading to high affinity (Kd 1.2 nM) and selectivity for NPY1-Rs. Additionally, these antagonists exhibit an excellent long-term stability and only slow radiolysis if storage is performed as TFA salt in ethanol. However, during longer periods of incubation at physiological pH the release of argininamide interferes with the efficiency of UR-MK114.
NG-carbamoylation instead of alkanoylation can avoid this disadvantage [123]. Therefore, BIBP-3226 (15) was substituted with a N-propionylated aminobutylcarbamoyl moiety.The newly synthetized NPY1 ligand, UR-MK136 (17) (Figure 3(a)) showed a Ki value similar to that of UR-MK114 [123].
Recently, the presentation of the 18F-labeled NPY1 antagonist Y1-973 (18) (Figure 3(b)) [124] was a breakthrough in NPY1 PET imaging. The tracer showed a IC50 of 0.13 nM for human Y1-R, a log P of 3.2 (Table 2) and a P-gp ratio of 1.3.These properties meet the requirements to peptide receptor ligands with Ki < 0.5 nM, log P < 3.5; and P-gp ratio < 3, which have been declared to be a generally necessary feature [120,124]. The authors emphasize the favourable kinetics in the brain, an accumulation in the striatum at 30 min with a binding potential of 1.7 and a high uptake also in cortical regions, whereas the uptake was moderate in thalamus and lowest in cerebellum as can be expected for a NPY1-R ligand Y1-718 (IC50 17 nM) (19) was used for investigation of reversibility and specifity of Y1-973 binding [124].
A first small-molecule Y2-R antagonist, JNJ-5207787 (20) (Figure 4), had been reported by Bonaventure et al. [108]. The compound showed a modest affinity and a receptor occupancy of 50%.
A further NPY2-R antagonist described by Seierstad et al. [125] and further characterized by Shoblock et al. [51], JNJ-31020028 (21) (Figure 4), possessed a high selectivity, an affinity close to that of BIIE-0246 [126] between 6 and 9 nM and a Hill coefficient of 1. Investigations with this ligand provided evidence that NPY2-Rs play rather a modulating role, but not a pivotal one in the occurrence of preponderance and obesity.
Also the role of NPY5-R has been discussed controversially. This was the case predominantly for its role in the occurrence of obesity in competition to NPY1.
CGP 71683 (22) (Figure 5) was the first NPY5-R antagonist reported in the literature [127,128] with a Ki of 1.4 and extremely high selectivity for NPY5-Rs.
Sato et al. [103] (23) (Figure 5) introduced the aryl pyrazole N-[5-(4-chlorophenyl)-1Hpyrazol-3-yl]-2-indane carboxamide (Ki 59 nM) as a lead structure to inhibit the NPY5-R. The scaffold was substituted with a chiral 3,3- dihydropental[α]naphthalene moiety resulting in a novel high affinity compound (Ki 3.5 nM). However, originally developed as a therapeutic drug it was not included in clinical studies because it was not effective enough following oral application in rats.
Kanatani et al. [129] concluded from experiments with the orally active, selective NPY5 receptor antagonist (L- 152,804) (24) (Figure 5) (Ki for displacement of PYY: 26 and 31 nM at human and rat NPY-Rs) that the NPY5-R contributes in part to the feeding response to NPY in their model. This contribution, however, was regarded as negligible. Recently, Nguyen et al. [130] using mice with individual NPY1- or NPY5-R knockout
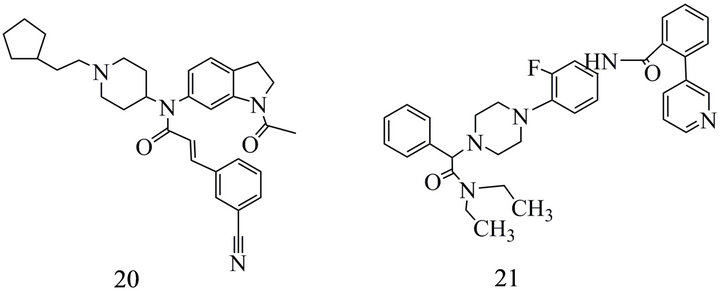
Figure 4. NPY2 receptor antagonists: (20) N-(1-Acetyl-2,3- dihydro-1H-indol-6-yl)-3-(3-cyano-phenyl)-N-[1-(2-cyclopentylethyl)-piperidin-4-yl]-acrylamide; (21) N-{4-[4-(Diethyl-carbamoyl-phenyl-methyl)-piperazin-1-yl]-3-fluoro-phenyl}-2- pyridin-3-yl-benzamide.
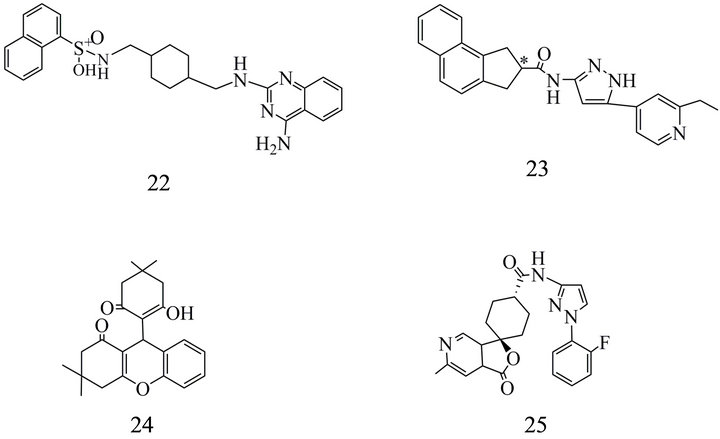
Figure 5. NPY5 receptor antagonists: (22) N-[[4-[[(4-aminoquinazolin-2-yl)amino]methyl]cyclohexyl]methyl]naphthalene- 1-sulfonamide; (23) 2,3 Dihydro-1H-cyclopental[a] naphthalene-2-carboxylic acid [5-(2-ethyl-pyridin-4-yl)-1H-pyrazol-3yl] amide; (24) 9-(2-Hydroxy-4,4-dimethyl-6-oxocyclohex-1-enyl)- 3,3-dimethyl-2,3,4,9-tetrahydro-xanthen-1-one; (25) trans-N-(1-(2- fluorophenyl)-3-pyrazolyl)-3-oxospiro(6-azaisobenzofuran-(3H), 1'-cyclohexane)-4'-carboxamide.
or combined one demonstrated that food intake requires the concerted actions of both receptors, though compensatory mechanisms induced by a general knockout have to take into account.
A series of NPY5-R antagonists developed five years after the study by Kanatani et al. starting with the report on MK0557 (25) (Figure 5) [131] based on spiroindol scaffolds and achieved Ki values of 0.57 nM [132]. A combination of high receptor affinity and suitable brain/ plasma ratio showed a spiroindol linked with a biphenyl. But with a log D > 4 and a poor bioavailability [132] it did not meet the criteria as a probe for in vivo receptor imaging. A further compound provided lower NPY5-R affinity (Ki 1.5 nM) but a log D7.4 of 2.79 and was therefore used as 11C-labeled radioligand. This tracer was not a substrate for the human P-gp transporter [132].
4. MELANOCORTIN RECEPTORS
4.1. Melanocortins, Agouti-Related Peptide and Their Receptors
Melanocortins, including ACTH and MSH, are characterized by the amino acid sequence His-Phe-Arg-Trp (HFRW) in their active core. The tetrapeptide motif has been recognized to be essential for their binding and agonistic activity at the melanocortin receptors (MCR) [9]. Such endogenous compounds are products of the cleavage of the 241 amino acid precursor pro-opiomelanocortin (POMC) by prohormone convertases (PC) acting in different parts of the pituitary gland as well as in the hypothalamus. Both subtypes of PC have been identified in anterior and intermediate pituitary. The expression of the distinct peptides has been suggested to be determined by the ratio between PC1 and PC2 (also referred to as PCSK1 or PC3 and PCSK2, respectively). PC1 in corticotroph cells supplies ACTH-related peptides (ACTH, ß-lipoprotein (LPH) and a K-peptide) in pars anterior distalis of the pituitary gland. In melanotrophs of the pars intermedia PC1 and PC2 reveal predominantly α-MSH-related peptides (α-MSH, corticotrophin like intermediate peptide; CLIP) as well as LPHprocessed to β-endorphin [55]. PC2 has been recognized to be responsible for the formation of the α-, ß- and y MSH [133]. A former review [134] refers also to the less studied peptids γ3-MSH and desacetyl- α-MSH.
Five receptors, MC1-R to MC5-R, are known as binding sites of the melanocortins. Two of them-the MC3-R and MC4-R -are localized also in the central nervous system. MC3-R is found mainly in the hypothalamus, cortex, thalamus and hippocampus [135,136] but also in kidney and gut. MC4-R has been observed in the hypothalamus, thalamus, hippocampus and other parts of the limbic system as well as in brainstem, with the highest affinity in the dorsal motor nucleus of the vagus nerve and spinal cord [137].
Important functions of the MC3-R are associated with the cardiovascular control as well as sodium and energy homeostasis. MC4-R is discussed predominantly for its direct role in the regulation of food intake and energy expenditure [138]. Mutations of the MC4-R have been shown to result in binge eating and obesity [11, 139-141]. MC3-R is presumed to be involved in regulation of food intake rather indirectly. Mutations induce a mild obesity related to an insulin-resistant phenotype [142]. ACTH and MSH act as agonists at the MC-Rs and mediate anorexic actions in normal individuals [20]. In obese individuals, however, the anti-obesity effect of agonists seems to be very limited, possibly because of downregulation of the melanocortin effects by the arrestin pathway [20].
MC-Rs have been found in contrast to many other seven-transmembrane-domain-receptor types to recognize not only endogenous agonists but also inverse agonists or endogenous antagonists [143,144].
One of these is agouti signaling protein (ASIP) which under normal physiological conditions antagonizes MC1- R effects [20,145] and controls skin pigmentation. It is a peptide triggering a switch in pigmentation from eumelanin to phaeomelanin [146]. The second antagonistic endogenous ligand, AgRP, acts at the hypothalamic MC4-Rs and MC3-Rs as has been shown in transgenic mice overexpressing AgRP. AgRP can be processed by PC1 resulting in a smaller peptide 6fold more potent at MC4-R than AgRP (131 a.a.r. (mice) and 132 a.a.r. (humans) [147]. ASIP (110 a.a.r.) and AgRP show similar CYS rich C-terminal domains which belong to the motif responsible for the interaction with MC-receptors. The cystine rich domains are presumed also to be responsible for a stereotypic tertiary structure defining a new structural class for vertebrate proteins [146] the cystine-knot peptides [148].
Only a small population of Arc neurons (3000 in mice) has been shown to be POMC positive but develops widespread extra-hypothalamic projections to brainstem, medulla and spinal cord [9,149] as second order neurons containing MC4-R and MC3-R.
In general the MC-Rs are regarded to be coupled to Gs protein signaling [20,21,150]. The MC4-R, however, is known also to couple to Gi/o and Gq proteins influencing cAMP or Ca2+ release. Moreover, MC4-R activation has been shown to interact with insulin signaling via MAPK including the c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK1/2) [133,151, 152].
4.2. MC-R Antagonists
Most of the MCR ligands have been developed as agonists for the treatment of obesity and erectile dysfunction
[37,153] although the success in the diminution of overweight remained markedly behind the expectations. Peptide MC4-R antagonists as HS-014, HS-024, HS-131 and MBP-10 have been known for longer time as helpful research tools. Among these MBP-10 [153,154] has been described as the most interesting due to its high affinity (Ki 0.5 nM) and a selectivity to MC4-R 125 fold higher than to the MC3-R [37]. However, only few MC4-R antagonists are available as potential drugs or as probes for in vivo investigations. Whereas for peptide compounds vulnerability to peptidases, low brain permeability and high costs of synthesis are obstacles, in non-peptidic antagonists the affinity in relation to the density of the receptors is in general too low for the use in PET or SPECT imaging approaches. Bednarek and Fong provided 2004 [37] an overview on ligands available in the years 2002-2003 and Blankeney et al. [39] included in 2007 MC receptor antagonists in their reviews on neuroreceptor ligands. Potential candidates for in vivo imaging could be based on piperazine templates or on a 3-iodo-4-chloro-aminoguanidine wich achieved in binding studies Ki of 10 nM at MC4-R (compared to 0.5 µM at MC1-R, 5.8 µM at MC3-R, 4.9 µM MC5-R) [37]. Several of the tetrahydroisoquinoline (THIQ) derivatives with MC4-R antagonizing properties and relatively high affinity could also penetrate the blood brain barrier e.g. compound (26) with a log D of 1.1 and a Ki of 1.8 nM, or compounds (27) and (28) (Figure 6) with log D of 1.8
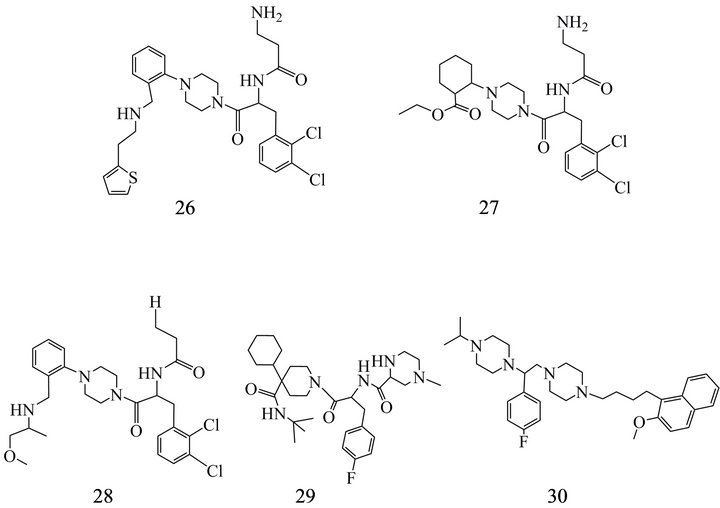
Figure 6. MC4-R antagonists: (26) 3-Amino-N-[1-(2,3-dichloro-benzyl)-2-oxo-2-(4-{2-[(2-thiophen2-yl-ethylamino)-methyl]-phenyl}-piperazin-1-yl-)-ethyl]-propionamide; (27) 2-{4- [2-(3-Amino-propionylamino)-3-(2,3-dichloro-phenyl)-propionyl]-piperazin-1-yl}-cyclohexanecarboxylic acid ethyl ester; (28) N-[1-(2,3-Dichloro-benzyl)-2-(4-{2-[(2-methoxy-1-methylethylamino)-methyl]-phenyl}-piperazin-1-yl)-2-oxo ethyl] propionamide; (29) (2S)-N-[(1R)-2-[4-cyclohexyl-4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperidinyl]-1-[(4-fluorophenyl) methyl]-2-oxoethyl]-4-methyl-2-piperazine-carboxamide; (30) 1- [(1S)-1-(4-fluorophenyl)-2-[4-[4-(2-meth-oxynaphthalen-1-yl) butyl]piperazin-1-yl]ethyl]-4-propan-2- ylpiperazine.
and Ki of 6.3 and 6.9 nM [155,156].
Wikberg & Mutulis [20] as well as Blankeney et al. [39] summarized derivatives (e.g. (29) (Figure 6) of the MC4-R agonist THIQ and the antagonist SHU9119 at the MC3-R and MC4R and emphasized further templates as potential scaffolds for MC4-R antagonists. Furthermore, a non-THIQ related low affinity piperazine had been reported in 2003 [157] as the first MC4-R antagonist. Modifications of this compound resulted in the synthesis of MCL-0129 (30) (Figure 6) which has been shown to improve stress induced depression and anxiety in rats [39, 54].
5. CART AND ITS POTENTIAL RECEPTOR
A receptor has been postulated also for CART peptides [158]. Rogge et al. proposed in 2008 [159] a signaling mechanism for such potential receptors. Due to the observation that signaling induced by CART can be inhibited by pertussis toxin a Gi/o protein coupled mechanism has been suggested but also activation of MEK and ERK ½ has been demonstrated in mouse pituitary cell lines [160]. CART itself is presumed to be involved in reward and reinforcement, feeding, stress and neuroendocrine control [161]. Active CART peptides occur by processing of a 89 and a 102 a.a.r. proCART peptide. The CART fragments 55 - 102, 62 - 102 and 42 - 89 [159] (with difference in the amino acid sequence of the active region in rats and humans) revealed biological activity also in vivo [162]. In humans has been observed only CART 42 - 89. The peptide can cross the BBB but is not subjected to mechanisms regulating saturation of its brain level [163]. The pro-peptides are predominantly expressed in hypothalamus, pituitary, adrenal gland and pancreas. A mutation has been found for the CART gene (Leu34Phe) in obese families [164,165] and the PACAP sequences 1 - 38 and 6 - 38 have been identified as low-affinity inhibitor of CART binding [166]. Moreover, the CART level has been observed to be markedly decreased in cerebrospinal fluid of patients suffering from dementia with Lewy bodies.
Rogge described for CART peptides a role in several parts of the stress axis including hypothalamus (retrochiasmatic nucleus, Arc, PVH), pituitary anterior and posterior, caudal raphe, rostral ventrolateral medulla, spinal cord as well as adrenal chromaffin cells [159].
6. OREXIN RECEPTORS
6.1. Endogenous Peptidic Ligands
Orexin A and B (33 and 28 a.a.r.) [167] also called hypocretin 1 and 2 according to the tissue where it has been identified [168] are both generated from pre-prohormone-orexin (pp-orexin; 130 - 131 a.a.r.). This is expressed in the lateral hypothalamic area particularly close to the median eminence, DMH and PFA [169,170]. Stability and lipophilicity of orexin A exceeds that of orexin B. That is the reason for a higher plasma concentration of orexin A and its ability to cross the blood brain barrier in difference to orexin B [163].
Additionally to their intrahypothalamic actions on feeding behavior orexin releasing neurons project to extrahypothalamic structures like the histaminergic tuberomamillary nucleus and the basal forebrain, as well in the posterior and pontal locus coeruleus and the median raphe nucleus, substantia nigra and ventral tegmental area. Moreover, there are strong interactions with signaling by neurons of the ventrolateral preoptic nucleus (VLPO) involved in the regulation of the ascending arousal system [171]. VLPO has been characterized to be in close relation to nucleus suprachiasmaticus which is one of the most powerful pacemakers of the circadian sleep/wake cycle and known as the internal clock of the mammalian body. Orexin (hypocretin) has been recognized as a wake-promoting agent [172,173].
Enormous boost for the presumption of a role of orexin in the sleep/wake cycle came from the discovery of natural animal models of narcolepsy in dogs [174] starting with investigations in a dog model with excitation-stimulated loss of muscle tone caused by a deficiency of the OX2-R [175-177]. Humans suffering from severe disturbances of sleep/wake cycle as narcolepsy show low or absent orexin in the cerebrospinal fluid [178,179].
The number of orexin-containing neurons has been reported to achieve 3000 - 4000 in rats and 50,000 - 80,000 in humans [173,180]. Orexin receptors have been found in the highest density in locus coeruleus.
OX2-R shows a nearly equal affinity to orexin A and B (34 nM and 60 nM), whereas OX1-R has a higher affinity to orexin A (30 nM vs 2500 nM for Orexin B) [167,168]. For OX1-R signal transduction via Gq/11 proteins is suggested, whereas OX2-R can act via Gq as well as via Gi and Gs proteins [167, 181,182]. However, it is not finally clear if this description is true also in different kinds of target cells [180]. Nevertheless, OX-R actions are mediated also by Na/Ca exchanger and GIRK.
6.2. Non-Peptide OX-R Ligands
OX-R antagonists have been developed predominantly as therapeutics for insomnia and dual orexin antagonists (DORA) attracted the highest attention because they could effectively promote sleep. Meanwhile, however, DORAs as well as selective orexin receptor antagonists (SORA) have been investigated in clinical trials [183]. Almorexant (31) (ACT-078573), SB-649868 (32) and MK-4305 (33) (Figure 7) as well as Merck-DORA 1 and Merck-DORA-5 belong to the first group. Almorexant and SB-649868 improved the natural sleep architecture by increasing the time spent in REM and non-REM sleep phase in contrast to the GABA modulator zolpidem which reduces the time in these sleep stages [48,49]. Both were subjected to clinical trials (Almorexant to phase III and SB-649868 to phase I) but the latter was delayed in further testing by preclinical toxicological findings.
The diazepane compound MK 4305 (33) with Ki values of 0.55 and 0.35 nM at OX1-R and OX2-R [49,180, 184] as well as a molecular weight of 450.19 g/mol has a log P of 4.24 and is included in a clinical phase III study. A volume distribution of 2.6 (rat) and 0.8 l/kg b. wt (dog) lets expect rather modest lipophilic properties. MK 4305 has been developed using 7-methyldiazepane and triazolyl-benzamide as core unit and is only one in a series of diazepanes highly potent as DORAs (see (34), (35) (Figure 7) [49,184].
The replacement of the benzoisoxazole moiety by benzopyrimidine and substitution in the 6-position by a fluorine atom resulted in a compound which was similarily to MK4305 not a substrate for the permeability glycoprotein (P-gp) and showed an excellent passive permeability. But the brain/plasma ratio between 0.4 and 0.6 excluded it from the list of candidates for in vivo imaging [49].
Although characterized by some favorable attributes the compound was shown to form electrophilic interme-
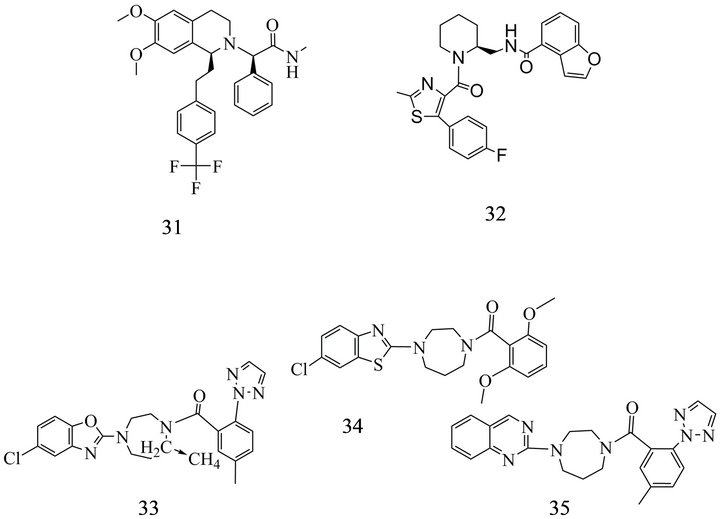
Figure 7. Dual orexin receptor antagonists: (31) 2- {6,7-Dimethoxy-1-[2-(4-trifluoromethyl-phenyl)-ethyl]-3,4-dihydro-1H-isoquinolin-2-yl}-N-methyl-2-phenylacetamide; (32) Benzofuran-4-carboxylic acid {1-[5-(4-fluoro-phenyl)-2- methyl-thiazole-4-carbonyl]-piperidin-2-ylmethyl}-amide; (33) (7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan- 1-yl]-[5-methyl-2-(2H-1,2,3-triazol-2-phenyl] methanone; (34) [4-(6-Chloro-benzothiazol-2-yl)-[1,4]diazepan-1-yl]-(2,6-dimethoxy-phenyl)- methanone; (35) (5-Methyl-2-[1,2,3]triazol-2-ylphenyl)-(4-quinazolin-2-yl-[1,4]diazepan-1-yl)-methanone.
diates which can be trapped by GSH if incubated in human or rat liver microsomes. Further investigations suggest a role of the fluor atom in the bioactivation. There was no marked alteration of the reactivity to GSH in double fluorinated molecules [49].
In general the DORAs available have higher affinities than the selective blockers. This is true also for the Merck-DORA-1 (0.2 nM and 3 nM for OX1-R and OX2- R, respectively) and Merck-DORA-5 (0.6 nM and 1.2 nM).
Nevertheless, selective OX1-R blockers have been found with SB 334867 (36) [185,186] and SB 674042 (37) and also OX2-R blockers have been characterized like JNJ-10394049 (38; 5 nM) and EMPA (39) (8 nM) [180,187] (Figure 8). Faedo et al. [182] subjected the newer orexin receptor antagonists not only to different in vitro activity tests but also to a sophisticated analysis of association and dissociation binding kinetics. The DORAs SB 649868 and MK6096 as well as the selective OX2-R antagonists ACT-078573, Roche-CP and JNJ10397049 were included. Short half-life of 0.19 min at OX1-R and 0.60 min binding to OX2-R where shown for JNJ10397049 associated with a surmountable antagonism with displacement of [3H] ACT-078573.
7. MELANIN-CONCENRATING HORMONE RECEPTOR
7.1. Endogenous Peptidic Ligands
The mammalian melanin-concentrating hormone (MCH)
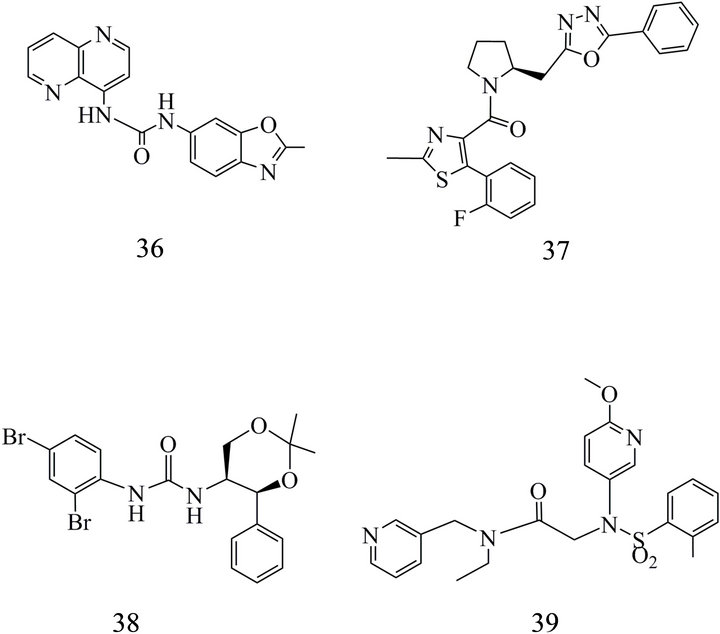
Figure 8. Selective orexin receptor antagonists (SORA) OX1-R: (36) 1-(2-Methyl-2,3-dihydro-benzooxazol-6-yl)-3- [1,5]naphthyridin-4-yl-urea; (37) [5-(2-Fluoro-phenyl)-2- methyl-thiazol-4-yl]-[2-(5-phenyl-[1,3,4]oxadiazol-2-ylmethyl)- pyrrolidin-1-yl]-methanone; OX2-R: (38) 1-(2,4-Dibromophenyl)-3-(2,2-dimethyl-4-phenyl-[1,3]dioxan-5-yl)-urea; (39) NEthyl-2-[(6-methoxy-pyridin-3-yl)-o-tolylsulfanyl-amino]-N-pyridin-3-yl-methyl-acetamide.
is a 19 a.a.r.cyclic peptide [188] which was discovered similar to the fish melanin-concentrating hormone (17 a.a.r.) earlier in extracts isolated from salmon pituitaries [189]. MCH supports the aggregation of melanin granules in melanophores resulting in a pale color of the skin. Its counterpart the melanocortin α-MSH disperses melanin pigments. MCH is formed by cleavage of a preprohormone which includes also amino-acid sequences of the neuropeptides E-I and G-E. Further splicing variants of the MCH gene can supply MCH gene overprinted polypeptide (MGOP)-14 and -17. MCH has been observed predominantly in lateral hypothalamus and zona incerta. Moreover, MCH has been detected in the periphery in testis and enteric neural system. In the peripheral tissue it is known to induce insulin hypersecretion and enhanced levels of the hepatocyte nuclear factor [190]. The absence of the peptide results in hypophagia and leaness [191]. MCH-Rs have been discovered in 1999 [192-197] and identified all over the brain but there are only few data available on the density of the receptors. These suggest a rather small cerebral receptor population compared with classical receptors well detectable by in vivo imaging methods. However, there are identified some regions with high expression of MCH-R mRNA [196-198] and it is known that fasting and obesity induce not only an upregulation of MCH but also of MCH1-R. The mechanism of this surprising response of the receptor is not completely understood [199,200]. The two receptor subtypes known in humans, MCH1-R and MCH2-R [201,202], are GPCRs. MCH1-R is acting via Gi/Go and Gq proteins [197,203]. Actions of MCH2-R are mediated via Gq proteins [204]. MCH1-R mRNA levels have been described to be high in the olfactoric system, VMH, Arc and zona incerta [205,206]. but it has been found also in hippocampus, PVH, subiculum, basolateral amygdala and in the shell of the nucleus accumbens which are involved in learning, addiction, emotion and motivated behavior [196]. The presence of MCH2-R has been confirm in human hippocampus as well as in the amygdala and is discussed controversially for hypothalamic regions.[201,202,207]. Nevertheless, it has been not confirmed that MCH2-R is essentially involved in feeding behavior or neuroendocrine function [205].
7.2. Non-Peptide Ligands of MCH-Receptors
The observations that mice with overexpression of MCH1-R are obese, MCH1-R knockout animals become lean and can develop a hypophagia [208,209] and leaness may occur also in consequence to ablation of melanin-concentrating hormone neurons [210] accelerated the efforts in search for suitable antagonistic agents up to date. Among these GW 856464 [211], AMG076 [212]
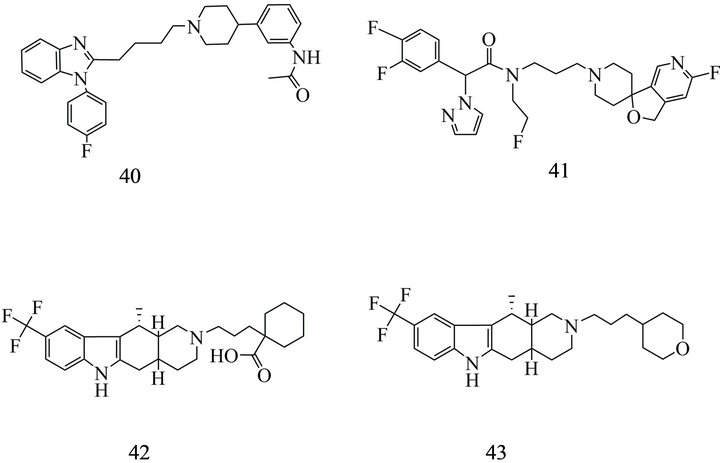
and NGD-4715 [213] achieved phase 1 clinical investigations. Trials to optimize quinazolines [214-219], as (40) (Figure 9(a)), revealed GW 803430 (GW3430; [214]), ATC0175 and ATC0065 [215] which were shown efficacious in rodent depression models [197]. However, ATC0175 and ATC0065 are binding with high affinity not only to MCH1-R but also to subtypes of the 5 HT receptor [197] suggesting that the antidepressiv effect of the MCH1-R antagonist is not due to MCH1-R alone.
 (a)
(a)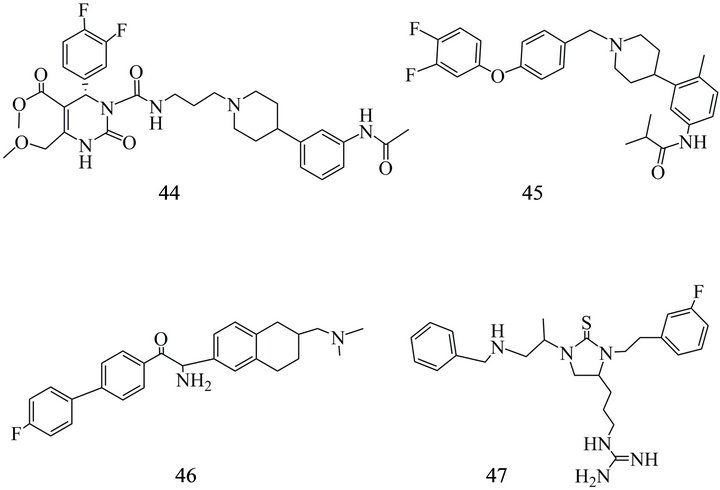 (b)
(b)
Figure 9. (a) MCH1 receptor antagonists: (40) N-[3-(1-{4-[1- (4-Fluoro-phenyl)-1H-benzoimidazol-2-yl]-butyl}-piperidin-4-yl)-phenyl]-acetamide; (41) 2-(3,4-difluorophenyl)-N-(3- (6- fluoro-1H-spiro[furo[3,4-c]pyridine-3,4'-piperidine]-1’-yl)propyl)-N-(2-fluoroethyl)-2-(1H-pyrazol-1-yl) acetamide; (42) 1-[3-(11- Methyl-9-trifluoromethyl-1,3,4,4a,5,6,11,11a-octahydro-pyrido[4,3-b] carbazol-2-yl)-propyl]-cyclohexane-carboxylic acid; (43) 11-Methyl-2-[3-(tetrahydro-pyran-4-yl)-propyl]-9-trifluoromethyl-2,3,4,4a,5,6,11,11a-octahydro-1H-pyrido [4,3-b]carbazole. (b) MCH1 receptor antagonists: (44) 3-{3-[4-(3-Acetylamino-phenyl)- piperidin-1-yl]-propylcarbamoyl}-4-(3,4-difluoro-phenyl)-6- methoxymethyl-2-oxo,2,3,4-tetrahydro-pyrimidine-5-carboxylic acid methyl ester; (45) N-[3-[1-[[4-(3,4-difluorophenoxy) phenyl]methyl] piperidin-4-yl]-4-methylphenyl]-2- methylpropanamide; (46) 2-Amino-2-(6-dimethylaminomethyl-5,6,7,8- tetrahydro-naphthalen-2-yl)-1-(4'-fluoro-biphenyl-4-yl)-ethanone; (47) {(4S)-1-[(1S)-2-(benzylamino)-methylethyl]-3-[2-(3-fluorophenyl) ethyl]-2-thioxoimidazolidin-4-yl} propyl) guanidine).
Furthermore, also aminopiperidine chromones [220], bicycle heptane derivatives [221], phenylpyridones [222] and benzazepine derivatives [223] recently have been reviewed by Johansson and Luthin [224,225]. However, no MCH1-R ligand is in use up to now for clinical imaging of cerebral receptors. The central reason for that should be the requirements arising to affinity and physicochemical properties by the low density of MCH-Rs in the brain. Moreover, the action of MCH1-R ligands as drugs for the treatment of obesity is frequently biased by interactions of the compounds with the hERG potassium channel (human ether-a-go-go-related gene channel) [226, 227], a channel associated with the long QT syndrome. Therefore, research for new therapeutically relevant selective MCH1-R ligands is steered in part by the necessity to find templates with warranty of low or no interaction with such channels.
Several compounds notable as candidates for in vivo MCH1-R imaging have been reported by Suzuki et al. [228] including spiro-fluorofuropyridine derivatives with Ki values of 0.49, 0.15 nM and 0.09 nM. Beside the reduction of the unspecific actions on hERG recent reports provide extended pharmakokinetic data including not only lipophilicity, solubility and further distribution parameters but also data on microsomal stability to P-gp, as well as SAR influencing the potency of binding to the MCH1-R receptor. Suzuki et al. [229] tested the susceptibility of their compounds to P-gp in human MDR1- and mouse mdr1α transfected porcine renal epithelial cells. Agents revealing transcellular transport ratio above 3 in these experiments are regarded to be P-gp substrates. This is true for the most of the compounds investigated by Suzuki et al. [229]. That property provides an additional important feature of exclusion and allowed to decide for a spirofuoropyridine substituted with an ethyl group and a further fluorine (41) (Figure 9(a)). This antagonist showed a MCH1-R affinity of 0.09 nM and a log D7.4 = 2.3. Two similar substances showed IC50 of 0.15 and 0.45 nM, respectively, but only one of it is not regarded as a P-gp substrate. Mihalcic et al. [205, 230] established the synthesis of isochonolin indols derived from alkaloid structures which were selected by means of an aequorin MCH1-R cellular assay using blockade of intracellular Ca2+ release by MCH. Derivatives with receptor affinities of 0.3 nM (42) and 0.6 nM (43) (Figure 9(b)) which can compete also with the affinity of the endogenous ligand of the MCH receptor (IC50 1 nM and 5 nM, respectively) were reported. Souers et al. and Vasudevan et al. proposed [231-234] some high affinity compounds with quinazoline, piperidine scaffolds. These include SNAP-7941 (44) [235] with Kd of 0.18 nM, SNAP-94847(45) [236] and T226296 (46) (Figure 9(b)) [56,197] (Table 2). Especially SNAP-94847 has been described to show a more rapid onset of anti-depressive action than a traditional antidepressant [236].
The thioxoimideazolimine TPI-1361-17 (47) (Figure 9(b)) has been selected among 800,000 compounds by data screening [188]. It could inhibit Ca2+ mobilization induced by 1 nM MCH with an IC50 of 6.1 nM and suppressed the food intake in vivo by 75% in rodents [188].
Particularly difficult is the identification of effective MCH2-R antagonists because the receptor had not been found in rodents. Nevertheless, Chen et al. [237] presented in 2012 a series of high affinity carbazol–derivatives with high selectivity for MCH2-R in comparison to MCH1-R.Two of the most effective compounds are (48) and (49) (Figure 10) with IC50 of 0.1 and 1 nM, respectively, in the CHO cells using Ca2+ FLIPR assay. (48) was obtained by replacement of hydroxy groups bound to the spiropiperidine region by a N-acetyl group which can mimic the hydrogen donating character of the hydroxyl moiety [237]. Substitution of the carbazole with an urea group improved further the pharmacokinetic properties. (49) showed in MCH2-R binding assays a Ki rather moderate for a ligand which should be a suitable tool for neuroimaging. But the compound has a good selectivity versus MCH1-R and no interactions with cardiac potassium channels providing sufficient reasons, that the authors selected this substance for further pharmacological evaluation.
8. CONCLUSIONS
The general handicap for brain neuropeptide receptor in vivo imaging is primarily due to the relative low density of many subtypes of peptide receptors. Additionally, some of them, e.g. MCH2-R, are not inherently expressed in species typically used as animal models for the evaluation of receptor ligands, especially for development of radioligands for SPECT and PET of cerebral “feeding receptors”. However, many reports on brain areas rich of neuropeptide receptor mRNA suggest the possibility to identify occasion-related changes of receptor expression if high affinity ligands are available. Furthermore, the pool of receptor ligands developed originally for therapeutic purposes and for labeling or blocking peripheral receptors revealed repeatedly compounds
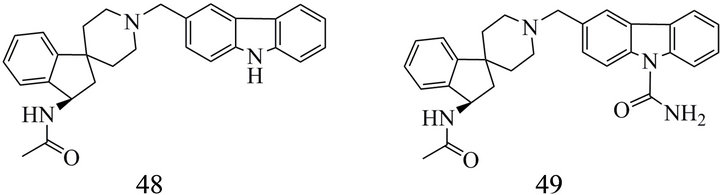
Figure 10. MCH2 receptor antagonists: (48) (R)-N-(1'-((9Hcarbazol-3-yl)methyl)-2,3-dihydrospiro[indene-1,4'-piperidine]-3-yl)acetamide; (49) (R)-3-((3-acetamido-2,3-dihy-drospiro[indene-1,4'-piperidine]-1'-yl)methyl)-9H-carbazole-9-carboxamide.
or templates appearing suitable for labeling with shortlived PET and SPECT isotopes and providing appropriate pharmacokinetic properties.
For the feeding receptors discussed here, at present, NPY-R ligands of the receptor subtypes Y1 and Y2 as well as CRF1-R ligands have the best perspective to become a diagnostic probe and to enter clinical applications due to relatively high receptor densities. MCH-R and MC-R as well as the hypothetic CART receptor should be the most challenging targets.
9. ACKNOWLEDGEMENTS
The authors would like to thank Dr. Ute Krügel, Beatrix Küven and Rike Preiß for their support in preparation of the manuscript as well as Dr. Johannes Ermert for advice on spiro-compounds.
![]()
![]()
REFERENCES
- Owen, M.J. and Nemeroff, C.B. (1999) Corticotropinreleasing factor antagonists in affective disorders. Expert Opinion on Investigational Drugs, 8, 1849-1858. doi:10.1517/13543784.8.11.1849
- Hökfelt, T., Broberger, C., Xu, Z.-Q.D., Sergeyev, V., Ubink, R. and Diez, M. (2000) Neuropeptides—An overview. Neuropharmacology, 39, 1337-1356. doi:10.1016/S0028-3908(00)00010-1
- Marchetti, B., Morale, M.C., Gallo, F., Lomeo, E., Testa, N., Tirolo, C. and Caniglia Garozzo, G. (2001) The hypothalamo-pituitary-gonadal axis and the immune system. In: Ader, R., Felten, D.L. and Cohen, N., Eds., Psychoneuroimmunology, 3rd Edition, Academic Press, San Diego, 363-389.
- Chaki, S. and Kanuma, K. (2007) Neuropeptide receptors: Novel therapeutic targets for depression and anxiety disorders. Drugs of the Future, 32, 809-822. doi:10.1358/dof.2007.032.09.1131450
- Nemeroff, C.B. (2008) Recent findings in the pathophysiology of depression. Focus, 6, 3-14.
- Schottelius, M. and Wester, H.-J. (2009) Molecular imaging targeting peptide receptors. Methods, 48, 161-177. doi:10.1016/j.ymeth.2009.03.012
- Appleyard, S., Hayward, M., Young, J.I., Butler, A.A., Cone, R.D., Rubinstein, M. and Low, M.J. (2003) A role for the endogenous opioid β-endorphin in energy homeostasis. Endocrinology, 14, 1753-1760. doi:10.1210/en.2002-221096
- Dunn, A.J., Swiergiel, A.H. and Palamarchouk, V. (2004) Brain circuits involved in corticotrophin releasing factornorepinephrine interactions during stress. Annals of the New York Academy of Sciences, 1018, 25-34. doi:10.1196/annals.1296.003
- Coll, A.P. (2007) Effects of pro-opiomelanocortin (POMC) on food intake and body weight: Mechanisms and therapeutic potential? Clinical Science, 113, 171-182. doi:10.1042/CS20070105
- Holmes, A., Heilig, M., Rupniak, N.M.J., Steckler, T. and Griebel, G. (2003) Neuropeptide systems as novel therapeutic targets for depression and anxiety disorders. Trends in Pharmacological Sciences, 24, 580-588. doi:10.1016/j.tips.2003.09.011
- Inui, A. (2003) Neuropeptide gene polymorphism and human behavioural disorders. Nature Reviews Drug Discovery, 2, 986-998. doi:10.1038/nrd1252
- Moosa, M.Y.H. and Jeenah, F.Y. (2008) Orexin—Does it have a role in mental illness? South African Journal of Psychiatry, 14, 63-64.
- Stahl, S.M. and Wise, D.D. (2008) The potential role of a corticotrophin-releasing factor receptor-1 antagonist in psychiatric disorders. CNS Spectrums, 13, 467-472, 476- 483.
- Giesbrecht, C.J., Mackay, J.P., Silveira, H.B., Urban, J.H. and Colmers, W.F. (2010) Countervailing modulation of Ih by neuropeptide Y and corticotrophin-releasing factor in basolateral amygdale as a possible mechanism for their effects on stress-related behavior. The Journal of Neuroscience, 30, 1670-16982. doi:10.1523/JNEUROSCI.2306-10.2010
- Zorilla, E.P. and Koob, G.F. (2010) Progress in corticotrophin-releasing factor-1 antagonist development. Drug Discovery Today, 15, 371-383. doi:10.1016/j.drudis.2010.02.011
- Refojo, D., Schweizer, M., Kuehne, C., Ehrenberg, S., Thoeringer, C., Schumacher, M., von Wolff, G., Avrabos, C., Touma, C., Engbliom, D., Schütz, E., Nave, K.-A., Eder, M., Wotjak, C.T., Sillaber, I., Holsboer, F., Wurst, W. and Deussing, J.M. (2011) Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science, 333, 1903-1907.
- Fahmy, H., Spyridaki, K., Kuppast, B. and Liapakis, G. (2012) The homeostasis hormone and its CRF1 receptor. From structure to function. Hormones, 11, 254-271.
- Schwartz, M.W., Woods, S.C., Porte Jr., D., Seeley, R.J. and Baskin, D.G. (2000) Central nervous system control of food intake. Nature, 404, 661-671.
- Stanley, S., Wynne, K., McGowan, B. and Bloom, S. (2005) Hormonal regulation of food intake. Physiological Reviews, 85, 1131-1158. doi:10.1152/physrev.00015.2004
- Wikberg, J.E.S. and Mutulis, F. (2008) Targeting melanocortin receptors: an approach to treat weight disorders and sexual dysfunction. Nature Reviews Drug Discovery, 7, 307-323. doi:10.1038/nrd2331
- Büch, T.R.H., Heling, D., Damm, E., Gudermann, T. and Reit, A. (2009) Pertussis toxin-sensitive signaling of melanocortin-4 receptor in hypothalamic GT1-7 cells defines Agouti-related protein as a biased agonist. The Journal of Biological Chemistry, 284, 26411-26420. doi:10.1074/jbc.M109.039339
- Bethge, N., Diel, F., Rösick, M., and Holz, J. (1981) Somatostatin half-life: A case report in one health volunter and a three month follow up. Hormone and Metabolic Research, 13, 709-710. doi:10.1055/s-2007-1019383
- Ehrström, M., Näslund, E., Levin, F., Kaur, R., Kitchgessner, A.L., Theodorsson, E. and Hellström, P.M. (2004) Pharmacokinetic profile of orexin A and effects on plasma insulin and glucagon in the rat. Regulatory Peptides, 119, 209-212. doi:10.1016/j.regpep.2004.02.004
- Kastin, A.J., Pan, W., Maness, L.M. and Banks, W.A. (1999) Peptides crossing the blood-brain barrier: some unusal observations. Brain Research, 848, 96-100. doi:10.1016/S0006-8993(99)01961-7
- Kastin, A., Akerstrom, V. and Hackler, L. (2000) Agoutirelated protein (83-132) aggregates and crosses the bloodbrain barrier slowly. Metabolism, 49, 1444-1448. doi:10.1053/meta.2000.16556
- Banks, W.A. (2006) Blood brain barrier and energy balance. Obesity, 14, 234S-237S. doi:10.1038/oby.2006.315
- Banks, W.A. (2008) Delivery of peptides to the brain: Emphasis on therapeutic development. Peptide Science, 90, 589-593.
- Zilow, G., Zilow, E.P., Burger, R. and Lindenkamp, O. (1993) Complement activation in newborn infants with early onset infection. Pediatric Research, 34, 199-203. doi:10.1203/00006450-199308000-00020
- Akopjan, T.N., Arutiunian, A.A., Laita, A. and Galoian, A.A. (1978) Breakdown of luliberin, somatostatin and substance P as an effect of hypothalamic endopeptidases. Voprosy Biokhimii Mozga, 13, 189-205.
- Grouzmann, E., Fathi, M., Gillet, M., de Torrentè, A., Cavadas, C., Brunner H. and Buclin, T. (2001) Disappearance rate of catecholamines, total metanephrines, and neuropeptide Y from the plasma of patients after resection of pheochromocytoma. Clinical Chemistry, 47, 1075- 1082.
- Foley, K.M., Kourides, I.A., Inturrisi, C.E., Kaiko, R.F., Zaroulis, C.G., Posner, J.B., Houde, R.W. and Li, C.H. (1979) β-Endorphin: Analgesic and hormonal effects in humans. Proceedings of the National Academy of Sciences of the United States of America, 76, 5377-5381. doi:10.1073/pnas.76.10.5377
- Lipinski, C.A. (2000) Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological and Toxicological Methods, 44, 235- 249. doi:10.1016/S1056-8719(00)00107-6
- Reubi, J.C. (1995) Neuropeptide receptors in health and disease: the molecular basis for in vivo imaging. Journal of Nuclear Medicine, 36, 1825-1835.
- Egleton R.D. and Davis, T.P. (2005) Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx Research, 2, 44-53. doi:10.1602/neurorx.2.1.44
- Reubi, J.C. (2003) Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocrine Reviews, 24, 389-427. doi:10.1210/er.2002-0007
- Patel, D. and Patel, M (2010) Review of NPY and NPY receptor for obesity. Internet Journal of Pharmacology, 8, 8. doi:10.5580/2531
- Bednarek, M.A. and Fong, T.M. (2004) Ligands of the melanocortin receptors, 2002-2003. Expert Opinion on Therapeutic Patents, 14, 327-336. doi:10.1517/13543776.14.3.327
- Lerner, E.N., van Zanten, E.H. and Stewart, G.R. (2004) Enhanced delivery of octreotide to the brain via transnasal iontophoretic administration. Journal of Drug Targeting, 12, 273-280. doi:10.1080/10611860400000938
- Blankeney, J.S., Reid, R.C., Le, G.T. and Fairlie, O.P. (2007) Nonpeptidic ligands for peptide-activated G protein-coupled receptors. Chemical Reviews, 107, 2960- 3041. doi:10.1021/cr050984g
- Betancur, C., Azzi, M. and Rostene, W. (1997) Nonpeptide antagonists of neuropeptide receptors: Tools for research and therapy. Treasury Inflation Protected Securities, 18, 372-386. doi:10.1016/S0165-6147(97)90666-0
- Klabunde, T. and Hessler, G. (2002) Drug design strategies for targeting G-Protein-coupled receptors. ChemBioChem, 3, 928-944. doi:10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5
- Chen, Y.L., Mansbach, R.S., Winter, S.M., Brooks, E., Collins, J., Corman, M.L., Dunaiskis, A.R., Faraci, W.S., Gallaschun, R.J., Schmidt, A. and Schulz, D.W. (1997) Synthesis and oral efficacy of a 4-butylethylamino)pyrrolo [2,3-d]pyrimidine: A centrally active corticotrophin-releasing factor 1receptor antagonist. Journal of Medicinal Chemistry, 40, 1749-1754. doi:10.1021/jm960861b
- Zobel, A.W., Nickel, T., Kunzel, H.E., Ackl, N., Sonntag, A., Ising, M. and Holsboer, F. (2000) Effect of high affinity corticotrophin-releasing hormone receptor 1 antagonist R121919 in major depression: The first 20 patients treated. Journal of Psychiatric Research, 34, 171- 181. doi:10.1016/S0022-3956(00)00016-9
- Gully, D., Geslin, M., Serva, L., Fontaine, E., Roger, P., Lair, C., Darre, V., Marcy, C., Rouby, P.-E., Simiand, J., Guitard, J., Gout, G., Steinberg, R., Rodier, D., Griebel, G., Soubrie, P., Pascal, M., Pruss, R., Scatton, B., Maffrand, G.-P. and Le Fur, G. (2002) 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro- 4-methylphenyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A): A potent and selective corticotrophin-releasing factor1 receptor antagonist. I. Biochemical and pharmacological characterization. Journal of Pharmacology and Experimental Therapeutics, 301, 322-332. doi:10.1124/jpet.301.1.322
- Gutman, D.A, Owens, M.J., Skelton, K.H., Thrivikraman, K.V. and Nemeroff, C.B. (2003) The corticotropin-releasing factor1 receptor antagonist R121919 attenuates the behavioral and endocrine responses to stress. Journal of Pharmacology and Experimental Therapeutics, 304, 874- 880. doi:10.1124/jpet.102.042788
- Barbosa, H.J., Collins, E.A., Hamdouchi, C., Hembre, E.J., Hipskin, P.A., Johnston, R.D., Lu, J., Rupp, M.J., Takakuwa, T. and Johnston, R.C. (2006) Imidazopyridazine compounds. WO2006102194.
- Yoon, T., de Lombaert, S., Brodbeck, R., Gulianello, M., Krause, J.E., Hutchinson, A., Horvath, R.F., Ge, P., Kehne, J., Hoffman, D., Chandrasekhar, J., Doller, D. and Hodgetts, K.-J. (2008) 2-Arylpyrimidines: Novel CRF-1 receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 18, 4438-4490. doi:10.1016/j.bmcl.2008.07.063
- Boss, C., Brisbare-Roch, C., Jenck, F., Aissaoui, H., Koberstein, R., Sifferlen, T. and Weller, T. (2008) Orexin receptor antagonism: A new principle in neuroscience. CHIMIA International Journal for Chemistry, 62, 974- 979. doi:10.2533/chimia.2008.974
- Cox, C.D., Breslin, M.J., Whitman, D.B., Schreier, J.D., McGaughey, G.B., Bogusky, M.J., Roecker, A.J., Mercer, S.P., Bednar, R.A., Lemaire, W., Bruno, J.G., Reiss, D.R, Harrell, C.M, Murphy, K.L., Garson, S.L., Doran, S.M., Prueksaritanont, T., Anderson, W.B., Tang, C., Roller, S., Cabalu, T.D., Cui, D., Hartman, G.D., Young, S.D., Koblan, K.S., Winrow, C.J., Renger, J.J. and Coleman, P.J. (2010) Discovery of the dual receptor antagonist [(7R)-4-(5-Chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone (MK-4305) for the treatment of insomnia. Journal of Medicinal Chemistry, 53, 5320-5332. doi:10.1021/jm100541c
- Dvorak, C.A., Swanson, D.M., Wong, V.D. (2009) Piperazinyl derivatives useful as modulators of the neuropeptide Y2 receptor. WO2009/006185, US2008/068289, 2010.
- Shoblock, J.R., Welty, N., Nepomuceno, D., Lord, B., Aluisio, L., Fraser, I., Motley, S.T., Sutton, S.W., Morton, K., Galici, R., Attack, J.R., Dvorak, L., Swanson, D.M., Carruthers, N.I., Dvorak, C., Lovenberg, T.W. and Bonaventure, P. (2010) In vitro and in vivo characterization of JNJ-31020028 (N-(4-{4-[2-(diethylamino)-2-oxo-1-phenylethyl]piperazin-1yl}-flurophenyl)-2pyridin-3-ylbenzamide), a selective brain penetrant small molecule antagonist of the neuropeptide YY2 receptor. Psychopharmacology, 208, 265-277. doi:10.1007/s00213-009-1726-x
- Burke, T.R., Rice, K.C. and Pert, C.B. (1985) Probes for narcotic receptor mediated phemomena. II. Synthesis of 17-methyl and 17-cyclopropylmethyl-3,14-dihydroxy-4,5- alpha-epoxy-6-beta-fluoromorphinans (foxy and cyclofoxy) as models of opioid ligands suitable for positron emission transaxial tomography. Heterocycles, 23, 69-99.
- Kask, A., Rägo, L., Korrovits, P., Wikberg, J.E.S. and Schiöth, H.B. (1998) Evidence that orexigenic effects of melanocortin C receptor antagonist HS014 are mediated by neuropeptide Y. Biochemical and Biophysical Research Communications, 248, 245-249. doi:10.1006/bbrc.1998.8961
- Chaki, S., Hirota, S., Funakoshi, T., Suzuki, Y., Suetake, S., Okubo, T., Ishii, T., Nakazato, A. and Okuyama, S. (2003) Anxiolytic-like and antidepressant-like activities of MCL0129 (1-[(S)-2-(4-fluorophenyl)-2-(4-iso-propylpiperadin-1-yl-)ethyl]-4-[4-(2-met hoxynaphthalen-1-yl)butyl] piperazine), a novel and potentnonpeptide antagonist of the melanocortin-4 receptor. Journal of Pharmacology and Experimental Therapeutics, 304, 818-826. doi:10.1124/jpet.102.044826
- Kato, H., Kuwako, K.I., Suzuki, M. and Tanaka, S. (2004) Gene expression patterns of pro-opiomelanocortin-processing enzymes PC1 and PC2 during postnatal development of rat corticotrophs. Journal of Histochemistry & Cytochemistry, 52, 943-957. doi:10.1369/jhc.4A6276.2004
- Takekawa, S., Asami, A., Ishihara, Y., Terauchi, J., Kato, K., Shimomura, Y., Mori, M., Murakoshi, H., Suzuki, N., Nishimura, O. and Fujino, M. (2002) T226296: A novel, orally active and selective melanin-concentrating hormone receptor antagonist. European Journal of Pharmacology, 438, 129-135. doi:10.1016/S0014-2999(02)01314-6
- Tokunaga, T., Hume, W.E., Nagamine, J., Kawamura, T., Taiji, M. and Nagata, R. (2005) Structure-activity relationships of the oxindole growth hormone secretagogues. Bioorg. Bioorganic & Medicinal Chemistry Letters, 15, 1789-1792. doi:10.1016/j.bmcl.2005.02.042
- Tomita, D., Yamatsugu, K., Kanai, M. and Shibasaki, M., (2009) Enantioselctive synthesis of SM-130686 based on the development of asymmetric Cu (I)F catalysis to access 2-oxindoles containing a tetrasubstituted carbon. Journal of the American Chemical Society, 131, 6946- 6948. doi:10.1021/ja901995a
- Vale, W., Spiess, J. and Rivier, C. (1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science, 213, 1394-1397. doi:10.1126/science.6267699
- [61] Huising, M.O., Vaughan, J.M., Shah, S.H., Grillot, K.L., Donaldson, C., Rivier, J., Flik, G. and Vale, W.W. (2008) Residues of corticotrophin releasing factor-binding protein (CRF-BP) that selectively abrogate binding to CRF but not to urocortin. The Journal of Biological Chemistry, 283, 8902-8912. doi:10.1074/jbc.M709904200
- [62] Sawchenko, P.E. and. Swanson, L.W. (1981) A method for tracing biochemically defined pathways in the central nervous system using combined fluorescence retrograde transport and immunohistochemical techniques. Brain Research, 210, 31-51. doi:10.1016/0006-8993(81)90882-9
- [63] Sullivan, G.M., Parsey, R.V., Kumar, J.S.D., Arango, V., Kassir, S.A., Huang, Y.-Y., Simpson, N.R., van Heertum, R.L. and Mann, J.J. (2007) PET imaging of CRF1 with [11C] R121919 and [11C]DMP696, is the target of sufficient density. Nuclear Medicine and Biology, 34, 353- 361. doi:10.1016/j.nucmedbio.2007.01.012
- [64] De Souza, E.B. (1995) Corticotropin releasing factor receptors: physiology, pharmacology, biochemistry and role in central nervous system and immune disorders. Psychoneuroendocrinology, 20, 789-819. doi:10.1016/0306-4530(95)00011-9
- [65] Kolakowski, L.F. (1994) GCRDb: A G-protein-coupled receptor database. Receptors & Channels, 2, 1-7.
- [66] Horn, F., Vriend, G. and Cohen, F.E. (2001) Collecting and harvesting biological data: The GPCRDB and NuclearDB information systems. Nucleic Acids Research, 29, 346-349. doi:10.1093/nar/29.1.346
- [67] Davies, M.N., Secker, A., Freitas, A.A., Mendao, M., Timmis, J. and Flowe, D.R. (2007) On the hierarchical classification of G protein coupled receptors. Bioinformatics, 23, 3113-3118. doi:10.1093/bioinformatics/btm506
- [68] Parthier, C., Reedtz-Runge, S., Rudolph, R. and Stubbs, M.T. (2009) Passing the baton in class B GPCRs: Peptide hormone activation via helix induction? Trends in Biochemical Sciences, 34, 303-310. doi:10.1016/j.tibs.2009.02.004
- [69] Suwa, M., Sugihara, M. and Ono, Y. (2011) Functional and structural overview of G-protein-coupled receptors comprehensively obtained from genom sequences. Pharmaceuticals, 4, 652-664. doi:10.3390/ph4040652
- [70] Unla, H. and Karnik, S.S. (2012) Domain coupling in GPCRs: The engine for induced conformational changes. Treasury Inflation Protected Securities, 33, 79-88. doi:10.1016/j.tips.2011.09.007
- [71] Dautzenberg, F.M., Kilpatrick, G.J., Hauger, R.L. and Moreau, J. (2001) Molecular biology of the CRH recaptors—In the mood. Peptides, 22, 753-760. doi:10.1016/S0196-9781(01)00388-6
- [72] Gutknecht, E., van der Linden, I., van Kolen, K., Verhoeven, K.F.C., Vauqelin, G. and Dautzenberg, F.M. (2009) Molecular mechanisms of corticotrophin-releasing factor receptor-induced calcium signalling. Molecular Pharmacology, 75, 648-657. doi:10.1124/mol.108.050427
- [73] Millan, M.A., Samra, A.B., Wynn, P.C., Catt, K.J. and Aguilera, G. (1987) Receptors and actions of corticotrophin-releasing hormone in the primate pituitary gland. The Journal of Clinical Endocrinology & Metabolism, 64, 1036-1041. doi:10.1210/jcem-64-5-1036
- [74] Hauger, R.L., Lorang, M., Irwin, M. and Aguilera, G. (1990) CRF receptor regulation and sensitization of ACTH response to acute ether stress during chronic intermittent immobilization stress. Brain Research, 532, 34-40. doi:10.1016/0006-8993(90)91738-3
- [75] Millan, M.A., Jacobowitz, D.M., Hauger, R.L., Catt, K.J. and Aguilera, G. (1986) Distribution of corticotrophinreleasing factor receptors in primate brain. Proceedings of the National Academy of Sciences of the United States of America, 83, 1921-1925. doi:10.1073/pnas.83.6.1921
- [76] Madaan, V. and Wilson, D. (2009) Neuropeptides: Relevance in treatment of depression and anxiety disorders. Drug News & Perspectives, 22, 319-324. doi:10.1358/dnp.2009.22.6.1395255
- [77] Werner, F.-M. and Covenas, R. (2010) Classical neurotransmitters and neuropeptides involved in major depression: A review. International Journal of Neuroscience, 120, 455-470. doi:10.3109/00207454.2010.483651
- [78] Hoare, S.R.J., Sullivan, S.K., Fan, J., Khongsaly, K. and Grigoriadis, D.E. (2005) Peptide ligand binding properties of the corticotrophin-releasing factor (CRF) type 2 receptor: Pharmacology of endogenously expressed receptors, G-protein-coupling sensitivity and determinants of CRF2 receptor selectivity. Peptides, 26, 457-470. doi:10.1016/j.peptides.2004.10.019
- [79] Lovenberg, T.W., Liaw, C.W., Grigoriadis, D.M., Clevenger, W., Chalmers, D., De Souza, E.B. and Oltersdorf, T. (1995) Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proceedings of the National Academy of Sciences of the United States of America, 92, 836-840. doi:10.1073/pnas.92.3.836
- [80] Chalmers, D.T., Lovenberg, T.W. and DeSouza, E.B. (1995) Localization of novel corticotrophin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: Comparison with CRF1 receptor mRNA expression. The Journal of Neuroscience, 15, 6340-6350.
- [81] Kostich, W.A., Chen, A., Sperle, K. and Largent, B.L. (1998) Molecular identification and analysis of a novel human corticotropin-releasing factor (CRF) receptor: The CRF2 receptor. Molecular Endocrinology, 12, 1077-1085. doi:10.1210/me.12.8.1077
- [82] Baigent, S.M. and Lowry, P.J. (2000) mRNA expression profiles for corticotrophin-releasing factor (CRF), urocortin, CRF-receptors and CRF-binding protein in peripheral rat tissues. Journal of Molecular Endocrinology, 25, 43-52. doi:10.1677/jme.0.0250043
- [83] Vaughan, J., Donaldson, C., Bittencourt, J., Perrin, M.H., Lewis, K., Sutton, S., Chan, R., Turnbull, A.V., Lovejoy, D., Rivier, C., Rivier, J., Sawchenko, P.E. and Vale, W. (1995) Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature, 378, 287-292. doi:10.1038/378287a0
- [84] Rühmann, A., Chapman, J., Higelin, J., Butscha, B. and Dautzenberg, F.M. (2002) Design, synthesis and pharmacological characterization of new highly selective CRF2 antagonist: Development of 123I-K31440 as a potential SPECT ligand. Peptides, 23, 453-460. doi:10.1016/S0196-9781(01)00640-4
- [85] Kehne, J.H. and Cain, C.K. (2010) Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression and stress-related disorders: evidence from animal models. Pharmacology & Therapeutics, 128, 460- 487. doi:10.1016/j.pharmthera.2010.08.011
- [86] Schulz, D.W. Mansbach, R.S., Sprouse, J., Braselton, J.P., Collins, J., Corman, M., Dunaiskis, A., Faraci, S., Schmit, A., Chen, Y. and Heym, J. (1996) CP-154,526: a potent and selective nonpeptide antagonist of corticotrophin releasinf factor receptors. Proceedings of the National Academy of Sciences of the United States of America, 93, 10477-10482. doi:10.1073/pnas.93.19.10477
- [87] Whitten, J.P., Xie, X.F., Erickson, P.E., Webb, T.R., DeSouza, E.B., Grigoriadis, D.E. and McCarthy, J.R. Rapid microscale synthesis, a new method for lead optimization using robotics and solution phase chemistry: Application to the synthesis and optimization of corticotropin releasing factor 1 receptor antagonists. Journal of Medicinal Chemistry, 39, 4354-4357. doi:10.1021/jm960148m
- [88] Gehlert, D.R., Cippitelli, A., Thorsell, A., D. Le, A.D., Hipskind, P.A., Hamdouchi, C., Lu, J., Hembre, E.J., Cramer, J., McKinzie, D., Morin, M., Ciccocioppo, R. and Heilig, M. (2007) 3-(4-Chloro-2-morpholin-4-ylthiazol-5-yl)-8-(1-ethylpropyl-2,6-dimethyl-imidazo[1,2-b] pyridazine: A novel brain penetrant, orally available corticotropin releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. The Journal of Neuroscience, 27, 2718-2726. doi:10.1523/JNEUROSCI.4985-06.2007
- [89] Chen, C., Wilcoxen, K.M., Huang, C.Q., Xie, Y.-F., McCarthy, J.R., Webb, T.R., Zhu, Y.-F., Saunders, J., Liu, X.-J., Chen, T.K., Bozigian, H. and Grigoriadis, D.E. (2004) Design of 2,5-Dimethyl-3-(6-dimethyl-4-methylpyridin-3-yl)-7-dipropylamino-pyrazolo[1,5-a]pyrimidine (NBI 30775/R121919) and structure activity relationships of a series of potent and orally active corticotrophin-releasing factor receptor antagonists. Journal of Medicinal Chemistry, 47, 4787-4798. doi:10.1021/jm040058e
- [90] Seymour, P.A., Schmidt, A. W. and Schulz, D.W. (2003) The pharmacology of CP-154, 526, a non-peptide antagonist of the CRH1 receptor: A review. CNS Drug Reviews, 9, 57-96. doi:10.1111/j.1527-3458.2003.tb00244.x
- [91] Hodgetts, K.J., Ge, P., Yoon, T., de Lombaert, S., Brodbeck, R., Gulianello, M., Kieltyka, A., Horvath, R.F., Kehne, J.H., Krause, J.E., Maynard, G.D., Hoffmann, D., Lee, Y., Fung, L. and Doller, D. (2011) Discovery of N-(1-ethylpropyl)-3-methoxy-5-(2-methoxy-4-trifluoromethoxyphenyl) 6-methyl-pyrazin-2yl]amine 59 (NGD 98- 2): An orally active corticotrophin releasing factor-1 (CRF-1) receptor antagonist. Journal of Medicinal Chemistry, 54, 4187-4206. doi:10.1021/jm200365y
- [92] Martarello, L., Kilts, C.D., Ely, T., Owens, M.J., Nemeroff, C.B., Camp, M. and Goodman, M.M. (2001) Synthesis and characterization of fluorinated and iodinated pyrrolopyrimidines as PET/SPECT ligands for the CRF1 receptor. Nuclear Medicine and Biology, 28, 187-195. doi:10.1016/S0969-8051(00)00199-2
- [93] Hsin, L.W., Webster, E.L., Chrousos, G.P., Gold, P.W., Eckelman, W.C., Contoreggi, C. and Rice, K.G. (2000) Synthesis and biological activity of fluoro-substituted pyrrolo[2,3-d] pyrimidines: The development of potential positron emission tomography imaging agents for the corticotrophin-releasing hormone type 1receptor. Bioorganic & Medicinal Chemistry Letters, 10, 707-710. doi:10.1016/S0960-894X(00)00071-8
- [94] Zuev, D., Mattson, R.J., Huang, H., Mattson, G.K., Zueva, L., Nielsen, M., Kozlowski, E.S., Huang, X.S., Dedong, W., Gao, Q., Lodge, N.J., Bronson, J.J. and Macor, J.E. (2011) Potential CRF1 R PET imaging agents: N-Fluoroalkyl-8-(6-methoxy-2-methyl-pyridin-3-yl)-2,7-dimethyl-N-alkylpyrazolo[1,5-a][1,3,5]triazin-4-amines. Bioorganic & Medicinal Chemistry Letters, 21, 2484-2488. doi:10.1016/j.bmcl.2011.02.050
- [95] Richardson, H.N., Zhao, Y., Fekete, E.M., Funk, C.K., Wirsching, P., Janda, K.D., Zorilla, E.P. and Koob, G.F. (2008) MPZP: A novel small molecule corticotrophinreleasing factor type 1 receptor (CRF1) antagonist. Pharmacology Biochemistry and Behavior, 88, 497-510. doi:10.1016/j.pbb.2007.10.008
- [96] Jagoda, E.M., Lang, L., McCullough, K., Contoreggi, C., Moon, K.B., Ma, Y., Rice, K.C., Szajek, L.P., Eckelman, W.C. and Kiesewetter, D.O. (2011) [76Br] BMK-152, a nonpeptide analogue, with high affinity and low nonspecific binding for the corticotrophin-releasing factor type 1 receptor. Synapse, 65, 910-918. doi:10.1002/syn.20919
- [97] Tatemoto, K., Carlquist, M. and Mutt, V. (1982) Neuropeptide Y—A novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature, 296, 659-660. doi:10.1038/296659a0
- [98] Herzog, H., Hort, Y.J., Ball, H.J., Hayes, G., Shine, J. and Selbie, L.A. (1992) Cloned human neuropeptide Y receptor couples to two different second messenger systems. Proceedings of the National Academy of Sciences of the United States of America, 89, 5794-5798. doi:10.1073/pnas.89.13.5794
- [99] Larhammar, D., Blomqvist, A.G., Yee, F., Jazin, E., Yoo, H. and Wahlestedt, C. (1992) Cloning and functional expression of a human neuropeptide Y/peptide YY receptor of the Y1 type. The Journal of Biological Chemistry, 267, 10935-10938.
- [100] Parill, A.L. and Bautista, D.L. (2011) GPCR conformations: Implications for rational drug design. Pharmaceuticals, 4, 7-43. doi:10.3390/ph4010007
- [101] Inui, A. (2003) Neuropeptide gene polymorphism and human behavioural disorders. Nature Reviews Drug Discovery, 2, 986-998. doi:10.1038/nrd1252
- [102] Kamiji, M.M. and Inui, A. (2007) Neuropeptide Y recaptor selective ligands in the treatment of obesity. Endocrine Reviews, 28, 664-684. doi:10.1210/er.2007-0003
- [103] Wisialowski, T., Parker, R., Preston, E., Sainsbury, A., Kraegen, E., Herzog, H. and Cooney, G. ( 2000) Adrenalectomy reduces neuropeptride Y-induced insulin release and NPY receptor expression in the rat ventromedial hypothalamus. The Journal of Clinical Investigation, 105, 1253-1259. doi:10.1172/JCI8695
- [104] Sato, N., Ogino, Y., Mashiko, S. and Ando, M. (2009) Modulation of neuropeptide Y receptors for the treatment of obesity. Expert Opinion on Therapeutic Patents, 19, 1401-1415. doi:10.1517/13543770903251722
- [105] Chee, M.J.S., Myers, M.G., Price, C.J. and Colmers, W.F. (2010) Neuropeptide Y suppresses anorexic output from the ventromedial nucleus of the hypothalamus. The Journal of Neuroscience, 30, 3380-3390. doi:10.1523/JNEUROSCI.4031-09.2010
- [106] Marston, O.J., Hurst, P., Evans, M.L., Burdakov, D.I. and Heisler, L.K. (2011) Neuropeptide Y cells represent a distinct glucose-sensing population in the lateral hypothalamus. Endocrinology, 152, 4046-4052. doi:10.1210/en.2011-1307
- [107] Yulyaningsih, E., Zhang, L., Herzog, H. and Sainsbury, A. (2011) NPY receptors as potential targets for anti-obesity drug development. British Journal of Pharmacology, 163, 1170-1202. doi:10.1111/j.1476-5381.2011.01363.x
- [108] Bowers, M.E., Choi, D.C. and Ressler, K.J. (2012) Neuropeptide regulation of fear and anxiety: Implications of cholecystokinin, endogenous opioids, and neuropeptide Y. Physiology & Behavior, 107, 699-710. doi:10.1016/j.physbeh.2012.03.004
- [109] Bonaventure, P., Nepomuceno, D., Mazur, C., Lord, B., Rudolph, D.A., Jablonoswki, J.A., Carruthers, N.I. and Lovenberg, T.W. (2004) Characterization of N-1-acetyl- 2,3-dihydro-1-H-indol-6-yl)-3-(3-cyano-phenyl)-N[1-(2- cyclopentyl-ethyl)-piperidin-4yl]acrylamide (JNJ-5207787), a small molecule antagonist of the neuropeptide Y Y2 receptor. Journal of Pharmacology and Experimental Thera- peutics, 308, 1130-1137. doi:10.1124/jpet.103.060459
- [110] Gehlert, D.R., Schober, D.A., Beavers, L., Gadski, R., Hoffmann, J.A., Smiley, D.L., Chance, R.E., Lundell, I. and Larhammar, D. (1996) Characterization of the peptide binding requirements for the cloned human pancreatic polypeptide-preferring receptor. Molecular Pharmacology, 50, 112-118.
- [111] Flood, J.F. and Morley, J.E. (1989) Dissociation of the effects of neuropeptide Y on feeding and memory: Evidence for preand postsynaptic Mediation. Peptides, 10, 963-966. doi:10.1016/0196-9781(89)90176-9
- [112] Thorsell, A. (2010) Brain neuropeptide Y and corticotrophin-releasing hormone in mediating stress and anxiety. Experimental Biology and Medicine, 235, 1163-1167. doi:10.1258/ebm.2010.009331
- [113] Weinberg, D.H., Sirinathsinghji, J.S., Tan, C.P., Shiao, L.-L., Morin, N., Rigby, M.R., Heavens, R.H., Rapoport, D.R., Bayne, M.L., Cascieri, M.A., Strader, C.D., Linemeyer, D.L. and MacNeil, D.J. (1996) Cloning and expression of a novel neuropeptide Y receptor. Journal of Biological Chemistry, 271, 16435-16438. doi:10.1074/jbc.271.28.16435
- [114] Griebel, G. and Holsboer, F. (2012) Neuropeptide recaptor ligands as drugs for psychiatric disease: The end of the beginning? Nature Reviews Drug Discovery, 11, 462- 478. doi:10.1038/nrd3702
- [115] Minor, R.K., Chang, J.N. and de Cabo, R. (2009) Hungry for life: How the arcuate nucleus and neuropeptide Y may play a critical role in mediating the benefits of calorie restriction. Molecular and Cellular Endocrinology, 299, 79-88. doi:10.1016/j.mce.2008.10.044
- [116] Daniels, A.J., Matthews, J., Slepetis, R.J., Jansen, M., Viveros, O.H., Tadapalli, A., Harrington, W., Heyer, D., Landavazo, A., Leban, J.J. and Spaltenstein, A. (1995) High-affinity neuropeptide Y receptor antagonists. Proceedings of the National Academy of Sciences of the United States of America, 92, 9067-9071. doi:10.1073/pnas.92.20.9067
- [117] Schober, D.A., Gackenheimer, S.L., Heiman, M.L. and Gehlert, D.R. (2000) Pharmacological characterization of 125I-1229U91 binding to Y1 and Y4 Neuropeptide Y/ peptide YY receptors. Journal of Pharmacology and Experimental Therapeutics, 293, 275-280.
- [118] Dumont, Y. and Quirion, R. (2000) [125I]-GR231118: A high affinity radioligand to investigate neuropeptide Y Y1 and YY4receptors. British Journal of Pharmacology, 129, 37-46. doi:10.1038/sj.bjp.0702983
- [119] Malmström, R.E., Alexandersson, A., Balmer, K.C. and Weilitz, J. (2000) In vivo characterization of the novel neuropeptide Y Y1 receptor antagonist H409/22. Journal of Cardiovascular Pharmacology, 36, 516-525. doi:10.1097/00005344-200010000-00016
- [120] Poindexter, G.S., Bruce, M.A., Breitenbucher, J.G., Higgins, M.A., Sit, S.-Y., Romine, J.L., Martin, S.W., Ward, S.A., McGovern, R.T., Clarke, W., Russell, J. and AntalZimanyi, I. (2004) Dihydropyridine neuropeptide YY1 receptor antagonist 2: Bioisosteric urea replacement. Bioorganic & Medicinal Chemistry, 12, 507-521. doi:10.1016/j.bmc.2003.10.016
- [121] Kameda, M., Ando, M., Nakama, C., Kobayashi, K., Kawamoto, H., Ito, S., Suzuki, T., Tani, T., Ozaki, S., Tokita, S. and Sato, N. (2009) Synthesis and evaluation of a series of 2,4-diaminopyridine derivatives as potential positron emission tomography tracers for neuropeptide Y Y1 receptors. Bioorganic & Medicinal Chemistry Letters, 19, 5124-5127. doi:10.1016/j.bmcl.2009.07.030
- [122] Serradeille Gal, C., Lafontan, M., Raufaste, D., Marchand, J., Pouzet, B., Casellas, P., Pascal, M., Maffrand, J.P. and Le Fur, G. (2000) Characterization of NPY receptors controlling lipolysis and leptin secretion in human adipocytes. FEBS Letters, 465, 150-156.
- [123] Keller, M., Pop, N., Hutzler, C., Beck-Sickinger, A.G., Bernhardt, G. and Buschauer, A. (2008) Guanidine-acylguanidine bioisosteric approach in the design of radioligands: Synthesis of a tritium-labeled N(G)-propionylargininamide ([3H]-UR-MK114) as a highly potent and selective neuropeptide Y Y1 receptor antagonist. Journal of Medicinal Chemistry, 51, 8168-8172. doi:10.1021/jm801018u
- [124] Keller, M, Bernhardt, G. and Buschauer, A. (2011) [3H] UR-MK 136: A Highly potent and selective radioligand for neuropeptide YY1 receptors. ChemMedChem, 6, 1566-1571. doi:10.1002/cmdc.201100197
- [125] Hostetler, E.D., Sanabria-Bohorquez, S., Fan, H., Zeng, Z., Gantert, L., Williams, M., Miller, P., O’Malley, S., Kameda, M., Ando, M., Sato, N., Ozaki, S., Tokita, S., Ohta, H., Williams, D., Sur, C., Cook, J.J., Burns, H.D. and Hargreaves, R. (2011) Synthesis, characterization, and monkey positron emission tomography (PET) studies of [18F]Y1-973, a PET tracer for the neuropeptide Y Y1 receptor. NeuroImage, 54, 2635-2642. doi:10.1016/j.neuroimage.2010.11.014
- [126] Seierstad, M., Bonaventure, P., Dvorak, L., Lord, B., Miller, K.L., Motley, S.T., Nepomuceno, D., Chai, W., Dvorak, C.A., Jablonowski, J. A. , Rudolph, D. A., Shah, C.R., Swanson, D.M., Wong, V.D., Axe, F.U., Lovenberg, T.M. and Carruthers, N.I. (2007) Small molecule Y2 receptor an tagonist: Identification of a novel potent series using pharmacophore-based virtual screening. 23th ACS National Meeting, Boston, 19-23 August 2007, 2009.
- [127] Doods, H., Gaida, W., Wieland, H.A., Dollinger, H., Schnorrenberg, G., Esser, F., Engel, E., Eberlein, W. and Rudolf, K. (1999) BIIE0246: A selective and high affinity neuropeptide YY2 receptor antagonist. European Journal of Pharmacology, 384, R3-R5. doi:10.1016/S0014-2999(99)00650-0
- [128] Criscione, L., Rigollier, P., Batzl-Hartmann, C., Rueger, H., Stricker-Krongrad, A., Wyss, P., Brunner, L., Whitebread, S., Yamaguchi, Y., Gerald, C., Heurich, R.O., Walker, M.W., Chiesi, M., Schilling, W., Hofbauer K.G., and Levens, N. (1998) Food intake in free-feeding and energy-deprived lean rats is mediated by the neuropeptide Y5 receptor. The Journal of Clinical Investigation, 102, 2136-2145. doi:10.1172/JCI4188
- [129] Rueeger, H., Rigollier, P., Yamaguchi, Y., Schmidlin, T., Schilling, W., Criscione, L., Whitebread, S., Chiesi, M., Walker, M.W., Dhanoa, D., Islam, I., Zhang, J. and Gluchowski, C. (2000) Design, synthesis and SAR of a series of 2-substituted 4-amino quinazo-line.neuropeptide Y Y5 receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 10, 1175-1179. doi:10.1016/S0960-894X(00)00177-3
- [130] Kanatani, A., Ishihara, A., Iwaasa, M., Nakamura, K., Okamato, O., Hidaka, M., Ho, J., Fukuroda, T., MacNeil, D.J., van der Ploeg, L.H., Ishii, Y., Okabe, T., Fukami, T. and Ihara, M. (2000) L-152,804: Orally active and selective neuropeptide Y Y5 receptor antagonist. Biochemical and Biophysical Research Communications, 272, 169-173. doi:10.1006/bbrc.2000.2696
- [131] Nguyen, A.D., Mitchell, N.F., Lin, S., Yulyanigsih, E., Baldock, P.A., Enriquez, R.F., Zhang, L., Shi, Y-C., Zolotukhin, S., Herzog, H. and Sainsbury, A. (2012) Y1 and Y5 receptors are both required for the regulation of food intake and energy homeostasis in mice. PLoS ONE, 7, e40191.
- [132] Iida, T., Satoh, H., Maeda, K., Yamamoto, Y., Asakawa, K.-I., Sawada, N., Wada, T., Kadowaki, C., Itoh, T., Mase, T., Weissmann, S.A., Tschaen, D., Krska, S. and Volante, R.P. (2005) Practical synthesis of a new neuropeptide y antagonist via stereoselective addition to a ketene. The Journal of Organic Chemistry, 70, 9222- 9229. doi:10.1021/jo0512709
- [133] Sakamoto, T., Moriya, M., Haga, Y., Takahashi, T., Shibata, T., Okamoto, O., Nonoshita, K., Kitazawa, H., Hidaka, M., Gomori, A., Iwaasa, H., Ishihara, A., Kanatani, A., Fukami, T., Gao, Y.-D., MacNeil, D.J. and Yang, L. (2009) Identification of novel and orally active spiroindoline NPY Y5 receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 19, 1564-1568. doi:10.1016/j.bmcl.2009.02.035
- [134] Tao, Y.-X. (2010) The melanocortin-4 receptor: Physiology, pharmacology and pathophysiology. Endocrine Reviews, 31, 506-543. doi:10.1210/er.2009-0037
- [135] Rodriguez, S., Gaunt, T.R., Abdollahi, M.R., Sonnenberg, S. and Day, N.M. (2007) Syndrome: Networks of genotype. Determined feedback loop setpoints. In: Batone, T.E., Ed., Metabolic Syndrome Research Trends, Nova Science Publishers, New York, 2007, pp. 1-63.
- [136] Gantz, I., Konda, Y., Tashiro, T., Shimoto, Y., Miwa, H., Munzert, G., Watson, S.J., DelValle, J. and Yamada, T. (1993) Molecular cloning of a melanocortin receptor. The Journal of Biological Chemistry, 268, 8246-8250.
- [137] Roselli-Rehfuss, L., Mountjoy, K.G., Robbins, L.S., Mortrud, M.T., Low, M.J., Tatro, J.B., Entwistle, M.L., Simerly, R.B. and Cone, R.D. (1993) Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proceedings of the National Academy of Sciences of the United States of America, 90, 8856-8860. doi:10.1073/pnas.90.19.8856
- [138] Mountjoy, K.G., Mortrud, M.T., Low, M.J., Simerly, R.B. and Cone, R.D. (1994) Localization of the melanocortin- 4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Molecular Endocrinology, 8, 1298-1308. doi:10.1210/me.8.10.1298
- [139] Mul, J.D., van Boxtel, R., Bergen, D.J.M., Brans, M.A.D., Brakkee, J.H., Toonen, P.W., Garner, K.M., Adan, R. A.H. and Cuppen, E. (2012) Melanocortin receptor 4 deficiency affects body weight regulation, grooming behavior, and substrate preference in the rat. Obesity, 20, 612-621. doi:10.1038/oby.2011.81
- [140] Branson, R., Potoczna, N., Kral, J.G., Lentes, K.-U., Hoehe, M.R. and Horber, F.F. (2003) Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. The New England Journal of Medicine, 348, 1096-1103. doi:10.1056/NEJMoa021971
- [141] Hinney, A., Bettecken, T., Tarnow, P., Brumm, H., Reichwald, K., Lichtner, P., Scherag, A., Nguyen, T.T., Schlumberger, P., Rief, W., Vollmert, C., Illig, T., Wichmann, H.-E., Schafer, H., Platzer, M., Biebermann, H., Meitinger, T. and Hebebrand, J. (2006) Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population based sample of obese adults from Germany. The Journal of Clinical Endocrinology & Metabolism, 91, 1761-1769. doi:10.1210/jc.2005-2056
- [142] Renè, P, Le Gouill, C., Pogozheva, I.D., Lee, G., Mosberg, H.I., Farooqi, I.S., Valenzano, K.J. and Beuvier, M. (2010) Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. Journal of Pharmacology and Experimental Therapeutics, 335, 520-532. doi:10.1124/jpet.110.172098
- [143] Mountjoy, K.G. (2010) Functions for pro-opiomelanocortin-derived peptides in obesity and diabetes. Biochemical Journal, 428, 305-324. doi:10.1042/BJ20091957
- [144] Boyce, R.S. and Duhl, D.M. (2004) Melanocortin 4 receptor agonists for the treatment of obesity. Current Opinion in Investigational Drugs, 5, 1063-1071.
- [145] Chai, B., Pogozheva, I., Lai, Y., Li, J., Neubig, R., Mosberg, H. and Gantz, I. (2005) Receptor antagonist interactions in the complex of agouti and agouti related protein with human melanocortin 1 and 4 receptors. Biochemistry, 44, 3418-3431. doi:10.1021/bi0478704
- [146] McNulty, J.C., Jackson, P.J., Thompson, D.A., Chai, B., Gantz, I., Barsh, G.S., Dawson P.E. and Millhauser, G.L. (2005) Structures of the Agouti signaling protein. Journal of Molecular Biology, 346, 1059-1070. doi:10.1016/j.jmb.2004.12.030
- [147] Wolff, G.L., Roberts, D.W. and Mountjoy, K.G. (1999) Physiological consequences of ectopic agouti gene expression: The yellow obese mouse syndrome. Physiological Genomics, 1, 151-163.
- [148] Yang, Y., Chen, M., Lai, Y., Gantz, I., Yagmurlu, A., Georgson, K.E. and Harmon, C.M. (2003) Molecular determination of Agouti-related protein binding to human melanocortin-4 receptor. Molecular Pharmacology, 64, 94-103. doi:10.1124/mol.64.1.94
- [149] Silverman, A.P., Kariolis, M.S. and. Cochran, J.R. (2011) Cystine-knot peptides engineered with specifities for aIIbβ3 or aIIbβ and avβ3 integrins are potent inhibitors of platelet aggregation. Journal of Molecular Recognition, 24, 127-135. doi:10.1002/jmr.1036
- [150] Elias, C.F., Saper, C.B., Maratos-Flier, E.,. Tritos, N.A., Lee, C., Kelly, J., Tatro, J.B., Hoffmann, G.E., Ollmann, M.M., Barsh, G.S., Sakurai, T., Yanagisawa, M. and Elmquist, J.K. (1998) Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. Journal of Comparative Neurology, 402, 442-459. doi:10.1002/(SICI)1096-9861(19981228)402:4<442::AID-CNE2>3.0.CO;2-R
- [151] Biebermann, H., Kühnen, P., Kleinau, G. and Krude, H. (2012) The Neuroendocrine circuit controlled by POMC. MSH, AGRP. In: Joost, H.-G., Ed., Appetite Control, Handbook of Experimental Pharmacology, Springer-Verlag, Berlin, 47-75.
- [152] Banno, R., Arima, H., Hayashi, M., Goto, M., Watanabe, M., Sato, I., Ozaki, N., Nagasaki, H., Ozaki, N. and Oiso, Y. (2007) Central administration of melanocortin agonist increased insulin sensitivity in diet-induced obese rats. FEBS Letters, 581, 1131-1136. doi:10.1016/j.febslet.2007.02.019
- [153] Chai, B., Li, J.Y., Zhang, W., Wang, H. and Mulholland, M.W. (2009) Melanocortin-4 receptor activation inhibits c-Jun N-terminal kinase activity and promotes insulin signalling. Peptides, 30, 1098-1104. doi:10.1016/j.peptides.2009.03.006
- [154] Martin, W.J. and Macintyre, D.E. (2004) Melanocortin receptors and erectile function. European Urology, 45, 706-713. doi:10.1016/j.eururo.2003.03.001
- [155] Bednarek, M.A., MacNeil, T., Kalyani, R.N., Tang, R., van der Ploeg, L.H. and Weinberg, D.H. (2001) Selective, high affinity peptide antagonists of alphamelanotropin action at human melanocortin receptor 4: Their synthesis and biological evaluation in vitro. Journal of Medicinal Chemistry, 44, 3665-3672.
- [156] Tucci, F.C., White, N.S., Markison, S., Joppa, M., Tran, J. A., Fleck, B.A., Madan, A., Dyck, B.P., Parker, J., Pontillo, J., Arellano, L.M., Marinkovic, D., Jiang, W., Chen, C.W., Gogas, K.R., Goodfellow, V.S., Saunders, J., Foster, A.C. and Chen, C. (2005) Potent and orally active non-peptide antagonists of human melanocortin-4 receptor based on a series of trans-2-disubstituted cyclohexylpiperazines. Bioorganic & Medicinal Chemistry Letters, 15, 4389-4395. doi:10.1016/j.bmcl.2005.06.071
- [157] Tran, J.A., Pontillo, J., Arellano, M., Fleck, B.A., Marinkovic, D., Tucci, F.C., Chen, C.W., Saunders, J., Foster, A.C. and Chen, C. (2005) Structure-activity relationship of a series of cyclohexylpiperidines bearing an amide side chain as antagonists of the human melanocortin-4 receptor. Bioorganic & Medicinal Chemistry Letters, 15, 3434-3438. doi:10.1016/j.bmcl.2005.05.017
- [158] Arasasingham, P.N., Fotsch, C., Ouyang, X., Norman, M. H., Kelly, M.G., Stark, K.L., Karbon, B., Hale, C., Baumgartner, J.W., Zambrano, M., Cheetham, J. and Tamayo, N.A. (2003) Structure-activity relationship of (1-aryl-2- piperazinylethyl)piperazines: Antagonists for the AGRP/ melanocortin receptor binding. Journal of Medicinal Chemistry, 46, 9-11. doi:10.1021/jm0255522
- [159] Douglass, J., McKinzie, A.A. and Couceyro, P. (1995) PCR differential display identifies a rat brain mRNA that is transcriptionally regulated by cocaine and amphetamine. The Journal of Neuroscience, 15, 2471-2481.
- [160] Rogge, G., Jones, D., Hubert, G.W., Lin, Y. and Kuhar, M.J. (2008) CART peptides: Regulators of body weight, reward and other functions. Nature Reviews Neuroscience, 9, 747-758. doi:10.1038/nrn2493
- [161] Lakatos, A., Prinster, S., Vicentic, A., Hall, R. and Kuhar, M.J. (2005) Cocaine-and amphetamine-regulated transcript (CART) peptide activates the extracellular signalregulated kinase (ERK) pathway in AtT20 cells via putative G-protein coupled receptors. Neuroscience Letters, 384, 198-202. doi:10.1016/j.neulet.2005.04.072
- [162] Jones, D.J. and Kuhar, M.J. (2008) CART receptor binding in primary cell culture of rat nucleus accumbens. Synapse, 62, 122-127. doi:10.1002/syn.20476
- [163] Maletinska, L., Maixnerova, J., Matyskova, R., Haugvicova, R., Sloncova, E., Elbert, T., Slaninova, J. and Zelezna, B. (2007) Cocaineand amphetamine-regulated transcript (CART) peptide specific binding in pheochromocytoma cells PC12. European Journal of Pharmacology, 559, 109-114. doi:10.1016/j.ejphar.2006.12.014
- [164] Kastin, A. and Akerstrom, V. (1999) Entry of CART into brain is rapid but not inhibited by excess CART or leptin. American Journal of Physiology, 277, E901-E904.
- [165] Del Guidice, E.M., Santoro, N., Cirillo, G., D’Urso, L., Di Toro, R. and Perrone, L. (2001) Mutational screening of the CART gene in obese children: Identifying a mutation (Leu34Phe) associated with reduced resting energy expediture and co-segregating with obesity phenotype in a large family. Diabetes, 50, 2157-2160. doi:10.2337/diabetes.50.9.2157
- [166] Hunter, R.G., Philpot, K., Vicentic, A., Dominguez, G., Hubert, G.W. and Kuhar, M.J. (2004) CART in feeding and obesity. Trends in Endocrinology & Metabolism, 15, 454-459. doi:10.1016/S1043-2760(04)00220-6
- [167] Lin, Y., Hall, R.A. and Kuhar, M.J. (2011) CART peptide stimulation of G-protein-mediated signalling in differentiated PC12 cells: Identification of PACAP 6-38 as a CART receptor antagonist. Neuropeptides, 45, 351-358 doi:10.1016/j.npep.2011.07.006
- [168] Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R.M., Tanaka, H., Williams, H.C., Richardson, J.A., Kozlowski, G.P., Wilson, S., Arch, J.R., Buckingham, R.E., Haynes, A.C., Carr, S.A., Annan, R.S., McNulty, D.E., Liu, W.S., Terrett, J.A., Elsbourbagy, N.A., Bergsma, D.J. and Yanasigawa, M. (1998) Orexins and orexin receptors: A family of hypothalamic neuropeptides and G-protein coupled receptors that regulate feeding behaviour. Cell, 92, 573-585. doi:10.1016/S0092-8674(00)80949-6
- [169] De Lecea, L. and Sutcliffe, J.G. (1999) The hypocretins/ orexins: Novel hypothalamic neuropeptides involved in different physiological systems. Cellular and Molecular Life Sciences, 56, 473-480. doi:10.1007/s000180050446
- [170] Spinazzi, R., Andreis, P.G., Rossi, G.P. and Nussdorfer, G.G. (2006) Orexins in the regulation of the hypothalamic-pituitary-adrenal-axis. Pharmacological Reviews, 58, 46-57. doi:10.1124/pr.58.1.4
- [171] Kilduff , T.S. and Peyron, C. (2000) The hyocretin/orexin ligand-receptor system: Implications for sleep and sleep disorders. Trends in Neurosciences, 23, 359-365. doi:10.1016/S0166-2236(00)01594-0
- [172] Espana, R.A. and Scammell, T.E. (2004) Sleep neurobiology for the clinician. Sleep, 27, 811-820.
- [173] Tsujino, N. and Sakurai, T. (2009) Orexin/hypocretin: A neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacological Reviews, 61, 162- 176. doi:10.1124/pr.109.001321
- [174] Bagdoyan, H.A. and Lydic, R. (2012) The neurochemistry of sleep and wakefulness in. In: Brady, T., Siegel, G.J., Albers, R.W. and Price, D.L., Eds., Basic Neurochemistry, 8th Edition, Elsevier, Amsterdam, 982-999.
- [175] Overeem, S., Lammers, G.J. and Tafti, M. (2007) Disorders of sleep and circadian rhythm. In: Waxman, S.G., Ed., Molecular Neurology, Elsevier, Academic Press, Burlington, San Diego, London, 409-426.
- [176] Foutz, A.S., Mitler, M.M., Cavalli-Sforza, L.L., Dement, W.C. (1979) Genetic factors in canine narcolepsy. Sleep, 1, 413-421.
- [177] Baker, T.L. (1985) Sleep apnea disorders. Introduction to sleep and sleep disorders. Medical Clinics of North America, 69, 1123-1152.
- [178] Lin, L., Faraco, F., Li, R., Kadotani, H., Rogers, W., Lin, X., Qiu, X., de Jong, P.J., Nishino, S. and Mignot, E. (1999) The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin receptor 2 gene. Cell, 98, 365-376. doi:10.1016/S0092-8674(00)81965-0
- [179] Nishino, S., Ripley, B., Overeem, S., Nevsimalova, S., Lammers, G.J., Vankova, J., Okun, M., Rogers, W., Brooks, S. and Mignot, E. (2001) Low cerebrospinal fluid hypocretin (orexin) and altered energy homeostasis in human narcolepsy. Annals of Neurology, 50, 381-388. doi:10.1002/ana.1130
- [180] Ripley, B., Overeem, S., Fujiki, N., Nevsimalova, S., Uchino, M., Yesavage, J., Di Monte, D., Dohi, K., Melberg, A., Lammers, G.J., Nishida, Y., Roelandse, F. W.C., Hungs, M., Mignot, E. and Nishino, S. (2001) CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology, 57, 2253-2258. doi:10.1212/WNL.57.12.2253
- [181] Scammell, T.E. and Winrow, C. (2011) Orexin receptors: Pharmacology and therapeutic opportunities. Annual Review of Pharmacology and Toxicology, 51, 243-266. doi:10.1146/annurev-pharmtox-010510-100528
- [182] Holmqvist, T., Johansson, L., Ostman, M., Ammoun, S., Akerman, K.E. and Kukkonen, J.P. (2005) OX1 orexin receptor couple to adenylyl cyclase regulation via multiple mechanisms. The Journal of Biological Chemistry, 280, 6570-6579. doi:10.1074/jbc.M407397200
- [183] Faedo, S., Perdona, E., Antolini, M., di Fabio, R., Pich, E.M. and Corsi, M. (2012) Functional and binding kinetic studies make a distinction between OX1 and OX2 orexin receptor antagonists. European Journal of Pharmacology, 692, 1-9.
- [184] Heifetz, A., Morris, G.B., Biggin, P.C., Barker, O., Fryatt, T., Bentley, J., Hallett, D., Manikowski, D., Pal, S., Reifegerste, R., Slack, M. and Law, R. (2012) Study of human orexin-1 and -2 G-protein-coupled receptors with novel and published antagonists by modeling, molecular dynamics simulation, and site-directed mutagenesis. Biochemistry, 51, 3178-3197. doi:10.1021/bi300136h
- [185] Whitman, D.B., Cox, C.D., Breslin, M.J., Brashear, K.M., Schreier, J.D., Bogusky, M.J., Bednar, R.A., Lemaire, W., Bruno, J.G., Hartman, G.D., Reiss, D.R., Harrell, C.M., Kraus, R.L., Li, Y., Garson, S.L., Doran, S.M., Prueksaritanont, T., Li, C., Winrow, C.J., Koblan, K.S., Renger, J.J. and Coleman, P.J. (2009) Discovery of a potent CNS penetrant orexin receptor antagonist based on an N,Ndisubstituted-1,4-diazepane scaffold that promotes sleep in rats. ChemMedChem, 4, 1069-1074. doi:10.1002/cmdc.200900069
- [186] Haynes, A.C., Jackson, B., Chapman, H., Tadayyon, M., Johns, A., Porter, R.A. and Arch, J.R. (2000) A selective orexin-1 receptor antagonist reduces food consumption in male and female rats. Regulatory Peptides, 96, 45-51. doi:10.1016/S0167-0115(00)00199-3
- [187] Putula, J. and Kukkonen, J.P. (2012) Mapping of the binding sites for OX1 orexin receptor antagonist, SB- 334867, using orexin/hypocretin receptor chimaeras. Neuroscience Letters, 506, 111-115. doi:10.1016/j.neulet.2011.10.061
- [188] Malherbe, P., Borroni, E., Gobbi, L., Knust, H., Nettenkoven, M., Pinard, E., Roche, O., Roger Evans, M., Wettstein, J.G. and Moreau,, J.-L. (2009) Biochemical and behavioral characterization of EMPA, a novel high-affinity, selective antagonist for the OX2 receptor. British Journal of Pharmacology, 156, 1326-1341. doi:10.1111/j.1476-5381.2009.00127.x
- [189] Nagasaki, H., Chung, S., Dooley, C.T., Wang, Z., Li, C., Saito, Y., Clark, S.D., Houghten, R.A. and Civelli, O. (2009) The pharmacological properties of a novel MCH1 receptor antagonist isolated from combinatorial libraries. European Journal of Pharmacology, 602, 194-202. doi:10.1016/j.ejphar.2008.10.068
- [190] Kawauchi, H., Kawazoe, I., Tsubokawa, M., Kishida, M., and Baker, B.I. (1983) Characterization of melanin-concentrating hormone in chum salmon pituitaries. Nature, 305, 321-323. doi:10.1038/305321a0
- [191] Pissios, P., Ozcan, U., Kokkotou, T., Liew, C.W., Liu, S., Peters, J.N., Dahlgren, G., Karamchandani, J., Kudva, Y. C., Kurpad, A.J., Kennedy, R.T., Marathos-Flier, E. and Kulkarni, R.N. (2007) Melanin-concentrating hormone is anovel regulator of islet function and growth. Diabetes, 56, 311-319. doi:10.2337/db06-0708
- [192] Shimada, M., Tritos, N.A., Lowell, B.B., Flier, J.S. and Maratos-Flier, E. (1998) Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature, 396, 670-674. doi:10.1038/25341
- [193] Kreienkamp, D.H., Weise, C., Buck, F. and Richter, D. (1999) Identification of melanin concentrating hormone as the natural ligand for the orphan somatostatin-like receptor (SLC-1). FEBS Letters, 457, 522-524. doi:10.1016/S0014-5793(99)01092-3
- [194] Chambers, J., Ames, R.S., Bergsma, D., Muir, A., Fitzgerald, L.R., Hervieu, G., Dytko, G.M., Foley, J.J., Martin, J., Liu, W.S., Park, J., Ellis, C., Gangully, S., Konchar, S., Cluderay, J., Leslie, R., Wilson, S. and Sarau, H.M. (1999) Melanin concentrating hormone ist the cognate ligand for the orphan G-protein coupled receptor SLC-1. Nature, 400, 261-265. doi:10.1038/22313
- [195] Lembo, P.M., Grazzini, E., Cao, J., Hubatsch, D.A., Pelletier, M., Hoffert, C., Stonge, S., Pou, C., Labrecque, J., Groblewski, T., O’Donnell, O., Payza, K., Ahmad, S. and Walker, P. (1999) The receptor for the orexigenic peptide melanine concentrating hormone in a G-protein coupled receptor. Nature Cell Biology, 1, 267-271. doi:10.1038/12978
- [196] Saito, Y., Nothacker, H.P., Wang, Z, Lin, S.H., Leslie, F. and Civelli, O. (1999) Molecular characterization of the melanin-concentrating-hormone receptor. Nature, 400, 265- 269. doi:10.1038/22321
- [197] Saito, Y. and Nagasaki, H. (2008) The melanin-concentrating hormone system and its physiological function. In: Civelli, O. and Zhou, Q.-Y., Eds., Results and Problems in Cell Differentiation (46) Orphan G-Protein Coupled Receptors and Novel Neuropeptides, Springer-Verlag, Berlin, 159-176. doi:10.1007/400_2007_052
- [198] Chung, S., Parks, G.S., Lee, C. and Civelli, O. (2011) Recent updates on the melanin-concentrating hormone (MCH) and its receptor system: Lessons from MCH1R antagonists. Journal of Molecular Neuroscience, 43, 115- 121. doi:10.1007/s12031-010-9411-4
- [199] Saito, Y., Cheng, M., Leslie, F.M. and Civelli, O. (2001) Expression of the melanin-concentrating hormone (MCH) receptor mRNA in the rat brain. Journal of Comparative Neurology, 435, 26-40. doi:10.1002/cne.1191
- [200] Kokkotou, E.G., Tritos, N.A., Mastaitis, J.W., Slieker, L., and Maratos-Flier, E. (2001) Melanin concentrating hormone receptor is a target of leptin action in the mouse brain. Endocrinology, 142, 680-686. doi:10.1210/en.142.2.680
- [201] Wermter, A.-K., Reichwald, K., Büch, T., Geller, F., Platzer, C., Huse, K., Hess, C., Remschmidt, H., Gudermann, T., Preibisch, G., Siegfried, W., Goldschmidt, H.-P., Li, W.-D., Price, R.A., Biebermann, H., Krude, H., Vollmert, C., Wichmann, H.E., Illig, T., Sorensen, T.I., Astrup, A., Hingstrup Larsen, L., Pedersen, O., Eberle, D., Clement, K., Blundell, J., Wabitsch, M., Schäfer, H., Platzer, M., Hinney, A. and Hebebrand, J. (2005) Mutation analysis of the MCHR1 gene in human obesity. European Journal of Endocrinology, 152, 851-862. doi:10.1530/eje.1.01917
- [202] Hill, J., Duckworth, M., Murdock, P., Rennie, G., SabidoDavid, C., Ames, R.S., Szekeres, P., Wilson, S., Bergsma, D.J., Gloger, I.S., Levy, D.S., Chambers, J.K. and Muir, A.I. (2001) Molecular cloning and functional characterization of MCH2, a novel human MCH receptor. The Journal of Biological Chemistry, 276, 20125-20129. doi:10.1074/jbc.M102068200
- [203] Sailer, A.W., Sano, H., Zeng, Z., McDonald, T.P., Pong, S.S., Feighner, S.D., Tan, C.P., Fukami, T., Iwaasa, H., Hreniuk, D.L., Morin, N.R., Sadowski, S.J., Ito, M., Bansal., A., Ky, B., Figueroa, D.J., Jiang, Q., Austin, C.P., MacNeil, D.J., Ishihara, A., Ihara, M., Kanatani, A., van der Ploeg, L.H., Howard, A.D. and Liu, Q. (2001) Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proceedings of the National Academy of Sciences of the United States of America, 98, 7564-7569. doi:10.1073/pnas.121170598
- [204] Hawes, B.E., Green, B., O’Neill, K., Fried, S. and Graziano, M. P. (2000) The melanin-concentrating hormone couples to multiple tG protein to activate diverse intracellular signaling pathways. Endocrinology, 141, 4524- 4532. doi:10.1210/en.141.12.4524
- [205] An, S., Cutler, G., Zhao, J.J., Huang, S.-G., Tian, H., Li, W., Liang, L., Rich, M., Bakleh, A., Du, J, Chen, J.-L. and Dai, K. (2001) Identification and characterization of a melanin concentrating hormone receptor. Proceedings of the National Academy of Sciences of the United States of America, 98, 7576-7581. doi:10.1073/pnas.131200698
- [206] Mihalic, J.T., Chen, X., Fan, P., Chen, X., Fu, Y., Liang, L., Reed, M., Tang, L., Chen, J.-L., Jaen, J., Li, L. and Dai, K. (2011) Discovery of a novel series of melaninconcentrating hormone receptor 1 antagonists for the treatment of obesity. Bioorganic & Medicinal Chemistry Letters, 21, 7001-7005. doi:10.1016/j.bmcl.2011.09.110
- [207] Hervieu, G.I., Cluderay, J.E., Harrison, D., Meakin, J., Maycox, P., Nasir, S. and Leslie, R.A. (2000) The distribution of the mRNA and protein products of the melanin concentrating hormone receptor gene, slc-1, in the central nervous system of the rat. European Journal of Neuroscience, 12, 1194-1216. doi:10.1046/j.1460-9568.2000.00008.x
- [208] Mori, M., Harada, M., Terao, Y., Sugo, T., Watanabe, T., Shimomura, Y., Abe, M., Shintani, Y., Onda, H., Nishimura, O. and Fujino, M. (2001) Cloning of a novel G protein coupled receptor, SLT, a subtype of melaninconcentrating hormone receptor. Biochemical and Biophysical Research Communications, 283, 1013-1018. doi:10.1006/bbrc.2001.4893
- [209] Qu, D., Ludwig, D.S., Gammeltoft, S., Piper, M., Pelleymounter, M.A., Cullen, M.J., Mathes, W.F., Przypek, J., Kanarek, R. and Maratos-Flier, E. (1996) A role for melanin concentrating hormone in the central regulation of feeding behaviour. Nature, 380, 243-247. doi:10.1038/380243a0
- [210] Ludwig, D.S., Mountjoy, K.G., Tatro, J.B., Gilette, J.A., Frederich, R.C., Flier, J.S. and Maratos-Flier, E. (1998) Melanin-concentrating hormone: A functional melanocortin antagonist in the hypothalamus. Endocrinology and Metabolism: American Journal of Physiology, 274, E627- E633.
- [211] Alon, T. and Friedman, J.M. (2006) Late-onset leaness in mice with targeted ablation of melanin concentrating hormone neurons. The Journal of Neuroscience, 26, 389- 397. doi:10.1523/JNEUROSCI.1203-05.2006
- [212] Mendez-Andino, J.L. and Wos, J.A. (2007) MCHR1 antagonists: What is keeping most research programs away from the clinic. Drug Discovery Today, 12, 972-979. doi:10.1016/j.drudis.2007.08.010
- [213] Andersen, D., Storz, T., Liu, P., Wang, X., Li, L., Fan, P., Chen, X., Allgeier, A., Burgos, A., Tedrow, J., Baum, J., Chen, Y., Crockett, R., Huang, L., Syed, R., Larsen, R.D. and Martinelli, M.J. (2007) Stereoselective synthesis of a MCH1R antagonist. The Journal of Organic Chemistry, 72, 9648-9655. doi:10.1021/jo701894v
- [214] Rokosz, L.L. (2007) Discovery and development of melanin-concentrating hormone receptor 1 antagonists for the treatment of obesity. Expert Opinion on Drug Discovery, 2, 1301-1327. doi:10.1517/17460441.2.10.1301
- [215] Gehlert, D.R., Rasmussen, K., Shaw, J., Li, X., Ardayfio, P., Craft, L., Coskun, T., Zhang, H.Y., Chen, Y. and Witkin, J.M. (2009) Preclinical evaluation of melanin-concentrating hormone receptor 1 antagonism for the treatment of obesity and depression. Journal of Pharmacology and Experimental Therapeutics, 329, 429-438.
- [216] Chaki, S., Funakoshi, T., Hirota-Okuno, S., Nishiguchi, M., Shimazaki, Iijima, M., Grottick, A.J., Kanuma, K., Omodera, K., Sekiguchi, Y., Okuyama, S., Tran, T.-A., Semple, G. and Thomsen, W. (2005) Anxiolyticand antidepressant-like profile of ATC0065 and ATC0175: Nonpeptidic and orally active melanin-concentrating hormone receptor 1 antagonists. Journal of Pharmacology and Experimental Therapeutics, 313, 831-839. doi:10.1124/jpet.104.081711
- [217] Sasmal, P.K., Sasmal, S., Rao, P.T., Venkatesham, B., Roshaiah, M., Abbineni, C., Khanna, I., Jadhav, V.P., Suresh, J., Talwar, R., Muzeeb, S., Receveur, J.M., Frimurer, T.M., Rist, O., Elster, L. and Högberg, T. (2010) Discovery of novel, orally available benzimidazoles as melanin concentrating hormone receptor 1 (MCHR1) antagonists. Bioorganic & Medicinal Chemistry Letters, 20, 5443-5448. doi:10.1016/j.bmcl.2010.07.086
- [218] Sasmal, P.K., Sasmal, S., Abbineni, C., Venkatesham, B., Rao, P.T., Roshaiah, M., Khanna, I., Sebastian, V.J., Suresh, J., Singh, M., Talwar, R., Shashikumar, D., Reddy, K.H., Frimurer, T.M., Rist, O., Elster, L. and Högberg, T. (2011) Synthesis and SAR studies of benzimidazol derivatives as melanin concentrating hormone receptor 1 (MCHR1) antagonists focus to detune hERG inhibition. MedChemComm, 2, 385-389. doi:10.1039/c1md00015b
- [219] Sasmal, S., Balaji, G., Kanna Reddy, H.R., Balasubrahmanyam, D., Srinivas, G., Kyasa, S.K., Khanna, I., Talwar, R., Suresh, J., Jadhav, V.P., Muzeeb, S., Shashikumar, D., Harinder Reddy, K., Sebastian, V.J., Frimurer, T.M., Rist, O., Elster, L. and Högberg, T.D., (2012) Design and optimization of quinazoline derivatives as melanin concentrating hormone receptor 1 (MCHR1) antagonists. Bioorganic & Medicinal Chemistry Letters, 22, 3157-3162. doi:10.1016/j.bmcl.2012.03.050
- [220] Samal, S., Balasubrahmanyam, D., Kanna Reddy, H.R., Balaji, G., Srinivas, G., Cheera, S., Abbineni, C., Sasmal, I., Khanna, P.K., Sebastian, V.J., Jahav, V., Singh, M.P., Talwar, R., Suresh, J., Shashikumar, D., Reddy, K.H., Shihorkar, V., Frimurer, T.M., Rist, O., Elster, K. and Högberg, T. (2012) Design and optimization of quinazoline derivatives as melanin concentrating hormone receptor 1 (MCHR1) antagonist: Part 2. Bioorganic & Medicinal Chemistry Letters, 22, 3163-3167. doi:10.1016/j.bmcl.2012.03.049
- [221] Iyengar, R.R., Lynch, J.K., Mulhern, M.M., Judd, A.S., Freeman, J.C., Gao, J., Souers, A.J., Zhao, G., Wodka, D., Falls, H.D., Brodjian, S., Dayton, B.D., Reilly, R.M., Swanson, S., Su, Z., Martzin, R.L., Leitza, S.T., Houseman, K.A., Diaz, G., Collins, C.A., Sham, H.L. and Kym, P.R. (2007) An evaluation of 3,4-methylenedioxy phenyl replacements in the aminoperidine chromone class of MCHr1 antagonists. Bioorganic & Medicinal Chemistry Letters, 17, 874-878. doi:10.1016/j.bmcl.2006.11.065
- [222] Su, J., Tang, H., McKittrick, B.A., Gu, H., Guo, T., Qian, G., Burnett, D.A., Clader, J.W., Greenlee, W.J., Hawes, B.E., O’Neill, K., Spar, B., Weig, B., Kowalski, T. and Sorota, S. (2007) Synthesis of novel bicycle [4.1.0] heptanes and bicycle[3.1.0]hexane derivatives as melaninconcentrating hormone receptor R1 anatgonists. Bioorganic & Medicinal Chemistry Letters, 17, 4845-4850. doi:10.1016/j.bmcl.2007.06.048
- [223] Ando, M., Sekino, E., Haga, Y., Moriya, M., Ito, M., Ito, J., Iwaasa, H., Ishihara, A., Kanatani, A. and Ohtake, N. (2009) Discovery of novel phenethylpyridone derivatives as potent melanin-concentrating hormone 1 receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 19, 5186-5190. doi:10.1016/j.bmcl.2009.07.023
- [224] Kasai, Kamaura, M., Kamata, M., Aso, K., Ogino, H., Nakano, Y., Watanabe, K., Kaisho, T, Tawada, M., Nagisa, Y., Takekawa, S., Kato, K., Suzuki, N. and Ishihara, Y. (2011) Melanin-concentrating hormone receptor 1 antagonists: Synthesis, structure-activity relationship, docking studies. Bioorganic & Medicinal Chemistry, 19, 6261- 6273. doi:10.1016/j.bmc.2011.09.007
- [225] Johansson A. (2011) Recent progress in the discovery of melanin-concentrating hormone 1-receptor antagonists. Expert Opinion on Therapeutic Patents, 21, 905-925. doi:10.1517/13543776.2011.575063
- [226] Luthin, D.R. (2007) Anti-obesity effects of small molecule melanin-concentrating hormone receptor1 (MCHR1) antagonists. Life Sciences, 81, 423-440. doi:10.1016/j.lfs.2007.05.029
- [227] Recanatini, M., Poluzzi, E., Masetti, M., Cavalli, A. and De Ponti, F. (2005) QT prolongation through hERG K+ channel blockade: Current knowledge and strategies for the early prediction during drug development. Medicinal Research Reviews, 25, 133-166.
- [228] Sanguinetti, M.C. and Tristani-Firouzi, M. (2006) hERG potassium channels and cardiac arrhythmia. Nature, 440, 463-469. doi:10.1038/nature04710
- [229] Suzuki, T., Kameda, M., Ando, M., Mayazoe, H., Sekino, E., Ito, S., Masutani, K., Kamijo, K., Takezawa, A., Moriya, M., Ito, M., Ito, J., Nakase, K., Matsushita, H., Ishihara, A., Takenaga, N., Tokita, S., Kanatani, A., Sato, N. and Fukami, T. (2009) Disvovery of novel diarylketoxime derivatives as selective and orally active melanin-concentrating hormone 1 receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 19, 5339-5345. doi:10.1016/j.bmcl.2009.07.132
- [230] Suzuki, T., Moriya, M., Sakamoto, T., Suga, T., Kishino, H., Takahashi, H., Ishikawa, M., Nagai, K., Imai, Y., Sekino, E., Ito, M., Iwaasa, H., Ishihara, A., Tokita, S., Kanati, A., Sato, N. and Fukami, T. (2009) Discovery of novel spiro-piperidine derivatives as highly potent and selective melanin-concentrating hormone 1 receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 19, 3072-3077. doi:10.1016/j.bmcl.2009.04.016
- [231] Mihalic, J.T., Fan, P., Chen, X., Fu, Y., Motani, A., Liu, L., Liang, L., Lindstrom, M., Tang, L., Chen, J.-L., Jaen, J., Dai, K. and Li, L. (2012) Discovery of a novel melanin-concentrating hormone receptor 1 (MCHR1) antagonist with reduced hERG inhibition. Bioorganic & Medicinal Chemistry Letters, 22, 3781-3785. doi:10.1016/j.bmcl.2012.04.006
- [232] Souers, A.J , Wodka, D., Gao, J., Lewis, J.C., Vasudevan, A., Gentles, R., Brodjian, S., Dayton, B., Ogiela, C.A., Fry, D., Hernandez, L.E., Marsh, K.C., Collins, C.A. and Kym, P.R. (2004) Synthesis and evaluation of 2-amino-8- alkoxy quinolines as MCHR1 antagonists, Part 1. Bioorganic & Medicinal Chemistry Letters, 14, 4873-4877. doi:10.1016/j.bmcl.2004.07.032
- [233] Souers, A.J., Iyengar, R.R., Judd, A.S., Gao, J., Zhao, G., Brune, M.E., Napier, J.J., Mulhern, M.M., Lynch, J.K., Freeman, J.C., Wodka, D., Chen, C.J., Falls, H.D., Brodjian, S., Dayton, B.D., Diaz, G.J., Bush, E.N., Shapiro, R., Droz, B.A., Knourek-Segel, V., Hernandez, L.E., Marsh, K.C., Reilly, R.M., Sham, H.L., Collins, C.A. and Kym, P.R. (2007) Constrained 7-fluorocarbochromone-4-aminopiperidine based melanin-concentrating hormone recaptor 1 antagonists. The effects of chirality on substituted indan-1-ylamines. Bioorganic & Medicinal Chemistry Letters, 17, 884-889. doi:10.1016/j.bmcl.2006.11.061
- [234] Vasudevan, A., Wodka, D., Verzal, M.K., Souers, A.J., Gao, J., Brodjian, S., Fry, D., Dayton, B., Marsh, K.C., Hernandez, L.E., Ogiela, C.A., Collins C. A. and Kym, P.R. (2004) Synthesis and evaluation of 2-amino-8- alkoxy quinolins as MCHR1antagonists. Part 2. Bioorganic & Medicinal Chemistry Letters, 14, 4879-4882. doi:10.1016/j.bmcl.2004.07.034
- [235] Vasudevan, A., Verzal, M.K., Wodka, D., Souers, A.J., Blackburn, C., Che, J.L., Lai, S., Brodjian, S., Falls, D.H., Dayton, B.D., Govek, E., Daniels, T., Geddes, B., Marsh, K.C. Hernandez, L.E., Collins, C.A. and Kym, P.R. (2005) Identification of aminopiperidine benzamides as MCHR1 antagonists. Bioorganic & Medicinal Chemistry Letters, 15, 3412-3416. doi:10.1016/j.bmcl.2005.05.023
- [236] Borowsky, B., Durkin, M.M., Ogozalek, K., Marzabadi, M.R., De Leon, J., Lagu, B., Heurich, R., L.ichtblau, H., Shaposhnik, Z., Daniewska, I., Blackburn, T.P., Branchek, T.A., Gerald, C., Vaysse, P.J. and Forray, C. (2002) Antidepressant, anxiolytic an anorectic effects of a melaninconcentrating hormone-1 receptor antagonist. Nature Medicine, 8, 825-830.
- [237] David, D.J., Klemenhagen, K.C., Holick, K.A., Saxe, M.D., Mendez, I., Santarelli, L., Craig, D.A., Zhong, H., Swanson, C.J., Hegde, L.G., Ping, X.L., Dong, D., Marzabadi, M.R., Gerald, C.P. and Hen, R. (2007) Efficacy of the MCHR1 antagonist N-[3-(1-{[4-(3,4-difluoro-phenoxy) phenyl]methyl}(4-piperidyl))-4-methylphenyl]-2-methylpropanamide (SNAP 94847) in mouse models of anxiety and depression following acute and chronic administration is independent of hippocampal neurogenesis. Journal of Pharmacology and Experimental Therapeutics, 321, 237-248. doi:10.1124/jpet.106.109678
- [238] Chen, X., Mihalic, J., Fan, P., Liang, L., Lindstrom, M., Wong, S., Ye, Q., Fu, Y., Jaen, J., Chen, J.-L., Dai, K. and Li, L. (2012) Discovery and characterization of a potent and selective antagonist of melanin-concentrating hormone receptor. Bioorganic & Medicinal Chemistry Letters, 22, 363-366. doi:10.1016/j.bmcl.2011.10.125
- [239] Statnick, M.A., Schober, D.A., Mayne, N.G., Burnett, J.P. and Gehlert, D.R. (1997) Analysis of NPY receptor subtypes in the human frontal cortex reveals abundant Y1 mRNA and binding sites. Peptides, 18, 137-143. doi:10.1016/S0196-9781(96)00246-X
- [240] Irani, B.G., Dunn-Meynell, A.A. and Levin, B.E. (2006) Altered hypothalamic leptin, insulin, and melanocortin binding associated with moderate-fat diet and predisposetion to obesity. Endocrinology, 148, 310-316. doi:10.1210/en.2006-1126
- [241] Audinot, V., Lahaye, S., Beauverger, P., Rodriguez, M., Galizzi, J.P., Fauchere, J.L. and Boutin, J.A. (2001) [125I]-S36057: A new and highly potent radioligand for the melanin-concentrating hormone receptor. British Journal of Pharmacology, 133, 371-378. doi:10.1038/sj.bjp.0704085
- [242] Arai, K., Ohata, H. and Shibasaki, T. (1998) Non-peptidic corticotrophin-releasing hormone receptor type 1 antagonist reverses restraint stress-induced shortening of sodium pentobarbital-induced sleeping time of rats: Evidence that an increase in arousal induced by stress is mediated through CRH receptor type 1. Neuroscience Letters, 255, 103-106. doi:10.1016/S0304-3940(98)00719-8
- [243] McCarthy, J.R., Heinrichs, S.C. and Grigoriadis, D.E. (1999) Recent advances with the CRF1 receptor: Design of small molecule inhibitors, receptor subtypes and clinical indications. Current Pharmaceutical Design, 5, 289- 315.
- [244] Jagoda, E., Contoreggi, C., Lee, M.J., Kao, C.H., Szajek, L.P., Listwak, S., Gold, P., Chrousos, G., Greiner, E., Kim, B.M., Jacobson, A.E., Rice, K.C. and Eckelman, W. (2003) Autoradiographic visualization of corticotrophin releasing hormone type 1 receptors with a nonpeptide ligand: synthesis of [76Br]MJL-1-109-2. Journal of Medicinal Chemistry, 46, 3559-3562. doi:10.1021/jm034077k
- [245] Chen, C., Dagnino Jr., R., De Souza, E.B., Grigoriadis, D.E., Huang, C.Q., Kim, K.I., Liu, Z., Moran, T., Webb, T.R., Whitten, J.P., Xie,Y.F. and McCarthy, J.R. (1996) Design and synthesis of a series of non-peptide highaffinity human corticotropin-releasing factor1 receptor antagonists. Journal of Medicinal Chemistry, 39, 4358- 4360. doi:10.1021/jm960149e
- [246] Whitten, J.P., Xie, Y.F., Erickson, P.E., Webb, T.R., DeSouza, E.B., Grigoriadis, D.E. and McCarthy, J.R. (1996) Rapid microscale synthesis, a new method for lead optimization using robotics and solution phase chemistry: Application to the synthesis and optimization of corticotropin releasing factor 1 receptor antagonists. Journal of Medicinal Chemistry, 39, 4354-4357. doi:10.1021/jm960148m
- [247] Griebel, G., Perrault, G. and Sanger, D.J. (1998) Characterization of the behavioral profile of the non-peptide CRF receptor antagonist CP-154,526 in anxiety models in rodents. Comparison with diazepam and buspirone. Psychopharmacology, 138, 55-66. doi:10.1007/s002130050645
- [248] Erondu, N., Wadden, T., Gantz, I, Musser, B., Nguyen, A.M., Bays, H., Bray, G., O’Neil, P.M.O., Basdevant, A., Kaufman, K.D., Heymsfield, S.B., Amatruda, J.M. (2007) Effect of NPY5R antagonist MK-0557 on weight regain after very-low-calorie diet-induced weight loss. Obesity, 15, 895-905. doi:10.1038/oby.2007.620
- [249] Chaki, S., Hirota, S., Funakoshi, T., Suzuki, Y., Suetake, S., Okubo, T., Ishii, T., Nakazato, A. and Okuyama,S. (2003) Anxiolytic-like and antidepressant-like activities of MCL0129 (1-[(S)-2-(4-fluorophenyl)-2-(4-isopropylpiperadin-1-yl-)ethyl]-4-[4-(2-methoxynaphthalen-1-yl)butyl] piperazine), a novel and potentnonpeptide antagonist of the melanocortin-4 receptor. Journal of Pharmacology and Experimental Therapeutics, 304, 818-826. doi:10.1124/jpet.102.044826
- [250] Kato, K., Terauchi, J., Mori, M., Suzuki, N., Shimomura, Y., Takekawa, S. and Ishihara, Y. (2001) Melanin concentrating hormone antagonist. PCT Application. WO/ 2001/021577. Takeda Chemical Industries, Ltd., March 29.
- [251] Okoyuma, S., Chaki, S., Kawashima, N., Suzuki, Y., Ogawa, S.-I., Nakazato, A., Kumagai, T., Okubo, T. and Tomisawa, K. (1999) Receptor binding, behavioral and electrophysiological profiles of nonpeptide corticotropinreleasing factor subtype 1 receptor antagonists CRA1000 and CRA1001. Journal of Pharmacology and Experimental Therapeutics, 289, 926-935.
- [252] Vidarsdottir, S., Smeets, P.A.M., Eichelsheim, D.L., van Osch, M.J.P., Viergever, M.A., Romijn, J.A., van der Grond, J. and Pijl, H. (2007) Glucose ingestion fails to inhibit hypothalamic neuronal activity in patients with Typ 2 diabetes. Diabetes, 56, 2547-2550. doi:10.2337/db07-0193
- [253] Podingbauer, A. and Ekmekcioglu, C. (2005) Regulation der nahrungsaufnahme: Physiologische mechanismen und klinische relevanz. Journal für Ernährungsmedizin, 7, 22-29.
- [254] Shimizu, H., Inoue, K. and Mori, M. (2007) The leptindependent and -independent melanocortin signaling system: Regulation of feeding and energy expenditure. Journal of Endocrinology, 193, 1-9. doi:10.1677/JOE-06-0144
- [255] Fei, H., Okano, H.J., Li, C., Lee, G.H., Zhao, C., Darnell, R. and Friedman. J.M. (1997) Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proceedings of the National Academy of Sciences of the United States of America, 94, 7001-7005.