Open Journal of Genetics
Vol.3 No.1(2013), Article ID:29373,12 pages DOI:10.4236/ojgen.2013.31008
Key genes of the interleukin 6 signaling pathway are not associated with coronary artery disease in a large European population
![]()
1Department of Internal Medicine III: Cardiology and Angiology, University of Kiel, Kiel, Germany
2Institute for Clinical Molecular Biology, University of Kiel, Kiel, Germany
3Department of Pediatric Neurology, University of Kiel, Kiel, Germany
4Institute of Medical Informatics and Statistics, University of Kiel, Kiel, Germany
5Department of Cardiovascular Surgery, University of Kiel, Kiel, Germany
6Department of Medicine, Christian-Albrechts-Universität Kiel, Kiel, Germany
7Imland Klinikum Rendsburg, Christian-Albrechts-Universität Kiel, Kiel, Germany
Email: #nour-eddine.el-mokhtari@imland.de
Received 3 February 2013; revised 6 March 2013; accepted 16 March 2013
Keywords: Coronary Artery Disease; Genetics; Single Nucleotide Polymorphism; Interleukin 6
ABSTRACT
Background: Recent studies indicate a strong functional relevance of the canonical inflammatory interleukin 6 signaling pathway in coronary artery disease (CAD). A genetic association of this signaling pathway with CAD has not been shown yet. We aimed to assess novel single nucleotide polymorphisms (SNPs) from genes of the Interleukin 6 signaling pathway. Results: To identify novel SNPs that are relevant for CAD, we employed a large-scale population-based case-control association study of 2199 cases and 1715 controls and assessed 73 SNPs from 12 genes out of the IL-6 signaling pathway. Results were adjusted to the CAD-related risk factors diabetes, hypertension, Body Mass Index, smoking and sex by logistic regression analysis. In a primary explorative study, we identified 5 SNPs that were significantly associated with CAD (MAPK1_rs6928, MAPK1_rs9340, MAPK1_ rs11913721, MAPK14_rs7757672, JAK1_rs310236). After adjustment to CAD-risk factors, MAPK1_ rs6928 showed the strongest association with CAD (P 0.0217, Odds Ratio 1.36, Confidence Interval 1.05 - 1.77). To reproduce this result, we performed a replication study employing independent patient and control panels. In this study we could not approve the association of rs6928 with CAD. Conclusion: In conclusion, we did not detect significant associations of SNPs from the IL-6 signaling pathway with CAD. Our investigation demonstrates the importance of independent replication studies to verify results from candidate-gene association studies in the quest to discover the underlying pathomechanism of CAD.
1. INTRODUCTION
Coronary artery disease (CAD) is one of the leading causes of death worldwide [1]. The cause of the disease, atherosclerosis is a systemic disease that is influenced by environmental and behavoural as well as genetic risk factors. The proportion of heritable factors for the individual risk of coronary artery disease has been estimated 30% - 60% [2]. Thus, unraveling the genetic sequence variants that are associated with an increased risk for CAD is an important goal of genetic research. Despite a multitude of studies, only a few gene variants could be credibly demonstrated to be associated with an increased risk of CAD, that would mostly comprise an effect on LDL-cholesterol [3]. Recent genome-wide association studies (GWAS) [4-6] and metanalyses [7] detected several novel sequence variants that could be linked to an increased risk of CAD. These studies demontrated that the analysis of large study populations is needed to discover candidate sequences with modest size effects [7]. Another possibility to discover novel CAD-related sequence variants is the target-oriented analysis of candidate genes in cardiovascular signaling pathways that may potentially mediate increased risk [3]. Overall, common disease variants could be mapped to 34 distinct loci [8]. These studies have proven to be an enormously valuable tool to detect frequent variants with a limited genetic effect [7]. However, GWAS are often restricted when pursuing the goal to discover rare variants with greater genetic effects [9]. This is mainly due to the fact that many GWAS are executed in an underpowered situation due financial limits [9]. In this regard, target-oriented analyses of candidate genes are an important complement to GWAS, since they can use much higher sample numbers and thus provide a more adequate study design to identify rare variants with a stronger genetic effect [9]. Genes that are involved in lipid metabolism [3], stress response [10,11] and inflammation [12,13] have been appealing targets of many studies, however a significant correlation could only be detected in few cases, possibly because study samples have been too small to deliver reproducible data [5].
The IL (interleukin)-6-like cytokines IL-6, IL-11, LIF (leukaemia inhibitory factor), OSM (oncostatin M), ciliary neurotrophic factor, cardiotrophin-1 and cardiotrophin-like cytokine are an essential family of cytokines that play a role as regulators of the acute phase response and inflammation [14]. They act through binding to membrane receptors containing gp130-like receptors, subsequently activating the canonical JAK (Janus Kinase)- STAT (signal transducers and activators of transcription)- pathway as well as the mitogen-activated protein (MAPK) cascade [14]. In the heart, the IL-6/JAK/STAT-signaling pathway plays a fundamental role in ischemia, stressinduced remodeling and cardiomyopathy [15,16]. Moreover, IL-6 seems to have an impact on plaque development and morphology in atherosclerosis [17]. Consistently, IL-6 serum levels are elevated in patients with CAD [18,19]. It thus seems possible that genetic variants of IL-6 are associated with an increased risk of CAD, however several reports on this correlation have provided inconclusive results: While Georges and coworkers could demonstrate an association between the interleukin-6 gene polymorphism (−174 G/C) and susceptibility to myocardial infarction [20], this association could not be confirmed in another study [21].
However, a novel report suggests that assessing the interaction between IL-6 and the coagulation factor 2 receptor (F2R) haplotypes modulates the risk of myocardial infarction [22]. These data implicate, that a candidate gene approach that comprises simoultaneous analysis of genetic variants of IL-6 and interacting molecules, might offer more conclusive information about the relevance of IL-6 in CAD-related risk prediction. We therefore performed a population based casecontrol study assessing the association of 73 Single nucleotide polymorphisms (SNPs) of 12 candidate genes from the IL-6 pathway with CAD. In a second approach, suspect SNPs were verified in a replication study on a different study and control cohorts.
2. MATERIALS AND METHODS
2.1. Study Population
CAD patients involved in this study were recruited from a population-representative collection of unrelated Germans (Table 1). The PopGen biobank (population based assessment of genetic defects [23]), which represents a large population from the northernmost German region Schleswig Holstein, was the source of recruitment. Due to small migration rates, this region offers the advantage of a homogenous genetic structure. Study subjects were identified by screening of all cardioangiogramms from any of the five cardiac catheterizations laboratories of the recruitment area. Subjects who were assigned to the patient cohort, had a significant CAD (at least 70% stenosis in one major epicardial coronary vessel), as demonstrated by coronary catheterization. Following catheterization, more than 90% of the patients had undergone a coronary revas-cularization procedure (percutaneous coronary intervention or coronary artery bypass grafting). The screening period ranged from 01/ 1997 until 10/2004. An age > 65 years at diagnosis was an exclusion criterion. The patient cohort was further subdivided into two groups: patients younger than 55 years (1096 patients) were obtained for an explorative study, patients 55 years or older (1103 patients) were obtained for an independent replication study.
Control subjects for the explorative study (636 subjects) were randomly identified on the basis of data that were supplied by local population registries. Sub-
Table 1. characteristics of patients in the study and control groups.
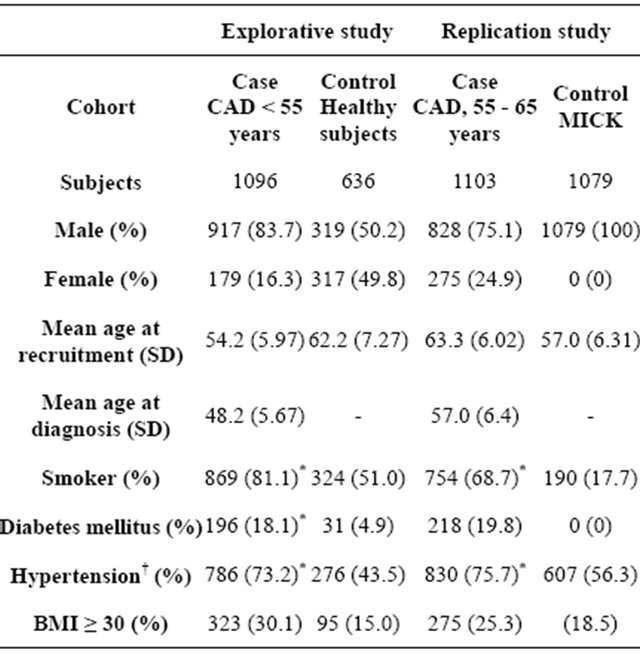
*P < 0.05, †Blood pressure ≥ 140 mmHg systolic or ≥90 mmHg diastolic. An SD: Standard deviation, BMI: Body mass index.
jects with known CAD were excluded from this group. Information on age, gender and general health status was obtained to adequately match controls and patients. For the second replication study, matched controls were obtained from the Metabolic intervention Cohort Kiel (MICK) [24], an independent cohort of the Max Rubner Federal Research Institute of Nutrition and Food. This representative population cohort (1079 subjects) was recruited between 2003 and 2006 in the area of Kiel, Schleswig Holstein, Germany and comprised male subjects that were between 45 and 65 years old. To adjust both studies to these risk factors, logistic regression analysis was applied. Of note, the replication study was not adjusted for diabetes, since this disease was an exclusion criterion for the MICK cohort. Written informed consent was obtained from all participants. Recruitment and experimental protocols were approved by the institutional ethics review board and data protection authorities.
2.2. Object of Investigation
In a population-based case-control study 73 tagging Single-Nucleotide-Polymorphisms (tSNPs) out of 12 candidate genes of the IL-6 pathway were assessed for association with CAD. Selection of tSNPs was aligned to linkage disequilibrium analyses and haplotype distribution models from HapMap CEU-genotypes, one of 11 populations in HapMap phase 3 from the Centre de’ Etude du Polymorphism Humain (CEPH, http://www.ncbi.nlm.nih.gov/SNP/snp_viewTable.cgi?pop=1409). The Haploview 3.2 software [25] was employed for this selection. Significant SNPs were assessed in a second replication study.
2.3. Genotyping
After extraction from blood samples (QIAGEN Flexi Gene DNA Kit, QIAGEN), genomic DNA was amplified by whole genome amplification (GenomiPhi, Amersham, Uppsala, Sweden). Genotyping was performed on an automated platform employing the TaqMan and SNPlex Genotyping Systems (Both Applied biosystems, Foster City, CA, USA). The process was facilitated by the use of Tecan Genesis RSP 150 pipetting robots (TECAN, Maennedorf, Switzerland). Detection of flourescence signals and generation of genotypes was carried out employing the ABI PRISM® 7900 HT Sequence Detector System (Applied Biosystems). The obtained data were further processed using the Data collection software v2.0 and analysed using the GeneMapper analysis Software v3.5.1 (Applied Biosystems) with default settings. Genotypes which gave significant association signals were additionally reviewed manually.
2.4. Statistical Analysis
To rule out possible systematic errors like insufficient genotyping [26] or diverging binding activities of probes in homozygos and heterozygos carriers of an allel, markers were assessed for deviations from the HardyWeinberg-equilibrium [27] in controls before employment in the study (http://ihg.helmholtz-muenchen.de/cqi-bin/hw/hwa2.pl,α=0.05). Association-case-control analysis was performed using Pearson’s chi-squared test [28]. Signficance was verified by a Wald test and by an odds ratio test. Logistic regression analysis was performed to assess the influence of covariants like sex, hypertension, body mass index (BMI), smoking and diabetes. Power calculation was performed for uncorrected χ2 tests as implemented for an independent and retrospective case-control study design in the software PS Power and Sample Size Calculations [29]. If the true odds ratio for disease in exposed subjects relative to unexposed subjects is 1.8, a putative risk allele of a minor allele frequency (MAF) of 5% given, in our explorative study, we were able to reject the null hypothesis that this odds ratio equals 1 with probability (power) 0.8. The Type I error probability associated with this test of this null hypothesis is 0.05. For more common alleles, e.g. 40% frequency in the general population, we were able to detect a genetic effect of 1.3 (Supplementary Figure S1).
3. RESULTS
3.1. Study Population
In this study we aimed to identify variants that were associated with CAD. In a primary explorative study 73 tagging SNPs from 12 candidate genes (Figure 1, Table 1) within the IL-6 signaling pathway were tested for association with CAD in a large population of 1096 early-onset CAD patients (age of disease onset < 55 years) and 636 ethnically matched healthy controls (Table 2). SNPs that gave significant evidence for an association with CAD were subsequently replicated in a second independent study population. This population comprised 1103 patients with age at disease onset between 55 and 65 years. The control cohort for this study population consisted of 1079 healthy, ethnically matched men (mean age of 57 years) who were tested free of diabetes. Collectively, we detected a significant preponderance of classical CAD-related risk factors (diabetes, hypertension, Body Mass Index, smoking and sex) in both study populations vs. control groups.
3.2. Explorative Study
In the primary explorative study, 1086 out of 1096 CAD patients and 631 out of 636 healthy controls were
Table 2. 73 tSNPs of 12 genes from the IL-6 pathway that were assesseds in the explorative study.

Continued

Continued
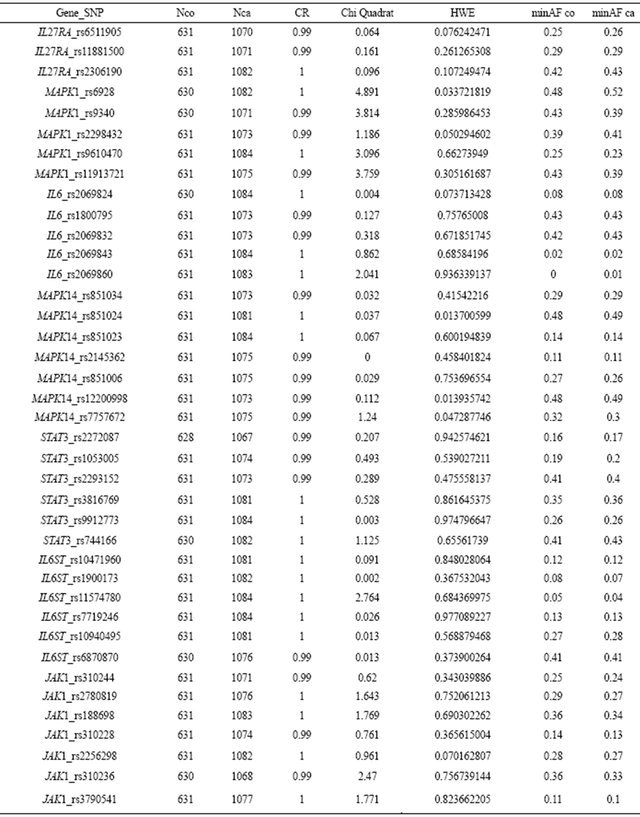
successfully genotyped. All 73 tested tSNPs reached a call rate of ≥95% and a PHWE > 0.01. Three out of these 73 SNPs (rs2069843, IL-6; rs2069860, IL-6; rs11574780, IL6ST) did not meet the required minor allele frequency (minAF) of >5% and were therefore excluded from further analysis (Table S1). In the next stage of the study, association analyses and subsequent adjustment for CAD-related risk factors resulted in the detection of 5 SNPs that showed significant association with CAD (Table 3). Interestingely, three out of these five SNPs were located in the MAPK1 gene encoding the Mitogen-activated protein kinase 1, a member of the extracellular signalregulated kinase (ERK) family. Significant associations before risk factor adjustment are shown in supplementary Table 2. After risk factor adjustment, rs6928, MAPK1 showed the strongest association with CAD under the dominant model both for homozygous and heterozygous carriers of the rare allele (P = 0.0217, OR 1.36 (95%
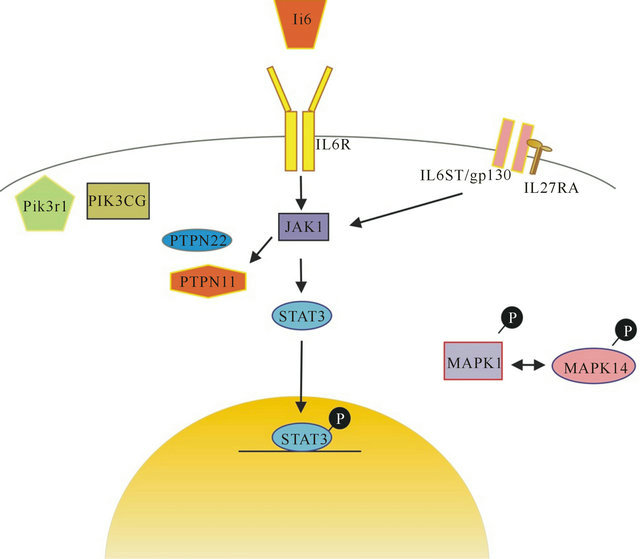
Figure 1. Proteins involved in interleukin 6 (IL-6)—related signaling that were assessed for genetic variants. IL-6: interleukin 6; IL6R: interleukin 6 receptor; IL6ST/gp130: Glycoprotein 130. IL27RA: Interleukin Receptor 27 subunit alpha; JAK1: Janus Kinase1; STAT3: Signal transductor and activator of transcription 3; PIK3r1: Phosphatidylinositol 3-kinase regulatory subunit alpha; PIK3CG: Phosphatidy-linositol-4, 5-bisphosphate 3-kinase catalytic subunit gamma isoform; PTPN11: Protein tyrosine phosphatase, non-receptor type 11; PTPN22: Protein tyrosine phosphatase, non-receptor type 22; MAPK1: Mitogen-activated protein kinase 1; MAPK14: Mitogen-activated protein kinase 14.
C.I. 1.05 - 1.77) after risk factor adjustment, Table 3). This association was accompanied by a higher frequency of homozygous and heterozygous carriers of the rare allele in the cases (27.0% and 50.4%, respectively) compared to the controls (25.4% and 45.7%, respectively; Table 4). The other 4 SNPs showed association in the recessive model (rs7757672, MAPK14) in the additive model (rs310236, JAK1), or in both models (rs9340, MAPK1 and rs11913721 MAPK1).
3.3. Replication Study
In the next step of the study, SNP rs6928 (MAPK1), which exhibited the strongest association signal in the explorative study was tested for association in a replication study that was carried out in an independent case-control population. In this study, all 1103 CAD patients and 1001 out of 1079 healthy controls were successfully genotyped. The results met the quality criteria CR ≥ 95%, PHWE > 0.01, MAF > 5% (Table 5). The significant association of rs6928 with CAD, that was detected in the explorative study, was not reproduced in the replication (Table 6). Consistently, highly similar allele frequencies between cases and controls contradicted a prominent genetic role of this SNP in CAD (Table 7).
4. DISCUSSION
In this multigenic candidate gene association study, we assessed the association of 12 candidate genes from the Interleukin-6 signaling pathway with coronary artery disease. Many studies have proven, that IL-6 plays a prominent role in the pathogenesis of atherosclerosis and CAD [15,17,30,31]. Consistently, genetic polymorphisms of IL-6 have been demonstrated to affect the onset and clinical course of CAD [32,33]. The functional relevance of two polymorphisms in the promoter region of the IL-6 gene, IL6-174G_C and -572G_were explained by their ability to increase serum IL6-levels in different situations of physiological stress, e.g. status after coronary bypass graft surgery [34]. These data imply that the inflammatory response to stress situations
Table 3. Explorative study: significant association of 5 tSNPs with coronary artery disease.
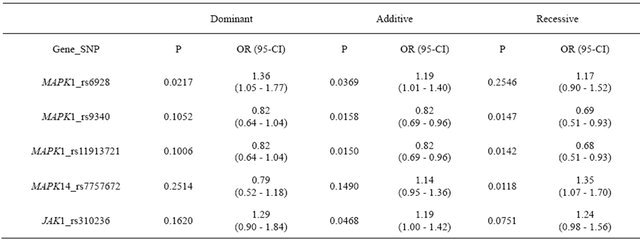
Association Statistics are shown for the case and control group of coronary artery disease (CAD). Given are the odds ratios (OR), their 95% confidence intervals (CI 95%) and the P values which were obtained from a Wald test (autosomal-dominant, additive and recessive models). Values are given after adjustment for the covariates diabetes, hypertension, BMI, smoking and gender in a logistic regression model. All other markers showed no significance at the 5% test level under either of the models. Siginficant associations are highlighted in yellow.
Table 4. Significant SNPs from the explorative study.
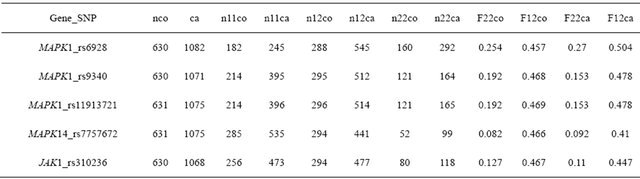
Numeric (N) and proportionate (F) frequency of the allelic genotypes 11, 12 and 22 in controls (co) and cases (ca), with 1 being the more frequent and 2 the less frequent allel.
Table 5. Replication study: quality criteria oft he most significant SNP from the explorative study.

nco: number of controls, nca: Number of cases, CR: call rate, HWE: Hardy Weinberg Equilibrium, minAFco: minor allel frequency in controls, minAFca: minor allel frequency in cases.
Table 6. Replication Study: No association of MAPK1_rs6928 with CAD.
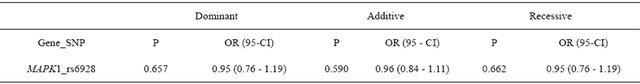
Association Statistics are shown for the case and control group of coronary artery disease (CAD). Given are the odds ratios (OR). Their 95% confidence intervals (CI 95%) and the P values which were obtained from a Wald test (autosomal-dominant, additive and recessive models). Values are given after adjustment for the covariates, hypertension, BMI, smoking and gender in a logistic regression model.
Table7. Numeric (N) and proportionate (F) frequency of the allelic genotypes 11, 12 and 22 of rs6928 in controls (co) and cases (ca), with 1 being the more frequent and 2 the less frequent allel.

in CAD is influenced by genetic variants, underlining the necessity of the candidate gene approach to systematically evaluate the functional relevance of a molecule in CAD. Since IL-6 is embedded in a canonical signaling pathway, and exerts it’s functions through many distinct molecules, we chose a multigenic approach to cover 12 important mediators of IL6-signaling in a single approach.
In the explorative study, we found five SNPs to be associated nominal significantly with CAD, with rs6928 (MAPK1) showing the strongest association. Additionally, two other MAPK1 SNPs showed significant associations in this part of the study (rs9340 and rs11913721).
While in our explorative study SNPs in the MAPK1 gene were significantly associated with CAD, we could not reproduce this result in the replication study. These conflicting results might not be caused by population stratification, based on undiscovered subpopulations, such as a different ethnic background or age [35]. The populations employed in this study were selected as genetically homogenous as previously described [36-38]. However, the population of CAD patients was on average almost nine years older in the replication study than in the explorative study. It is generally believed that genetic influences on the onset and course of a disease are overbalanced by environmental and behavioural factors in older patients more than in younger patients [37]. The lack of association of rs6928 with CAD in the replication study could be due to this effect. For common alleles, we were able to detect a genetic effect of 1.3 (Figure S1). Thus, we might have missed associations of common variants which confer a genetic risk of <1.3.
Recently, genome wide association studies (GWAS) have discovered several novel variants that were associated with CAD [6,7,39]. The high relevance of these investigations is based on their ability to discover novel associations in loci that were not assumed to be functionally relevant for CAD or even not protein-coding. However, the variants discovered explain only 4% of interindividual variants in disease risk and only up to 13% of the total heredity of CAD [8], underlining the need for further genetic studies. The limited size of study populations assessed in GWAS leads to the identification of mainly frequent variants, leaving a “gap” in the search for rare variants [9]. “Classical” candidate gene studies like ours maintain a strong supplemental value, since large study populations can easily be assessed to discover genetic variants of potentially higher impact. Moreover, by simultaneously addressing IL-6 and interacting molecules, we believe that we can offer an integrated view on the genetic relevance of this canonical signaling pathway in CAD. Our study might be a starting point for the genetic assessment of other IL6-related molecules in CAD to finally provide new insights in the pathogenetic mechanisms of this complex disease.
5. ACKNOWLEDGEMENTS
This study was supported by a research grant of the “Research Center Inflammation Medicine” of the Medical Faculty, Christian-AlbrechtsUniversity, University Medical Center Schleswig-Holstein, Campus Kiel, (Arne S. Schaefer) and of the German Ministry of Education and Research through the POPGEN biobank project (01GR0468).
![]()
![]()
REFERENCES
- World Health Organization (2011) The top 10 causes of death.
- Marenberg, M.E., Risch, N., Berkman, L.F., Floderus B., and de Faire, U. (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. New England Journal of Medicine, 330, 1041-1046. doi:10.1056/NEJM199404143301503
- Mayer, B., Erdmann, J. and Schunkert, H. (2007) Genetics and heritability of coronary artery disease and myocardial infarction. Clinical Research in Cardiology, 96, 1-7. doi:10.1007/s00392-006-0447-y
- Erdmann, J., Grosshennig, A., Braund, P.S., Konig, I.R., Hengstenberg, C., Hall, A.S., Linsel-Nitschke, P., Kathiresan, S., Wright, B., Tregouet, D.A., Cambien, F., Bruse, P., Aherrahrou, Z., Wagner, A.K., Stark, K., Schwartz, S.M., Salomaa, V., Elosua, R., Melander, O., Voight, B.F., O’Donnell, C.J., Peltonen, L., Siscovick, D.S., Altshuler, D., Merlini, P.A., Peyvandi, F., Bernardinelli, L., Ardissino, D., Schillert, A., Blankenberg, S., Zeller, T., Wild, P., Schwarz, D.F., Tiret, L., Perret, C., Schreiber, S., El Mokhtari, N.E., Schafer, A., Marz, W., Renner, W., Bugert, P., Kluter, H., Schrezenmeir, J., Rubin, D., Ball, S.G., Balmforth, A.J., Wichmann, H.E., Meitinger, T., Fischer, M., Meisinger, C., Baumert, J., Peters, A., Ouwehand, W.H., Deloukas, P., Thompson, J.R., Ziegler, A., Samani, N.J. and Schunkert, H. (2009) New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nature Genetics, 41, 280-282. doi:10.1038/ng.307
- Schunkert, H., Erdmann, J. and Samani, N.J. (2010) Genetics of myocardial infarction: a progress report. European Heart Journal, 31, 918-925. doi:10.1093/eurheartj/ehq038
- Tregouet, D.A., Konig, I.R., Erdmann, J., Munteanu, A., Braund, P.S., Hall, A.S., Grosshennig, A., LinselNitschke, P., Perret, C., DeSuremain, M., Meitinger, T., Wright, B.J., Preuss, M., Balmforth, A.J., Ball, S.G., Meisinger, C., Germain, C., Evans, A., Arveiler, D., Luc, G., Ruidavets, J.B., Morrison, C., van der Harst, P., Schreiber, S., Neureuther, K., Schafer, A., Bugert, P., El Mokhtari, N.E., Schrezenmeir, J., Stark, K., Rubin, D., Wichmann, H.E., Hengstenberg, C., Ouwehand, W., Ziegler, A., Tiret, L., Thompson, J.R., Cambien, F., Schunkert, H. and Samani, N.J. (2009) Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nature Genetics, 41, 283-285. doi:10.1038/ng.314
- Schunkert, H., Konig, I.R., Kathiresan, S., Reilly, M.P., Assimes, T.L., Holm, H., Preuss, M., Stewart, A.F., Barbalic, M., Gieger, C., Absher, D., Aherrahrou, Z., Allayee, H., Altshuler, D., Anand, S.S., Andersen, K., Anderson, J.L., Ardissino, D., Ball, S.G., Balmforth, A.J., Barnes, T.A., Becker, D.M., Becker, L.C., Berger, K., Bis, J.C., Boekholdt, S.M., Boerwinkle, E., Braund, P.S., Brown, M.J., Burnett, M.S., Buysschaert, I., Carlquist, J.F., Chen, L., Cichon, S., Codd, V., Davies, R.W., Dedoussis, G., Dehghan, A., Demissie, S., Devaney, J.M., Diemert, P., Do, R., Doering, A., Eifert, S., Mokhtari, N.E., Ellis, S.G., Elosua, R., Engert, J.C., Epstein, S.E., de Faire, U., Fischer, M., Folsom, A.R., Freyer, J., Gigante, B., Girelli, D., Gretarsdottir, S., Gudnason, V., Gulcher, J.R., Halperin, E., Hammond. N., Hazen, S.L., Hofman, A., Horne, B.D., Illig, T., Iribarren, C., Jones, G.T., Jukema, J.W., Kaiser, M.A., Kaplan, L.M., Kastelein, J.J., Khaw, K.T., Knowles, J.W., Kolovou, G., Kong, A., Laaksonen, R., Lambrechts, D., Leander, K., Lettre, G., Li, M., Lieb, W., Loley, C., Lotery, A.J., Mannucci. P.M., Maouche, S., Martinelli, N., McKeown, P.P., Meisinger, C., Meitinger, T., Melander, O., Merlini, P.A., Mooser, V., Morgan, T., Muhleisen, T.W., Muhlestein, J.B., Munzel, T., Musunuru, K., Nahrstaedt, J., Nelson, C.P., Nothen, M.M., Olivieri, O., Patel, R.S., Patterson, C.C., Peters, A., Peyvandi, F., Qu, L., Quyyumi, A.A., Rader, D.J., Rallidis, L.S., Rice, C., Rosendaal, F.R., Rubin, D., Salomaa, V., Sampietro, M.L., Sandhu, M.S., Schadt, E., Schafer, A., Schillert, A., Schreiber, S., Schrezenmeir, J., Schwartz, S.M., Siscovick, D.S., Sivananthan, M., Sivapalaratnam, S., Smith, A., Smith, T.B., Snoep, J.D., Soranzo, N., Spertus, J.A., Stark, K., Stirrups, K., Stoll, M., Tang, W.H., Tennstedt, S., Thorgeirsson, G., Thorleifsson, G., Tomaszewski, M., Uitterlinden, A.G., van Rij, A.M., Voight, B.F., Wareham, N.J., Wells, G.A., Wichmann, H.E., Wild, P.S., Willenborg, C., Witteman, J.C., Wright, B.J., Ye, S., Zeller, T., Ziegler, A., Cambien, F., Goodall, A.H., Cupples, L.A., Quertermous, T., Marz, W., Hengstenberg, C., Blankenberg, S., Ouwehand, W.H., Hall, A.S., Deloukas, P., Thompson, J.R., Stefansson, K., Roberts, R., Thorsteinsdottir, U., O’Donnell, C.J., McPherson, R., Erdmann, J. and Samani, N.J. (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nature Genetics, 43, 333-338. doi:10.1038/ng.784
- Peden, J.F. and Farrall, M. (2011) Thirty-five common variants for coronary artery disease: The fruits of much collaborative labour. Human Molecular Genetic, 20, R198-R205. doi:10.1093/hmg/ddr384
- Schafer, A.S., Jepsen, S. and Loos, B.G. (2011) Periodontal genetics: a decade of genetic association studies mandates better study designs. Journal of Clinical Periodontology, 38, 103-107. doi:10.1111/j.1600-051X.2010.01653.x
- Holmer, S.R., Bickeboller, H., Hengstenberg, C., Rohlmann, F., Engel, S., Lowel, H., Mayer, B., Erdmann, J., Baier, C., Klein, G., Riegger, G.A. and Schunkert, H. (2003) Angiotensin converting enzyme gene polymerphism and myocardial infarction a large association and linkage study. International Journal of Biochemistry & Cell Biology, 35, 955-962. doi:10.1016/S1357-2725(02)00261-3
- Lieb, W., Mayer, B., Konig, I.R., Borwitzky, I., Gotz, A., Kain, S., Hengstenberg, C., Linsel-Nitschke, P., Fischer, M., Doring, A., Wichmann, H.E., Meitinger, T., Kreutz, R., Ziegler, A., Schunkert, H. and Erdmann, J. (2008) Lack of association between the MEF2A gene and myocardial infarction. Circulation, 117,185-191. doi:10.1161/CIRCULATIONAHA.107.728485
- Brull, D.J., Serrano, N., Zito, F., Jones, L., Montgomery, H.E., Rumley, A., Sharma, P., Lowe, G.D., World, M.J., Humphries, S.E. and Hingorani, A.D. (2003) Human CRP gene polymorphism influences CRP levels: Implications for the prediction and pathogenesis of coronary heart disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 23, 2063-2069. doi:10.1161/01.ATV.0000084640.21712.9C
- Wang, L., Lu, X., Li, Y., Li, H., Chen, S. and Gu, D. (2009) Functional analysis of the C-reactive protein (CRP) gene -717A>G polymorphism associated with coronary heart disease. BMC Medical Geneticas, 10, 73. doi:10.1186/1471-2350-10-73
- Heinrich, P.C., Behrmann, I., Haan, S., Hermanns, H.M., Muller-Newen, G. and Schaper, F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochemical Journal, 374,1-20. doi:10.1042/BJ20030407
- Fuchs, M., Hilfiker, A., Kaminski, K., Hilfiker-Kleiner, D., Guener, Z., Klein, G., Podewski, E., Schieffer, B., Rose-John, S. and Drexler, H. (2003) Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB Journal, 17, 2118-2120.
- Hilfiker-Kleiner, D., Hilfiker, A., Fuchs, M., Kaminski, K., Schaefer, A., Schieffer, B., Hillmer, A., Schmiedl, A., Ding, Z., Podewski, E., Poli, V., Schneider, M.D., Schulz, R., Park, J.K., Wollert, K.C. and Drexler, H. (2004) Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circulation Research, 95, 187-195. doi:10.1161/01.RES.0000134921.50377.61
- Schieffer, B., Selle, T., Hilfiker, A., Hilfiker-Kleiner, D., Grote, K., Tietge, U.J., Trautwein, C., Luchtefeld, M., Schmittkamp, C., Heeneman, S., Daemen, M.J. and Drexler, H. (2004) Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation, 110, 3493-3500. doi:10.1161/01.CIR.0000148135.08582.97
- Biasucci, L.M., Vitelli, A., Liuzzo, G., Altamura, S., Caligiuri, G., Monaco, C., Rebuzzi, A.G., Ciliberto and G., Maseri, A. (1996) Elevated levels of interleukin-6 in unstable angina. Circulation, 94, 874-877. doi:10.1161/01.CIR.94.5.874
- Ikonomidis, I., Lekakis, J.P., Nikolaou, M., Paraskevaidis, I., Andreadou, I., Kaplanoglou, T., Katsimbri, P., Skarantavos, G., Soucacos, P.N. and Kremastinos, D.T. (2008) Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation, 117, 2662-2669. doi:10.1161/CIRCULATIONAHA.107.731877
- Georges, J.L., Loukaci, V., Poirier, O., Evans, A., Luc, G., Arveiler, D., Ruidavets, J.B., Cambien, F. and Tiret, L. (2001) Interleukin-6 gene polymorphisms and susceptibility to myocardial infarction: The ECTIM study. Etude Cas-Temoin de l’Infarctus du Myocarde. Journal of Molecular Medicine, 79, 300-305. doi:10.1007/s001090100209
- Lieb, W., Pavlik, R., Erdmann, J., Mayer, B., Holmer, S.R., Fischer, M., Baessler, A., Hengstenberg, C., Loewel, H., Doering, A., Riegger, G.A. and Schunkert, H. (2004) No association of interleukin-6 gene polymorphism (-174 G/C) with myocardial infarction or traditional cardiovascular risk factors. International Journal of Cardiology, 97, 205-212. doi:10.1016/j.ijcard.2003.07.038
- Gigante, B., Bennet, A.M., Leander, K., Vikstrom, M. and de Faire, U. (2010) The interaction between coagulation factor 2 receptor and interleukin 6 haplotypes increases the risk of myocardial infarction in men. PLoS One, 5, e11300. doi:10.1371/journal.pone.0011300
- Krawczak, M., Nikolaus, S., von Eberstein, H., Croucher, P.J., El Mokhtari, N.E. and Schreiber, S. (2006) PopGen: Population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Journal of Community Genetics, 9, 55-61. doi:10.1159/000090694
- Rubin, D., Helwig, U., Nothnagel, M., Folsch, U.R., Schreiber, S. and Schrezenmeir, J. (2010) Association of postprandial and fasting triglycerides with traits of the metabolic syndrome in the Metabolic Intervention Cohort Kiel. European Journal of Endocrinology, 162,719-727. doi:10.1530/EJE-09-0446
- Barrett, J.C., Fry, B., Maller, J. and Daly, M.J. (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263-265. doi:10.1093/bioinformatics/bth457
- Xu, J., Turner, A., Little, J., Bleecker, E.R., Meyers and D.A. (2002) Positive results in association studies are associated with departure from Hardy-Weinberg equilibrium: Hint for genotyping error? Human Genetics, 111, 573-574. doi:10.1007/s00439-002-0819-y
- Wigginton, J.E., Cutler, D.J. and Abecasis, G.R. (2005) A note on exact tests of Hardy-Weinberg equilibrium. American Journal Human Genetics, 76, 887-893. doi:10.1086/429864
- Ott, J. (1985) A chi-square test to distinguish allelic association from other causes of phenotypic association between two loci. Genetic Epidemiology, 2, 79-84. doi:10.1002/gepi.1370020108
- Dupont, W.D. and Plummer, W.D., Jr. (1998) Power and sample size calculations for studies involving linear regression. Controlled Clinical Trials, 19, 589-601. doi:10.1016/S0197-2456(98)00037-3
- Schuett, H., Oestreich, R., Waetzig, G.H., Annema, W., Luchtefeld, M., Hillmer, A., Bavendiek, U., von Felden, J., Divchev, D., Kempf, T., Wollert, K.C., Seegert, D., Rose-John, S., Tietge, U.J., Schieffer, B. and Grote, K. (2011) Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 32, 281-290.
- Wassmann, S., Stumpf, M., Strehlow, K., Schmid, A., Schieffer, B., Bohm, M. and Nickenig, G. (2004) Interleukin-6 induces oxidative stress and endothelial dys= function by overexpression of the angiotensin II type 1 receptor. Circulation Research, 94, 534-541. doi:10.1161/01.RES.0000115557.25127.8D
- Basso, F., Lowe, G.D., Rumley, A., McMahon and A.D., Humphries, S.E. (2002) Interleukin-6 -174G>C polymorphism and risk of coronary heart disease in West of Scotland coronary prevention study (WOSCOPS). Arteriosclerosis, Thrombosis, and Vascular Biology, 22, 599- 604. doi:10.1161/01.ATV.0000013283.84306.1A
- Vakili, H., Ghaderian, S.M., Akbarzadeh Najar, R., Tabatabaei Panah, A.S. and Azargashb, E. (2011) Genetic polymorphism of interleukin-6 gene and susceptibility to acute myocardial infarction. Coronary Artery Disease, 22, 299-305. doi:10.1097/MCA.0b013e328346b848
- Brull, D.J., Montgomery, H.E., Sanders, J., Dhamrait, S., Luong, L., Rumley, A., Lowe, G.D. and Humphries, S.E. (2001) Interleukin-6 gene -174g>c and -572g>c promoter polymorphisms are strong predictors of plasma interleukin-6 levels after coronary artery bypass surgery. Arteriosclerosis, Thrombosis, and Vascular Biology, 21, 1458-1463. doi:10.1161/hq0901.094280
- Cardon, L.R. and Palmer, L.J. (2003) Population stratifycation and spurious allelic association. Lancet, 361, 598-604. doi:10.1016/S0140-6736(03)12520-2
- Auinger, A., Helwig, U., Pfeuffer, M., Rubin, D., Luedde, M., Rausche, T., Eddine El Mokhtari, N., Folsch, U.R., Schreiber, S., Frey, N. and Schrezenmeir, J. (2011) A variant in the heart-specific fatty acid transport protein 6 is associated with lower fasting and postprandial TAG, blood pressure and left ventricular hypertrophy. British Journal of Nutrition, 107, 1422-1428.
- Schaefer, A.S., Richter, G.M., Groessner-Schreiber, B., Noack, B., Nothnagel, M., El Mokhtari, N.E., Loos, B.G., Jepsen, S. and Schreiber, S. (2009) Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genetics, 5, e1000378. doi:10.1371/journal.pgen.1000378
- Schunkert, H., Gotz, A., Braund, P., McGinnis, R., Tregouet, D.A., Mangino, M., Linsel-Nitschke, P., Cambien, F., Hengstenberg, C., Stark, K., Blankenberg, S., Tiret, L., Ducimetiere, P., Keniry, A., Ghori, M.J., Schreiber, S., El Mokhtari, N.E., Hall, A.S., Dixon, R.J., Goodall, A.H., Liptau, H., Pollard, H., Schwarz, D.F., Hothorn, L.A., Wichmann, H.E., Konig, I.R., Fischer, M., Meisinger, C., Ouwehand, W., Deloukas, P., Thompson, J.R., Erdmann, J., Ziegler, A. and Samani, N.J. (2008) Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation, 117, 1675-1684. doi:10.1161/CIRCULATIONAHA.107.730614
- Erdmann, J., Willenborg, C., Nahrstaedt, J., Preuss, M., Konig, I.R., Baumert, J., Linsel-Nitschke, P., Gieger, C., Tennstedt, S., Belcredi, P., Aherrahrou, Z., Klopp, N., Loley, C., Stark, K., Hengstenberg, C., Bruse, P., Freyer, J., Wagner, A.K., Medack, A., Lieb, W., Grosshennig, A., Sager, H.B., Reinhardt, A., Schafer, A., Schreiber, S., El Mokhtari, N.E., Raaz-Schrauder, D., Illig, T., Garlichs, C.D., Ekici, A.B., Reis, A., Schrezenmeir, J., Rubin, D., Ziegler, A., Wichmann, H.E., Doering, A., Meisinger, C., Meitinger, T., Peters, A. and Schunkert, H. (2011) Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. European Heart Journal, 32, 158-168. doi:10.1093/eurheartj/ehq405
Appendix
Supplementary Figure
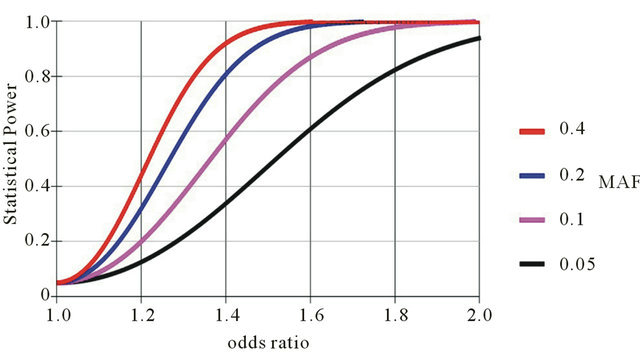
Figure S1. Statistical power of the finemapping study. The guaph shows the statistical power of the case-control population of the explorative study (1096 cases, 636 controls) of the finemapping in relation to the minor allele frequency (MAF) of the putative risk alleles in the controls and different genetic effects (odds ratio). A power of 0.8 is regarded as statistically significant.
Supplementary Table
Table S1. Quality criteria of the primary study for all 73tested tSNPs. tSNPs that did not meet all quality criteria are marked in yellow rows.
Continued
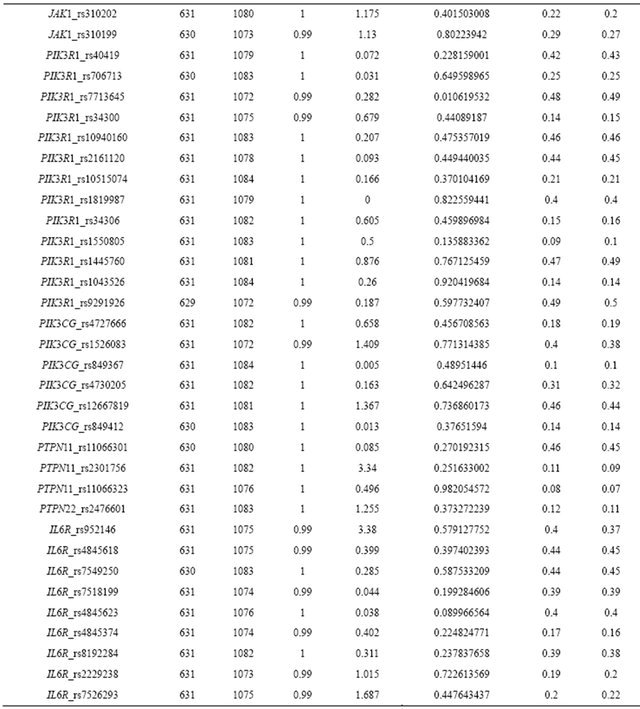
nco: number of controls, nca: Number of cases, CR: call rate, HWE: Hardy Weinberg Equilibrium, minAFco: minor allel frequency in controls, minAFca: minor allel frequency in cases.
NOTES
*Both authors contributed equally to this work.
#Corresponding author.