Open Journal of Genetics
Vol.1 No.3(2011), Article ID:8894,8 pages DOI:10.4236/ojgen.2011.13013
Screening for variants in the MUTYH gene in Saudis
Department of Biochemistry, Genome Research Chair, College of Science, King Saud University, Riyadh, Kingdom of Saudi Arabia.
Email: *naltassan@kfshrc.edu.sa
Received 26 September 2011; revised 11 November 2011; accepted 22 November 2011.
Keywords: MUTYH; Allele Frequencies; Single Nucleotide Polymorphism; Mutations; Colorectal Cancer
ABSTRACT
MUTYH is a base excision repair glycosylase responsible for correcting the G:A mismatches that arise from replication of a damaged DNA, such impairment results from attacks by reactive oxygen species that are produced from different sources including cigarette smoking. The produced reactive oxygen species trigger the oxidation of guanine in to 8-oxo-G and the latter mispairs with adenine. Alterations in the MUTYH gene can affect its glycosylase function and hence the DNA repair capacity that is tightly linked to cancer development. Defects in the MUTYH were found to be associated with predisposition to colorectal cancer; the third most common cancer in the world. Although some studies suggested the existence of an ethnic differentiation among MUTYH mutations, there are no documented reports regarding this gene in Saudi cases or controls so far. 153 healthy Saudi individuals including smokers were screened for the IVS1 + 5 G/C, V22M, Y165C, R231C, H324Q and G382D MUTYH variants using either ARMS or RFLP or direct sequencing. Allelic frequencies were calculated and were found to be as follows: IVS1 + 5 G/C = 1/0, V22M = 0.99/0.01, Y165C = 1/0, R231C = 1/0, H324Q = 0.29/0.71 and G382D = 0.997/0.003. Comparison of the allele frequencies between Saudis and other populations revealed a significant difference between the Saudis and Europeans for the V22M (p-value: 0.0003, OR: 5.899, 95% CI: 1.999 - 17.408); and between the Saudis and Asians for the H324Q (p-value: <0.0001, OR: 0.473, 95% CI: 0.341-0.6559). No significant differences were found between smokers and non-smokers groups. These data document the allele frequencies of different MUTYH variants in Saudi populations and support the existence of an ethnic difference between MUTYH variants which is beneficial as some MUTYH common polymorphisms in certain populations may correlate with disease predisposition in other rare populations.
1. INTRODUCTION
Genomic DNA damage occurs as a result of chemical events that are triggered by endogenous processes or the exposure to exogenous substances [1]. The most frequent DNA damage is caused by Reactive Oxygen Species (ROS) [2-3]. These species are generated by normal cellular metabolism, exogenous chemicals, ionizing radiation [4] and chronic infection that triggers inflammatory response [3].
Although ROS are capable of oxidizing the DNA, guanine is the most susceptible base to oxidation [5]. The oxidized 8-hydroxyguanosine (8-oxo-G) has the ability to mispair with Adenine and results in G:C to T:A and A:T to C:G transversions after replication. MUTYH enzyme is a member of the mammalian Base Excision Repair (BER) glycosylases that excise adenines misincorporated opposite to 8-oxo-G [6-9] thereby correcting the G: A mismatches arising from replication of a damaged DNA and protecting the cell from the mutagenic effect of guanine oxidation [10].
Defects in the BER pathway contribute to tumorigenesis as these defects may increase the frequency of mutations in tumour suppressor genes and oncogenes [11]. Earlier, a recessive familial form of colorectal cancer (CRC) known as the MUTYH associated polyposis (MAP) was linked to the base excision repair gene MUTYH [12], this was pursued by the identification of several polymorphic forms of MUTYH that were most likely associated with cancer susceptibility and were present in different human populations [4]. For instance, different studies reported on CRC patients that were compound heterozygous for the two missence variants Y165C and G382D of MUTYH and it is now an accepted fact that biallelic germline mutations in the MUTYH gene significantly increase the risk for colorectal cancer [13-18]. Yamaguchi et al., 2002 described a lung cancer cell line with an alternative splicing event in MUTYH intron 1 triggered by the IVS1 + 5 G/C variant. This variant was also found in an oesophageal cancer, a skin squamouse cell cancer, and two colon adenocarcinoma patients [19-20]. Additionally, Miyaki et al., 2005 reported a patient with multiple colorectal adenomas to be homozygous for the MUTYH R231C (891 C > T) mutation [21]. Moreover, the 972C allele of the MUTYH common polymorphism H324Q (972 C > G) was suggested to be inversely associated with the risk of proximal CRC in the Japanese population due to its interaction with other alleles [21,22]. On the other hand, V22M (66G > A) polymorphism was not associated directly with any disease state but rather it may be associated with localization of the MUTYH to the site of DNA replication [23].
Although carriers of biallelic MUTYH mutations have a 60% risk to develop CRC [24] it is now clear that some genes which cause autosomal recessive cancers predisposition are related to fairly increased risk of adult cancer in monoallelic carriers [25]. Such monoallelic mutations may be strongly related to cancer when combined with other genes mutations [26] or carcinogen exposure [27] and they also can influence genetic pathways in CRC [19].
The wide range of variation in the MUTYH reported frequencies for control populations [28] and the considerable inter-individuals variation in DNA repair capacity in the general population, emphasizes on the importance of screening for MUTYH genetic variants in the normal Saudi population.
This study aimed to determine allele frequencies for different MUTYH gene variants in Saudis to investigate the possible role of these variants in increasing the susceptibility to cancer and to compare the results with reported frequencies of this gene in other populations. Additionally, the study compared allele frequencies between smokers and non-smokers since cigarette smoke may induces production of 8-oxo-G that is considered the natural cause of genomic diversity and SNPs in organisms because of its mutagenic capability [5].
2. METHODS
2.1. Subjects, Samples and Polymerase Chain Reaction
Subjects enrolled in this study were healthy Blood donors attending local hospitals in Riyadh, Saudi Arabia. The study group included 153 healthy unrelated Saudi subjects (113 non-smokers and 40 smokers); peripheral blood was drawn by venepuncture in EDTA tubes after obtaining written informed consent. Blood samples were stored at –20˚C until required for analysis. DNA was extracted from the whole blood using the QIAmp blood mini kit (Qiagen) according to the manufacture’s instruction. Amplifications were performed in a DNA thermal cycler, using 2.5 units of pureTaq DNA polymeras, 5 pmole primer, 10 - 60 ng genomic DNA, 200 µM dNTP, 50 mM KCl and 1.5 mM MgCl2.
Cycling parameters were 95˚C for 10 minutes, 34 - 40 cycles of 94˚C for 30 seconds, annealing temperature from 52˚C - 60˚C for 30 seconds and 72˚C for 30 seconds followed by a final elongation step at 72˚C for 10 minutes.
2.2. Restriction Endonuclease Digestion
Exon 2 enclosing the V22M (66 G > A) polymorphism was amplified at 58˚C for 35 cycles using primers reported before [12]. The 285 bp PCR product was digested with NcoI restriction digest, 10 µl of the PCR product of each sample was mixed with 10U (1µl) of the Nco1 restriction enzyme (10U/µl), followed by incubation at 37˚C for 3 hours and finally analyzed by agarose gel electrophoresis.
Exon 13 harbouring the G382D (1145 G > A) mutation was amplified at 56˚C for 35 cycles using primers described before [12]. The 242 bp PCR product was digested with BglII restriction enzyme. 10 µl of exon 13 PCR product of each sample was mixed with 10U (0.1667 µl) of the BglII restriction enzyme (60 U/µl) and incubated at 37˚C for 3 hours and finally analyzed by agarose gel electrophoresis.
2.3. Sequencing
Sequencing was carried out using DYEnamic™ ET Terminator Cycle Sequencing Kit from Amersham Biosciences UK Limited (Littele Chalfont Buckinghamshire, UK). The mixture was combined with 5 µM of MUTYH exon 9 PCR product and 5 µM forward primer then thermo cycled (95˚C for 30 seconds, followed by 24 cycles of 95˚C for 20 seconds, 50°C for 15 seconds and 60˚C for 1 minute) then the products were passed through a gel-filtration resin and finally resuspended in a formamide loading buffer, the sequencing were preformed in the MegaBACE 1000 genetic analyzer from Amersham and analyzed using the lasergene6 package (DNAStar, WI, USA).
2.4. Amplification Refractory Mutation System (ARMS)
IVS1+5 G < C variant in intron 1 was assayed using a common (IVS1 + 5 C) primer and a normal (IVS1 + 5 N) or mutant (IVS1 + 5 M) ARMS primers as previously reported [12]. In addition, an internal control primer SETX (exon 20) was co-amplified in a di-plex reaction to ensure that the ARMS PCRs were efficient in each reaction tube and to avoid any false negative results. Annealing temperature of 58˚C and 33 cycles were the optimum conditions selected for equal amplification.
Y165C (494 A > G) in exon 7 was assayed using a common (IVS1 + 5 C) primer and a normal (165N) or mutant (165M) ARMS primers [12]. The internal control primer SETX (exon 20) was amplified together with the ARMS primers. Annealing temperature of 58˚C and 35 cycles were the optimum conditions selected for equal amplification.
H324Q (972 C > G) in exon 12 was assayed using a common (324C) and a normal (324N) or mutant (324M) ARMS primers [12]. The internal control primer DBTE (exon 2) was amplified together with the ARMS primer. Annealing temperature of 62˚C and 39 cycles were the optimum conditions selected for equal amplification. All products were visualized by regular agarose gel electrophoresis .
2.5. Statistical Analysis
Allele frequencies were obtained by allele counting and errors were assigned. Differences in allele frequencies between populations and between smokers and nonsmokers were tested using Fisher’s exact test, a P-value of ≤0.05 was considered statistically significant. For sample size larger than 1000, χ2 test was performed and in both cases 95% CI was calculated, all computations were undertaken using the statistical software GraphPad InStat version 2.04 (Ralf Stahlman, Purdue Univ. 931897S).
3. RESULTS
3.1. MUTYH Genotypes Assays
Agarose gel images for exon 2 digests showed that the wild type allele remained undigested and that the V22M allele was digested generating 207 and 78 bp products. In exon 13, the wild type allele remained undigested, whilst the G382D allele was digested generating 160bp and 82bp fragments (Figure 1(a)).
Exon 9 was amplified then sequenced and the electropherograms showed all the samples to be homozygous for the wild type allele (R231/R231) harbouring a C at position 891 and lacking the T at this position, no sample was found to carry the mutant allele T (Figure 1(b)).
Intron 1 ARMS primers generated a 178 bp product only in the normal reaction and a 300 bp product was generated for the internal control. Similarly, exon 7 ARMS primers generated a 148 bp product only in the normal reaction and a 300 bp product was generated for the internal control.
Finally, exon12 ARMS primers generated a 150 bp product in some normal and some mutant reactions and a 300bp product was generated for the internal control (Figure 1(c)).
3.2. MUTYH Genotype Frequencies
The genotype frequencies for all the genetic variants of the MUTYH are summarized in Table 1." target="_self">Table 1. Allele frequent- cies were as follows: IVS1 + 5 G > C 1/0, V22M (66 G > A) 0.99/0.01, Y165C (494 A > G) 1/0, R231C (891 C<T) 1/0, H324Q (972 C > G) 0.29/0.71 and G382D (1145 G > A) 0.997/0.003.
4. DISCUSSION
Several defence mechanisms that avoid accumulation of oxidative DNA damages exist in mammalian cells. The two BER glycosylases; MUTYH and OGG1 play important roles in the repair of modified bases including 8-oxo-G [29-32] MUTYH removes the mismatched adenine and helps in base excision repair of 8-oxo-G:A and G:A, while OGG1 removes 8-oxo-G from duplex DNA.
Different variants of the MUTYH gene can affect the protein function and hence the DNA repair capacity that is tightly linked to cancer development. Biallelic germline mutations in the MUTYH lead to MUTYH associated polyposis (MAP) and provide a significant risk for colorectal cancer [24]. Different studies supported the role of MUTYH in colorectal adenoma and carcinoma predisposition [33,34].
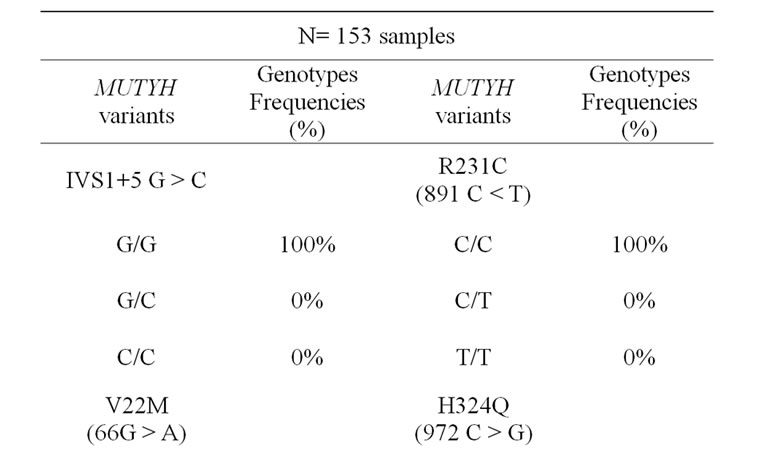
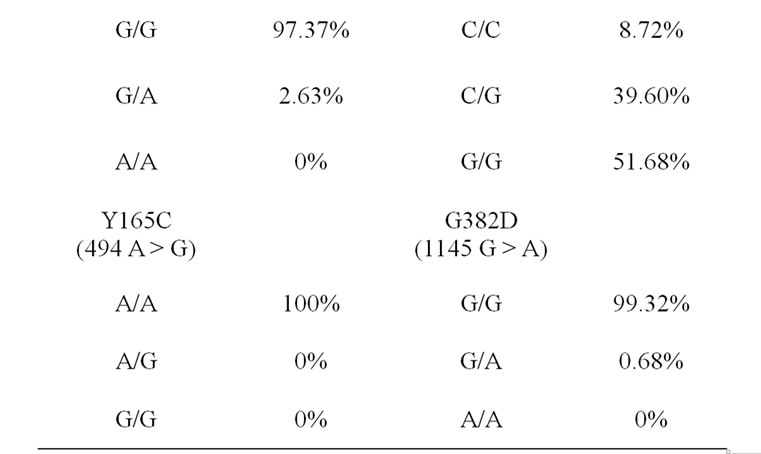
Table 1. The genotype frequencies for the MUTYH variants in Saudi samples.
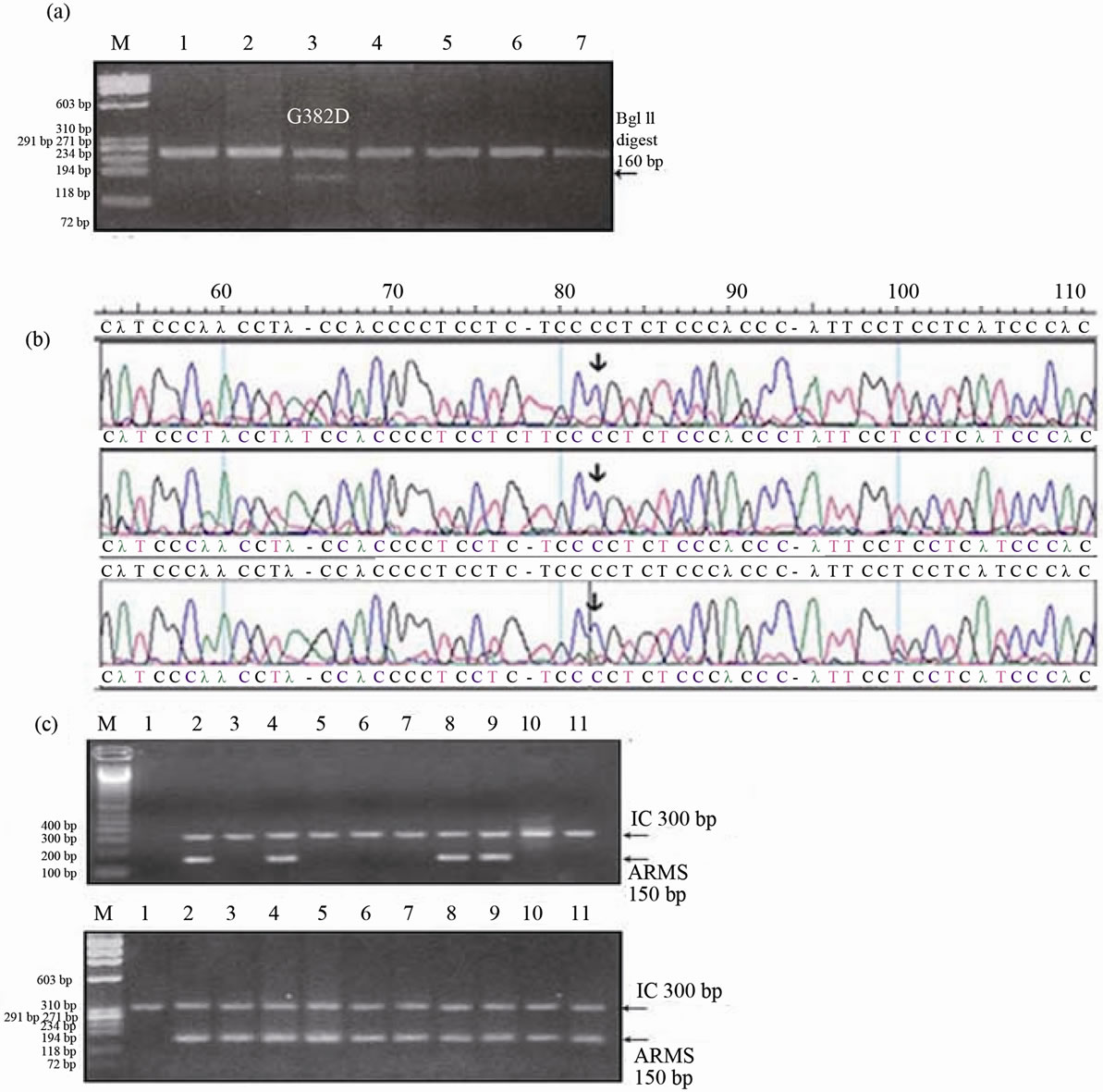
Figure 1. (a) RFLP assay for G382D (1145 G > A) polymorphism in Saudi controls. BglII restriction digests of the PCR product for different samples, lane 3 shows the G382D allele in a heterozygous sample, the wild type allele remained undigested (242 bp) and the mutant allele was digested generating a 160bp and 82 bp, all the other lanes had only the wild type allele. Lane 1: FX 174/HaeIII DNA size marker (M). (b) Electropherograms obtained by sequencing of the MUTYH exon 9 using the reverse primer and complimentary conting. The samples were screened for the R231C (891 C < T) mutation. Arrows point to the wild type allele (C) found in all the samples, mutant allele (T) was absent.(c) Agarose gel pictures resulting from the ARMS assay of the H324Q polymorphism in Saudi controls. Top: The gel pattern using the normal primer: No normal allele bands were detected in the different samples in lanes 3, 5, 6, 7, 10 and 11. Different individual samples in lanes 2, 4, 8, and 9 had a normal allele band, lanes 1 contained 100bp DNA size markers. Bottom: The gel pattern using the mutant primer: All except the first lane had mutant bands, lane 1 contained F X 174/HaeIII DNA size markers. Internal Control: SETX exon 20, 300 bp (M).
The IVS1+5 G/C SNP is located in intron 1 at the splice donor site where a G nucleotide is replaced by a “C”. Studies from lung cancer cell lines showed that such replacement causes the retention of the first 237 bp of intron 1 sequence between exon 1 and exon 2 of the transcript. The SNP was found to cause the activation of a cryptic splice donor site and the alternative splicing in intron 1 [20]. Comparison of the allele frequencies for IVS1+5 G/C mutation between the Saudi samples and an Australian study [19] revealed no significant difference (Fisher’s p-value = 0.0765, OR = 0.07559, 95% CI = 0.003596 - 1.589).
The V22M (66G > A) is located in exon 2 at the RPA binding domain of the MUTYH protein. This SNP may be associated with localization of MUTYH to the site of DNA replication [23] and is associated with substitution of hydrophobic valine for hydrophobic methionine resulting in a conservative change.
The study sample presented a frequency of 0.99 for the normal allele and of 0.01 for the mutant allele with no homozygous samples for the mutant allele. Interestingly, when the Saudi V22M allele frequencies were compared with four different populations (Europeans, Asians, Sub-Saharan Africans and African Americans), there was a significant difference between the Saudis and Europeans but a statistical similarity between the Saudis and the three other populations. Moreover, when compared with the same four populations, the Saudi H324Q allele frequencies were similar to Europeans, SubSaharan Africans and African Americans but were signifycantly different from the Asian population. These results are summarized in Table 3.
The Y165C (494 A > G) located in exon 7 of the MUTYH, causes the substitution of the hydrophobic tyrosine residue for a polar uncharged cysteine and results in a non-conservative mutation. This variation resides in the HhH motif of the MUTYH protein which is part of the catalytic core, and hence the mutation completely abolishes the adenine glycosylase activity towards the A.8-oxo-G mispairs. The G382D (1145 G > A) mutation located in exon 13 involves a substitution of a nonpolar uncharged glycine for a negatively charged aspartic acid, resulting in a non conservative pathogenic mutation. This mutation is located in the MutT like domain and it gives rise to decreased adenine removal [35]. In the present study on normal Saudis the allele frequencies for Y165C and G382D are similar to controls of Caucasians, Australians, Canadians, British populations [28]. Other studies showed that Y165C and G382D mutations were identified commonly in British, Italian, American, Portuguese and Dutch populations and never detected in East Asians including Japanese populations [21,22,36-38]. The low and similar Y165C and G382D allele frequencies between the Saudis and the other control populations further confirms that they are disease causing mutations.
The R231C (891 C > T) mutation located in exon 9 of the MUTYH protein causes the substitution of a charged arginine for an uncharged polar cysteine resulting in a non conservative mutation; the R231C appears to affect the interaction between the MUTYH and MSH6 and is assumed to be pathogenic [21]. No significant difference was observed in the allele frequencies for the R231C mutation between the study sample and that conducted by Miyaki et al. 2005 [21]. These results are summarized in Table 2.

Table 2. Comparison of Saudi IVS1 + 5 G/C and R231C MUTYH variations with other populations.
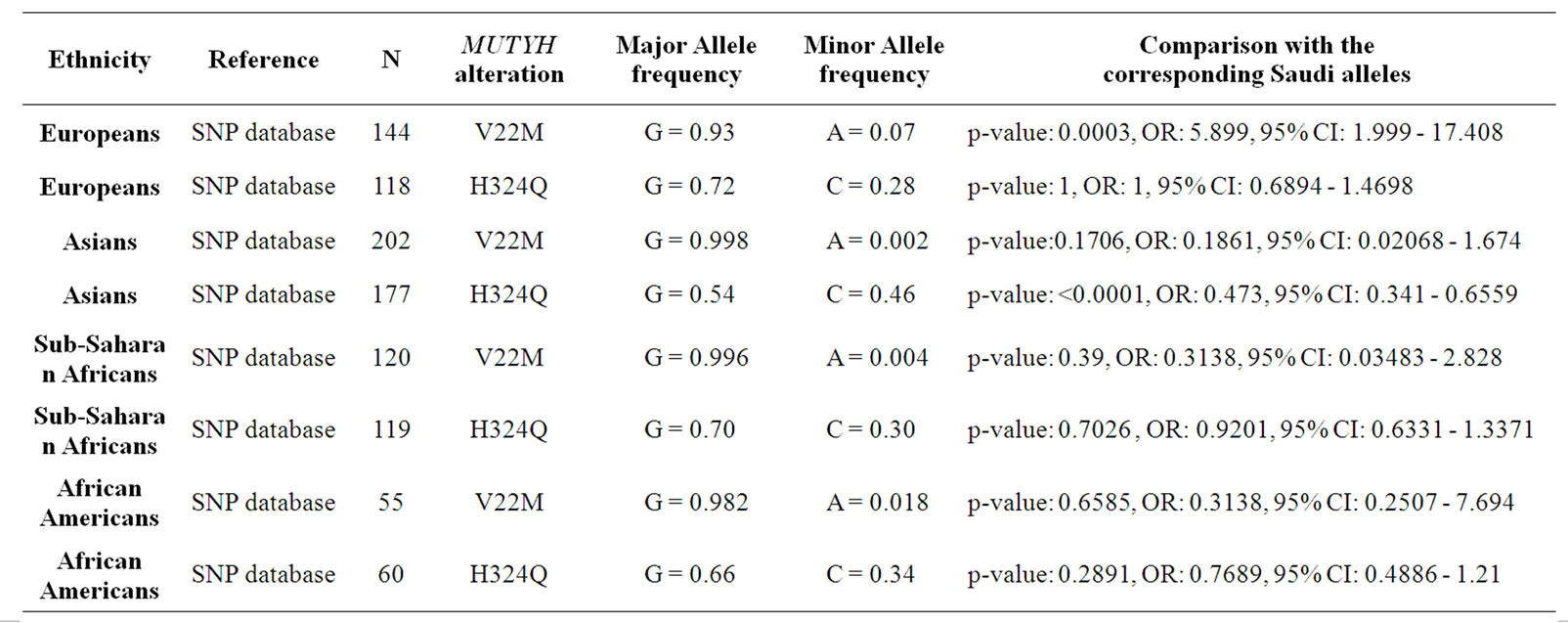
Table 3. Comparison of Saudi V22M and H324Q MUTYH variations with other populations.
The H324Q (972C > G) variation in exon 12 is associated with amino acid substitution of histidine to glutamine, where the replacement leads to a non-conservative polymorphism as a charged residue is substituted for an uncharged polar residue. There is no difference in the repair activities for the two types of polymorphic MUTYH proteins. This polymorphism is located near the APE1 binding site which is not a key functional site. The Saudi population H324Q allele frequency was similar to the Europeans, Sub-Saharan-Africans and African Americans. However, the Saudi H324Q allele frequency was significantly different compared to the Asians. These results are summarized in Table 3.
Genetic drift possibly contributed to the difference in allele frequency between the Asians and Saudis. The alleles subjected to random genetic drift would eventually reach fitness and hence the H324Q event in the Asians may be more recent than the Saudis and thus later in approaching 100% fitness.
Cigarette smoke is a rich source of ROS that induce a variety of DNA damage including oxidative DNA damages that can result in gene mutations, since guanine is the most susceptible base to oxidation resulting in 8- oxoG production; the 8-oxo-G has been recently considered the natural cause of genomic diversity and SNPs in organisms because of its mutagenic capability [5].
The allele frequencies for V22M, H324Q and G382D were compared between smoker and non-smoker subdivisions of the study group (113 smokers and 40 nonsmokers). No significant difference was observed in any of these variants allele frequencies between smokers and non-smokers, the comparison resulted in the following statistical parameters (Fisher’s p-value = 0.5762, OR = 0.3043, 95% CI = 0.01619 - 5.720), (Fisher’s p-value = 0.5563, OR = 0.8209, 95% CI = 0.4659 - 1.4463), (Fisher’s p-value = 1, OR = 0.954, 95% CI = 0.03852 - 23.746) for V22M, H324Q and G382D, respectively.
In a recent study it was found that smokers carrying the wild type of OGG1 S326C and variant genotype of XRCC1 R194W and variant genotype of MUTYH H324Q had a 31.86 fold increased risk for bladder cancer [39]. Another study suggested that the MUTYH H324Q polymorphism appears to play an important role in lung cancer risk in the Japanese population [40]. Moreover, the Saudis were found in this study to be statistically different from Asians (including Japanese) in the H324Q polymorphism as the mutant form was more common in the Asians. Also the Saudis were found to be statistically different from Europeans in the V22M polymorphism and the mutant form was more common in Saudis.
Although different studies showed a significant effect between smoking and genetic polymorphisms in the MUTYH yet there were no significant difference in any of the MUTYH allele frequency between smokers and non-smokers in this study.
In terms of future work, it is important to determine the allele frequencies for these MUTYH variations in the Saudi Colorectal cancer patients, endometrial cancer and liver cancer patients as some MUTYH mutations may be associated with increased susceptibility to these cancers.
5. CONCLUSIONS
This detailed report on MUTYH variants in Saudis and comparison with other populations highlights the existence of ethnic differences between MUTYH variants and further confirms the pathogenic role of some MUTYH mutations previously linked to disease.
6. ACKNOWLEDGEMENTS
This work was supported by grants from SABIC via research centre at King Saud University, Riyadh, Saudi Arabia. The authors declare no conflict of interests.
REFERENCES
- De Bont, R. and van Larebeke, N. (2004) Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis, 19, 169-185. doi:10.1093/mutage/geh025
- Lindahl, T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709-715. doi:10.1038/362709a0
- Jackson, A.L. and Loeb, L.A. (2001) The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutation Research, 477, 7-21. doi:10.1016/S0027-5107(01)00091-4
- Nohmi, T., Kim, S.R. and Yamada, M. (2005) Modulation of oxidative mutagenesis and carcinogenesis by polymorphic forms of human DNA repair enzymes. Mutation Research, 591, 60-73. doi:10.1016/j.mrfmmm.2005.03.033
- Ohno, M., et al. (2006) A genome-wide distribution of 8- oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Research, 16, 567-575. doi:10.1101/gr.4769606
- Slupska, M.M., et al. (1999) Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. Journal of Bacteriology, 181, 6210-6213.
- Ohtsubo, T., et al. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Research, 28, 1355-1364. doi:10.1093/nar/28.6.1355
- Shinmura, K., et al. (2000) Adenine excisional repair function of MYH protein on the adenine: 8-hydroxyguanine base pair in double-stranded DNA. Nucleic Acids Research, 28, 4912-4918. doi:10.1093/nar/28.24.4912
- Tsuzuki, T., Nakatsu, Y. and Nakabeppu, Y. (2007) Significance of error-avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Science, 98, 465-470. doi:10.1111/j.1349-7006.2007.00409.x
- Michaels, M.L., et al. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proceedings of the National Academy of Sciences of the USA, 89, 7022-7025. doi:10.1073/pnas.89.15.7022
- Charames, G.S. and Bapat, B. (2003) Genomic instability and cancer. Current Molecular Medicine, 3, 589-596. doi:10.2174/1566524033479456
- Al-Tassan, N., et al. (2002) Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nature Genetics, 30, 227-232. doi:10.1038/ng828
- Cheadle, J.P. and Sampson, J.R. (2007) MUTYH-associated polyposis from defect in base excision repair to clinical genetic testing. DNA Repair, 6, 274-279. doi:10.1016/j.dnarep.2006.11.001
- Enholm, S., et al. (2003) Proportion and phenotype of MYH-associated colorectal neoplasia in a populationbased series of Finnish colorectal cancer patients. American Journal of Pathology, 163, 827-832. doi:10.1016/S0002-9440(10)63443-8
- Halford, S.E., et al. (2003) Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. American Journal of Pathology, 162, 1545-1548. doi:10.1016/S0002-9440(10)64288-5
- Jones, S., et al. (2002) Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Human Molecular Genetics, 11, 2961-2967. doi:10.1093/hmg/11.23.2961
- Sampson, J.R., et al. (2003) Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet, 362, 39-41. doi:10.1016/S0140-6736(03)13805-6
- Sieber, O.M., et al. (2003) Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. The New England Journal of Medicine, 348, 791-799. doi:10.1056/NEJMoa025283
- Kambara, T., et al (2004) Role of inherited defects of MYH in the development of sporadic colorectal cancer. Genes Chromosomes Cancer, 40, 1-9. doi:10.1002/gcc.20011
- Yamaguchi, S., et al. (2002) A single nucleotide polymorphism at the splice donor site of the human MYH base excision repair genes results in reduced translation efficiency of its transcripts. Genes Cells, 7, 461-474. doi:10.1046/j.1365-2443.2002.00532.x
- Miyaki, M., et al. (2005) Germline mutations of the MYH gene in Japanese patients with multiple colorectal adenomas. Mutation Research, 578, 430-433. doi:10.1016/j.mrfmmm.2005.01.017
- Tao, H., et al. (2008) Association between genetic polymerphisms of the base excision repair gene MUTYH and increased colorectal cancer risk in a Japanese population. Cancer Science, 99, 355-360. doi:10.1111/j.1349-7006.2007.00694.x
- Parker, A.R. and Eshleman, J.R. (2003) Human MutY: Gene structure, protein functions and interactions, and role in carcinogenesis. Cellular and Molecular Life Sciences, 60, 2064-2083. doi:10.1007/s00018-003-3053-4
- Nielsen, M., et al. (2007) Cost-utility analysis of genetic screening in families of patients with germline MUTYH mutations. BMC Medical Genetics, 8, 42. doi:10.1186/1471-2350-8-42
- Rahman, N. and Scott, R.H. (2007) Cancer genes associated with phenotypes in monoallelic and biallelic mutation carriers: new lessons from old players. Human Molecular Genetics, 16, 60-66. doi:10.1093/hmg/ddm026
- Sulova, M., et al. (2007) Mutation analysis of the MYH gene in unrelated Czech APC mutation-negative polyposis patients. European Journal of Cancer, 43, 1617- 1621. doi:10.1016/j.ejca.2007.04.010
- Shields, P.G. and Harris, C.C. (2000) Cancer risk and low-penetrance susceptibility genes in gene-environment interactions. Journal of Clinical Oncology, 18, 2309- 2315.
- Peterlongo, P., et al. (2005) Colorectal cancer risk in individuals with biallelic or monoallelic mutations of MYH. International Journal of Cancer, 114, 505-507. doi:10.1002/ijc.20767
- Boiteux, S. and Radicella, J.P. (2000) The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Archives of Biochemistry and Biophysics, 377, 1-8. doi:10.1006/abbi.2000.1773
- Nakabeppu, Y., et al. (2004) The defense mechanisms in mammalian cells against oxidative damage in nucleic acids and their involvement in the suppression of mutagenesis and cell death. Free Radical Research, 38, 423- 429. doi:10.1080/10715760410001688348
- Bruner, S.D., Norman, D.P. and Verdine, G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859- 866. doi:10.1038/35002510
- Shinmura, K. and Yokota, J. (2001) The OGG1 gene encodes a repair enzyme for oxidatively damaged DNA and is involved in human carcinogenesis. Antioxidants and Redox Signaling, 3, 597-609. doi:10.1089/15230860152542952
- Croitoru, M.E., et al. (2004) Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. Journal of the National Cancer Institute, 96, 1631-1634. doi:10.1093/jnci/djh288
- Fleischmann, C., et al. (2004) Comprehensive analysis of the contribution of germline MYH variation to earlyonset colorectal cancer. International Journal of Cancer, 109, 554-558. doi:10.1002/ijc.20020
- Kairupan, C.F., et al. (2005) Mutation analysis of the MYH gene in an Australian series of colorectal polyposis patients with or without germline APC mutations. International Journal of Cancer, 116, 73-77. doi:10.1002/ijc.20983
- Tao, H., et al. (2004) A novel splice-site variant of the base excision repair gene MYH is associated with production of an aberrant mRNA transcript encoding a truncated MYH protein not localized in the nucleus. Carcinogenesis, 25, 1859-1866. doi:10.1093/carcin/bgh206
- Zhang, Y., et al. (2006) Germline mutations and polymorphic variants in MMR, E-cadherin and MYH genes associated with familial gastric cancer in Jiangsu of China. International Journal of Cancer, 119, 2592-2596. doi:10.1002/ijc.22206
- Kim, I.J., et al. (2004) Mutational analysis of OGG1, MYH, MTH1 in FAP, HNPCC and sporadic colorectal cancer patients: R154H OGG1 polymorphism is associated with sporadic colorectal cancer patients. Human Genetics, 115, 498-503. doi:10.1007/s00439-004-1186-7
- Huang, M., et al. (2007) High-order interactions among genetic variants in DNA base excision repair pathway genes and smoking in bladder cancer susceptibility. Cancer Epidemiology, Biomarkers and Prevention, 16, 84-91. doi:10.1158/1055-9965.EPI-06-0712
- Miyaishi, A., et al. (2009) MUTYH Gln324His gene polymerphism and genetic susceptibility for lung cancer in a Japanese population. Journal of Experimental and Clinical Cancer Research, 28, 10. doi:10.1186/1756-9966-28-10