Open Journal of Pediatrics
Vol.3 No.1(2013), Article ID:28806,3 pages DOI:10.4236/ojped.2013.31008
A C1-inhibitor rare mutation: Early diagnosis of hereditary angioedema in a paediatric patient
![]()
1Department of Health, Motherhood and Childhood Advancement Sciences, University of Palermo, Palermo, Italy
2Operative Unit of Molecular Genetic Laboratory, Ospedali Riuniti Villa Sofia-Cervello, Palermo, Italy
Email: *mariacristina.maggio@unipa.it, piero.sammarco@libero.it, carmelofabiano@alice.it, giocors@alice.it
Received 6 September 2012; revised 1 November 2012; accepted 3 December 2012
Keywords: Hereditary Angioedema; C1-Inhibitor Deficiency
ABSTRACT
Hereditary angioedema secondary to C1-inhibitor deficiency is a rare autosomal dominant disorder characterized by a deficiency of C1 esterase inhibitor. An eight-year-old girl showed periorbital painless swelling, diagnosed as ethmoiditis. A craniofacial scan did not evidence a paranasal sinus involvement, C1INH levels were undetectable, with low C4 levels: 7.6 mg/dl and C1INH: <8.46 mg/dl. The genetic study identified a rare mutation of the C1INH gene. This clinical report is of relieve because paediatric cases described in literature are rare, did not presented a positive family history, and received a diagnosis after many attacks. Furthermore our girl received a prompt diagnosis of HAE at the first attack of angioedema.
1. BACKGROUND
Hereditary angioedema (HAE), secondary to C1-inhibitor deficiency is a rare autosomal dominant disorder, characterized by a deficiency of C1 esterase inhibitor (C1INH) protein or function. Two types of the disorder are described. Type I, representing 85% of patients, characterized by serum levels of C1INH less than 35% of normal; type II defined by C1INH levels normal or elevated, with a non-functional protein. The 2 types are clinically indistinguishable [1].
Guidelines regarding diagnostic criteria or routine testing of family members of patients with HAE are not clear. Laboratory data for diagnosis include complement factor 4 level; C1INH antigenic protein level, which is reduced in approximately 85% of patients with HAE; C1INH functional assay, which is considered an unreliable test in the United States secondary to inconsistent standardization of the assays.
HAE is a disabling condition characterized by intermittent episodes of bradykinin-mediated angioedema, involving the skin, the larynx and the intestinal mucosa, with abdominal pain indistinguishable from acute abdomen [2].
The risk of life-treating attacks is real in 50% of the patients and is lethal in 25% - 30% of undiagnosed cases, when airway obstruction occurs.
C1INH replacement therapy is the treatment of choice for acute edema attacks in patients with HAE. C1INH should be considered the first-line treatment for acute oedema attacks, because of its fast onset of action and effectiveness. It is not clear whether fixed or weightbased dosing is preferred. Ecallantide (a kallikrein inhibitor) or Icatibant (a selective bradykinin B2 receptor antagonist) can be considered as a second-line treatment choice. A recombinant human C1INH is available in Europe [3]. In patients with air obstruction, when these drugs are not available, fresh frozen plasma can be used [4].
In paediatric patients early diagnosis is possible in children with an index case in the family.
However in the other patients early diagnosis is not easy for the frequent atypical presentation, and for the rare incidence of the disease, witch induces the physiccians to other diagnosis.
2. CASE PRESENTATION
We describe the clinical case of an eight year old girl, admitted to our division for the onset of periorbital rubor and oedema, started the day before and diagnosed as ethmoiditis.
She underwent an otolaryngologist evaluation and a craniofacial scan without any evidence of paranasal sinuses involvement.
Because of an increasing face and neck swelling, the girl was admitted to our unit for the definition of the diagnosis.
She showed a severe facial painless swelling, causing facial disfigurement, with difficulty to open eyes. The parents revealed a mild facial trauma that occurred the day before. However she did not show any subcutaneous haematoma of the region, despite more than 24 hours occurred from the injury.
The anamnestic familial records revealed that the father and the grandfather, during their youth, often showed legs painless swelling, not related to fever or to other etiological events, but subsequent to occasional traumatisms. They showed skin oedema, without visceral involvement.
The haematological assays of our patient were in the normal range; she only showed low C4 levels: 7.6 mg/dl (n.v.: 18 - 50), with C1NH undetectable: <8.46 mg/dl (n.v.: 15 - 35).
Genetic analysis of C1NH gene was sequenced. The use of glucocorticoids, epinephrine and antihistamines was ineffective, as described in HAE [5,6]. Since human C1-esterase concentrate was not available, she received C1INH by 1 unit of fresh plasma, repeated for two consecutive days, with a clear resolution of the swelling [4].
2.1. Materials and Methods
The C1INH gene, which maps on chromosome 11q12- q13, consists of eight exons and seven introns distributed over a stretch of DNA of 17 kb. The genetic analysis was carried out from genomic DNA isolated from peripheral white blood cells by QIAamp DNA blood minikit, Qiagen. DNA was subjected to 30 cycles of PCR amplification under the following conditions: an initial denaturation of 5 min at 95˚C followed by cycles of denaturetion at 95˚C for 30 sec, annealing at 60˚C for 30 sec and extension at 72˚C for 40 sec in a Perkin Elmer 9700 thermocycler. PCR was carried out in a reaction volume of 25 ml containing 200 ng DNA, 200 mM of each primer, 2 mM MgCl2, 1 × PCR Taq buffer, and 1 U of Taq DNA polymerase (Invitrogen). Primers used (MWG) were selected from genomic DNA sequence by our self in intronic regions flanking all 8 exons including promoter region and intron/exon boundaries (see Table 1).
The PCR products were purified and sequenced using Big Day terminator cycle sequencing kit (PerkinElmer, Oak Brook, IL, USA) and analyzed on ABI Prism 3100 DNA Sequencer Avant Genetic Analyzer (Perkin Elmer, Applied Biosystems) following the manufacture’s protocols.
2.2. Results
Our patient was heterozygous for IVS4 + 1 (G > A) mutation in intron 4. The same mutation was found in her father but neither in her brother nor in her mother (see Figures 1 and 2). This mutation was described in literature only twice [7,8]: it alters the invariant G nucleotide of the acceptor splice site of intron 4. The consequence of this splice-site mutation results in a defect of C1INH product, through a rarely described splice-site at intron 4/exon 5 junction, which has been found in the propositus and her father.
In literature a mutation in intron 4 (G > T) is more frequently described [9].
3. CONCLUSIONS
This clinical report is of relieve: in fact there are just a few paediatric cases of C1INH deficiency described in literature. In these cases children did not present a positive family history and they received the correct diagno sis after many attacks. Furthermore our girl received a prompt diagnosis of HAE at the first attack of angioedema, with an adequate treatment.
However the genetic mutation, detected in our patient
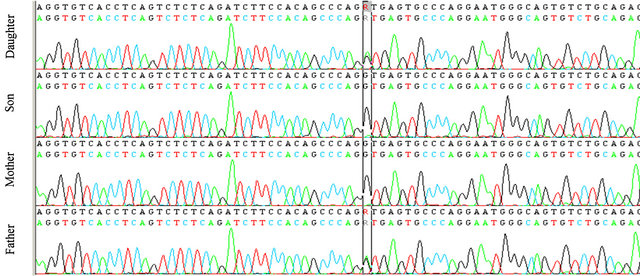
Figure 1. The genetic analysis of C1INH gene in the family.
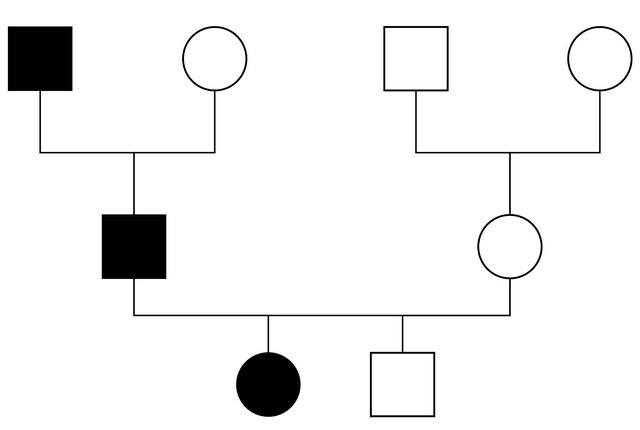
Figure 2. The patient’s family pedigree.
Table 1. The primers used in the study of C1INH gene.

and in her father, is a rare mutation, only described, as referenced above, in only two papers.
4. CONSENT
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
REFERENCES
- Bowen, B., Hawk, J.J., Sibunka, S., Hovick, S. and Weiler, J.M. (2001) A review of the reported defects in the human C1 esterase inhibitor gene producing hereditary angioedema including four new mutations. Clinical Immunology, 98, 157-163. doi:10.1006/clim.2000.4947
- Verdi, M. and Shaker, M. (2011) An update on hereditary angioedema. Advanced Emergency Nursing Journal, 33, 163-178.
- Bork, K. (2012) Current management options for hereditary angioedema. Current Allergy and Asthma Reports, 12, 273-280. doi:10.1007/s11882-012-0273-4
- Prematta, M., Gibbs, J.G., Pratt, E.L., Stoughton, T.R. and Craig, T.J. (2007) Fresh frozen plasma for the treatment of hereditary angioedema. Annals of Allergy, Asthma & Immunology, 98, 383-388. doi:10.1016/S1081-1206(10)60886-1
- Farkas, H. (2010) Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma & Clinical Immunology, 6, 18. doi:10.1186/1710-1492-6-18
- Krishnamurthy, A., Naguwa, S.M. and Gershwin, M.E. (2008) Pediatric angioedema. Clinical Reviews in Allergy and Immunology, 34, 250-259. doi:10.1007/s12016-007-8037-y
- Gösswein, T., Kocot, A., Emmert, G., Kreuz, W., Martinez-Saguer, I., Aygören-Pürsün, E., Rusicke, E., Bork, K., Oldenburg, J. and Müller, C.R. (2008) Mutational spectrum of the C1INH (SERPING1) gene in patients with hereditary angioedema. Gytogenetic and Genome Research, 121, 181-188. doi:10.1159/000138883
- Pappalardo, E., Caccia, S., Suffritti, C., Tordai, A., Zingale, L.C. and Cicardi, M. (2008) Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: Functional and structural correlates. Molecular Immunology, 45, 3536-3544. doi:10.1016/j.molimm.2008.05.007
- Kalmar, L., Hegedus, T., Farkas, H., Nagy, M. and Tordai, A. (2005) HAEdb: A novel interactive, locus-specific mutation database for the C1 inhibitor gene. Human Mutation, 25, 1-5. doi:10.1002/humu.20112
NOTES
*Corresponding author.