Open Journal of Physical Chemistry
Vol. 2 No. 1 (2012) , Article ID: 17542 , 9 pages DOI:10.4236/ojpc.2012.21008
A Review of the Aqueous Aerosol Surface Chemistry in the Atmospheric Context
Department of Chemical Engineering, Louisiana State University, Baton Rouge, USA
Email: Valsaraj@lsu.edu
Received October 24, 2011; revised November 21, 2011; accepted December 22, 2011
Keywords: Aerosols; Water Film; Adsorption; Reaction
ABSTRACT
In this review the surface chemistry and properties of aqueous atmospheric aerosols are explored. Water plays a major role in scavenging pollutants. Reactions occur on thin water films in atmospheric aerosols. The study of the aerosol water surface is important to properly account for chemical transformations in the troposphere. The thermodynamics of adsorption of organic molecules and oxidant species on the aqueous surface and, the techniques employed to quantify the adsorption isotherms are summarized. Experimental techniques for elucidating the reactions on the water surface are described. Field and laboratory data for oxidation reactions of compounds at the air-water interface are summarized. The Langmuir-Hinshelwood reaction mechanism is useful in quantifying the reaction rate on the aqueous aerosol surface. A hypothesis for the large heterogeneous reaction rate on the water surface over the homogeneous bulk aqueous phase reaction is presented.
1. Introduction
Atmospheric aerosols are colloidal systems with one phase (solid or liquid) dispersed in another phase (gas). Solid aerosols are generally organic or inorganic particles dispersed in air ranging from sub-micron to a few microns in diameter; they may either be dry or wet. Liquid aerosols comprise of rain, fog, mist, cloud droplets, snow and ice. Table 1 provides the essential characteristic dimensions and properties of atmospheric aerosols. It is noteworthy that they have lifetimes ranging from a few minutes to days. The presence of surfaces such as cloud, fog and fine particulates can influence atmospheric processes with characteristic times of the order of hours.
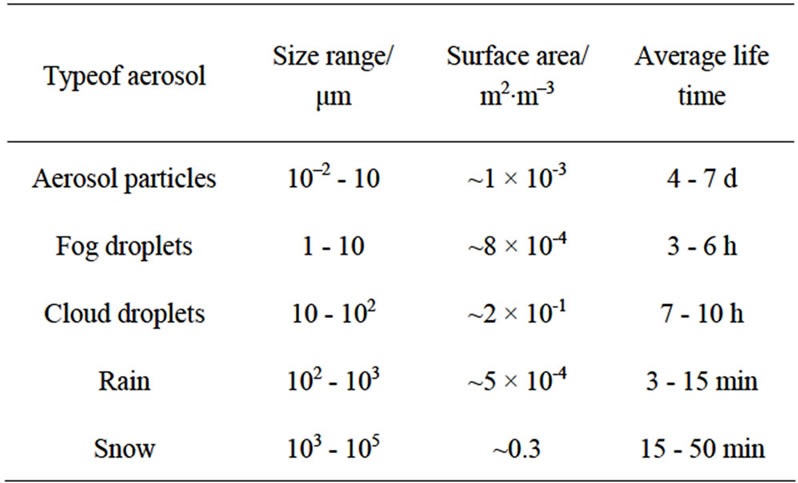
Table 1. Typical properties of atmospheric aerosols [1,2].
Aerosols play a large role in tropospheric chemistry and climate. Aerosols take part in numerous heterogeneous reactions with gases (organic and inorganic) and atmospheric oxidants (ozone, hydroxyl radical, singlet oxygen, nitrate radical, chlorine). Aerosols absorb and reflect sunlight and contribute to direct and indirect effects on climate forcing. The 2007 IPCC assessment [3] on global climate change stated that the largest uncertainty is the effect of aerosols on climate forcing. Over the last few decades an impressive array of data has been accumulated on the role of atmospheric aerosols.
Atmospheric aerosols consist of a core of inorganic or organic nucleus (e.g., soluble salts, insoluble black carbon, soil or soot) that forms the condensation site for water as well as organic and inorganic molecules. Water effects the aerosol properties in various ways: 1) it determines the nature of aerosol (solid, liquid, or glassy/ amorphous), 2) the hygroscopic growth and size of the aerosol, 3) uptake of gases and oxidants, and 4) multiphase reactions in aerosol droplets. The water content of the aerosol is determined by environmental factors such as the type of condensation nucleus (CN), the relative humidity, temperature, and oxidation state of molecules on the surface of the aerosol. The water either plays the role of a competitive adsorbate, or acts as a substrate to provide the surface area for reaction. If the water content is substantially less than that required for a monomolecular layer, then the individual water molecules compete for adsorption sites on the surfaces of CN. If, on the other hand, the water forms a thin layer (1 nm to 50 µm) of high specific surface area it provides the site for adsorption and reaction on the aerosol. For water films of ~1 - 2 nm on silica, the surface-induced modifications to the adsorbed film propagate to ~1 - 4 nm from the solid-water interface [4]. When the water layer thickness exceeds few monolayers (>100 µm), the bulk water properties predominate.
Thin layers of water are important from the atmospheric chemistry perspective. The thin water layer in aerosols may have properties different from those of bulk water. Questions that are relevant in this regard include the following: 1) Is there any difference between “bound” and “unbound” water? 2) Is the H-bonding characteristic of water near surfaces different from bulk water? 3) Are the activity coefficients of solutes in thin layers of water (γi) different from their infinite dilute activity coefficients (γi∞)?
Figure 1 is the schematic of an aqueous aerosol. The aqueous phase consists of dissolved inorganic ions, small particles and dissolved organic species, some of which are surface active and of large molecular weight that form a distinct phase on the water surface. The adsorption and reaction of trace gases and oxidants in the atmospheric aerosol will be influenced by the nature and composition of the aqueous phase. There have been several reports that describe the relevance of the aqueous surface in atmospheric droplets. For example, one of the earliest reports involved the conversion of sulfur dioxide in fog and cloud droplets to sulfates in the context of the so-called “acid rain” issue [2]. More recently, aqueous aerosol surface reaction was invoked for the depletion of ozone near sea surface aerosols [5]. There is also evidence for the real-time production of secondary organic aerosols in fog droplets [6]. We have provided evidence for the formation of both oxygenated PAHs and other compounds in fog sampled in the United States [7]. A variety of questions remain on the mechanisms of transport, adsorption and reaction on aqueous aerosol droplets [8].
In the following review I will summarize the various physico-chemical aspects pertaining to the adsorption and reaction of organic molecules on aqueous aerosol droplets and water thin films.
2. The Air-Water Interface
The surface of water is unique in its energy characteristics. The surface tension of water (72 mN·m–1) is one of the highest for any liquid. Hence, the air-water interface is considered “the most hydrophobic, non-polar surface known” [9]. The “hyper hydrophobicity of the air-water interface” attracts hydrophobic moieties to the surface
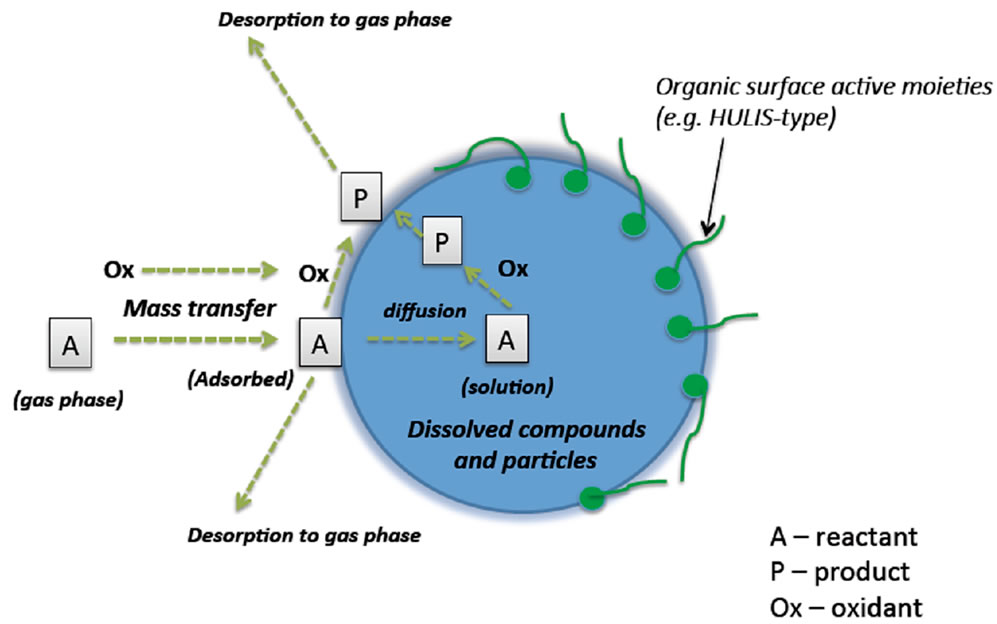
Figure 1. A pictorial representation of an atmospheric aqueous aerosol and its interactions with the gas-phase molecule and oxidant species.
[10]. The hydrophobic interactions are the basis for a number of well-known phenomena such as [11]: 1) the low aqueous solubility of hydrophobic molecules in water, 2) the ability of proteins to fold in water, 3) the formation of micelles by surfactants in water, 4) the inability of water to spread on hydrophobic surfaces, 5) the ability to separate hydrophobic particles and compounds by attachment to air bubbles (foam flotation) and, 6) dimerization of hydrophobic chains in water.
The separation of hydrophobic organic compounds (HOC) from water by adsorption on air bubbles, viz. solvent sublation, was developed in our laboratory for wastewater treatment [12]. It was observed that the inverse process, viz., water droplets in air (as in a spray column), was able to remove high concentrations of HOC by adsorption [13]. The latter process was also invoked to explain the high concentrations of HOC observed in fog and dew waters collected from urban areas [14]. Further there has been several observations regarding the unique reactions of gas-phase HOC in fog and cloud droplets leading to the formation of products that are precursors to secondary organic aerosols (SOA) in the aqueous phase [15]. Field data on real-time SOA formation in a London fog event was recently reported [6].
Two aspects concerning trace gas interactions with atmospheric water surfaces need study [16]. Firstly, we need data on the thermodynamics of adsorption at the air-water interface. Secondly, we need information on the reactivity of adsorbed molecules towards gas-phase and liquid-phase oxidant species. In the following discussion, we focus on both aspects.
2.1. Thermodynamics of Adsorption at the Air-Water Interface
Utilizing the thermodynamic criteria of equal fugacities between the air and the air-water interface, we can derive the following equation for the free energy of partitioning for an HOC [17]
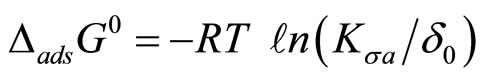 (1)
(1)
where Kσa is the Kemball-Rideal standard state surface thickness (=6 × 10–10 m). Experimental values of Kσa can be obtained from a variety of techniques. These include direct and indirect methods and are summarized in Table 2. The first two methods in Table 2 are direct methods that are suitable for compounds that have high vapor pressure. The last two indirect methods are suitable for compounds with low vapor pressure. They are especially useful for obtaining thermodynamic adsorption parameters (free energy, enthalpy and entropy) on thin water films of a few microns in thickness.
The experimental methods described above are complimentary to correlations with compound properties such as sub-cooled liquid vapor pressure, octanol-air partition constant, and molecular properties (e.g., molecular connectivity index, molecular surface area, molar volume) [23,24]. Further, the correlations were extended to include polar and non-polar compounds via the use of H-bond acceptor and donor indices [25,26]. Ab-initio and quantum chemistry based calculations of the partition constants using a Universal Surface Area and Solvation Model [27] and the use of the Conductor-Like Screening Model (COSMOTherm) [28] were also reported to provide good comparisons to experimental data.
Molecular dynamics (MD) and Monte Carlo (MC) simulations have been performed for the transfer of HOCs from air to water to determine the free energy of adsorption and solvation of molecules [29-33]. MD simulations provide good estimates of the basic thermodynamic properties, which can be compared to the experimental values. These calculations have been made not only for the HOCs but also for various gas-phase oxidant species (OH, O3, H2O2) at the interface [34]. MD simulations for the air-to-water transfer of a series of PAHs showed that a free energy minimum exists at the air-water interface which is conducive to surface adsorption. The free energy difference was larger between the bulk air and the interface than between the bulk air and bulk water phases, indicating a large energy for complete solvation of the molecule. The free energy values obtained from the calculations agree with the experimental values obtained (Table 3). The presence of surfactants at the interface increased the free energy minimum [33]. The MD and MC simulations also allowed an understanding of the differences in surface orientation of the HOC in the presence of the surfactant [33]. It is clear that the adsorption at the air-water interface is a favorable process, but that the complete solvation (solution) of the molecule involves considerable energy penalty for HOCs. However, for the small oxidant species the two free energy values are similar, although there is a slight preference for the interfacial adsorption.

Table 2. Methods suitable for determining the partition constants of HOC from the gas phase to the aqueous surface.
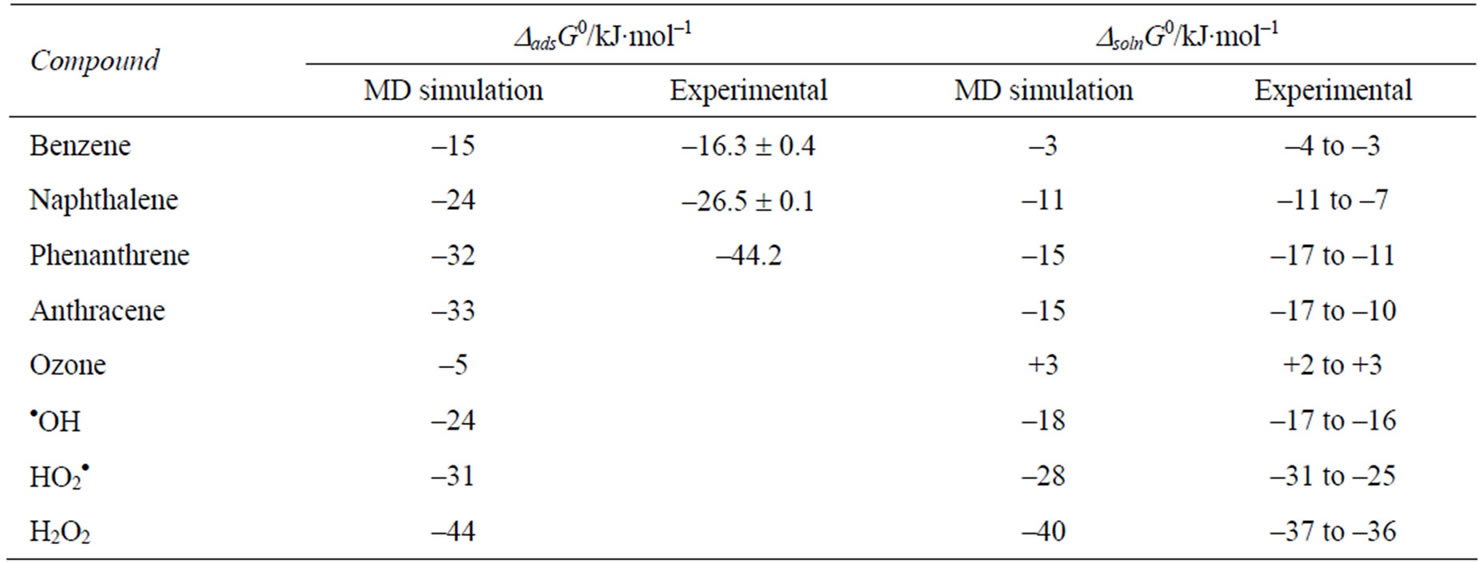
Table 3.Experimental and MD simulated values of free energy of adsorption and solution for HOC and Oxidant species [29,35,36].
2.2. Reactions of HOCs at the Air-Water Interface
As described above, both gas-phase oxidants and gaseous HOCs show a surface free energy minimum. We conclude that surface interactions between them should be feasible. The question is whether “surface” water is different from “bulk” water in supporting surface interactions. For example, theoretical work show that there is an increase in energy of surface water molecules of approximately 10 - 15 kcal·mol–1 over bulk water molecules [37] and, hence surface contains, on average, far more reactive sites than the bulk.
A number of techniques have been employed to study the reactions of gaseous HOC molecules with typical gas-phase oxidants at the air-water interface, which are summarized in Table 4. The air-water interface is difficult to probe compared to a gas-solid interface due to features peculiar to the former. For example, techniques suitable to study reactions of molecules on solids under vacuum are not useful since water has a high partial pressure. Unlike a solid surface, a water surface is constantly renewed and highly fluxional. The mobility of molecules adsorbed on water surfaces is larger than on solid surfaces with sub-nanosecond time scales for surface to bulk exchange of molecules. The delineation of a true surface phase for water is problematic since composition and density are not necessarily sharp and extend over a significant depth. Thus, the probing of water surfaces had to wait till suitable non-linear techniques were available to selectively amplify the molecular signals. One such technique is the sum frequency generation (SFG) method. This technique has been utilized to study the orientation of molecules at the water surface as well as the potential for configurational changes upon adsorption [46,47]. SFG has also been used to explore the dynamics of adsorption of molecules on the water surface [48].
A useful method to simultaneously study the adsorption and reaction involves the tracking of a molecular signal from the adsorbed species. For example, the adsorption and reaction of PAHs can be explored by following their characteristic fluorescence signals. Glancing angle laser induced fluorescence (LIF) of PAHs adsorbed at the air-water interface and its changes as a result of exposure and reaction with gas-phase oxidants (e.g., ozone) can be followed to obtain the progress of the reaction at the interface [40]. LIF can be also used to directly sample other reactions at the surface such as singlet oxygen with PAH molecules [48]. A recent innovation is the use of droplet electrospray mass spectroscopy [40], which involves the introduction of droplets of water containing the probe molecule into an electron spray mass spectrometer. The droplets are then exposed for a very short burst of an oxidant and the surface concentration directly ascertained. The principle of exposing water droplets in a falling droplet reactor to gas phase PAH and ozone and liquid analysis by HPLC/MS was used in our laboratory to study the surface reactions as a function of droplet size [43]. The reaction of aqueous surface adsorbed halide species to gas phase chlorine was reported recently using glancing angle Raman spectroscopy [44]. The pseudo-first order reaction rate constants for various molecules with gas phase oxidants have been explored using the above methods. Two examples are shown in Figures 2 and 3. An interesting observation is the saturation-type rate behavior. This can be justified by invoking a Langmuir-Hinshelwood (L-H) type surface reaction mechanism. Figure 3 shows the schematic of the same. The mechanism involves the reaction of adsorbed organic molecules with the oxidant on the surface of the water. The overall reaction rate is given by
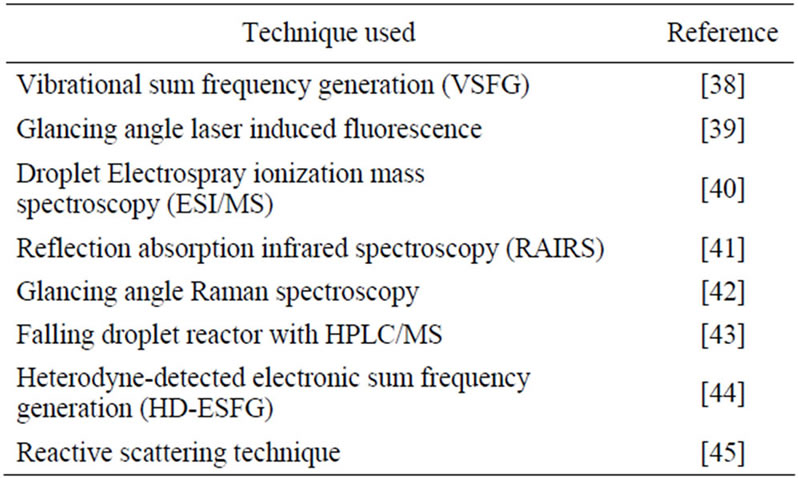
Table 4. A sampling of various techniques used for studying the adsorption and reaction at the water surface.
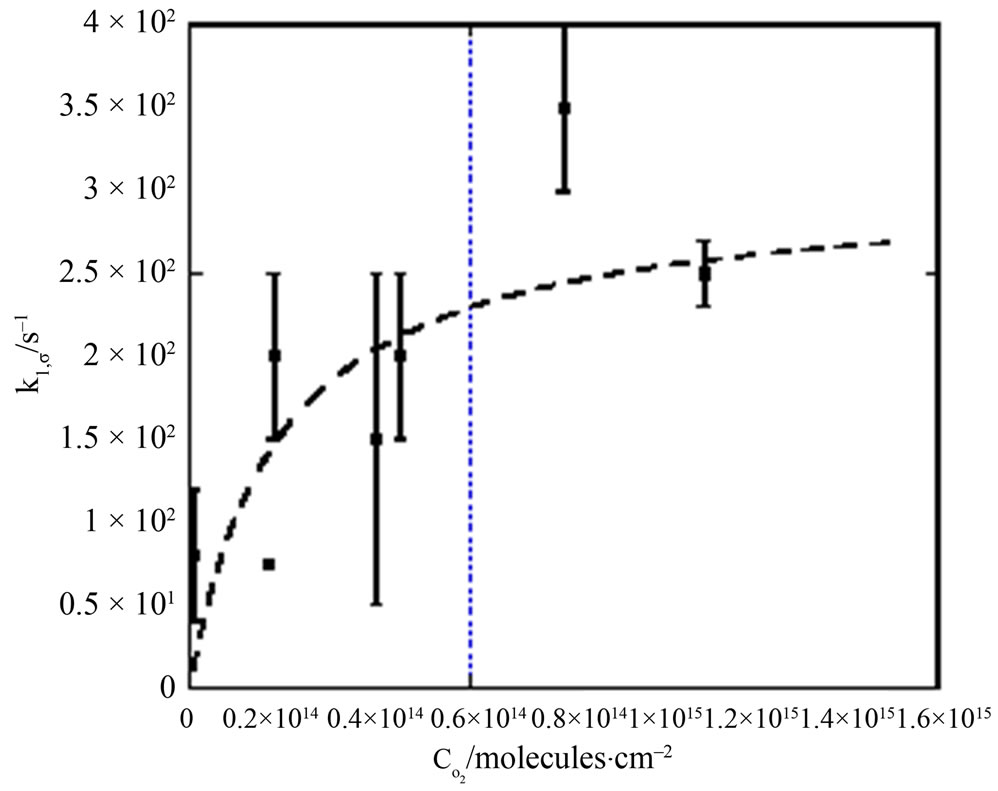
Figure 2. The pseudo first order reaction rate of naphthalene with gaseous ozone on aqueous droplets of 91 mm diameter in a falling droplet reactor. Data obtained from Raja et al [43]. The data fit gave kmax = 303 s–1 and COx,1/2 = 1.92 × 1014 molecules.cm-3. Note the asymptotic approach to a constant rate constant at high ozone concentration in the gas phase.
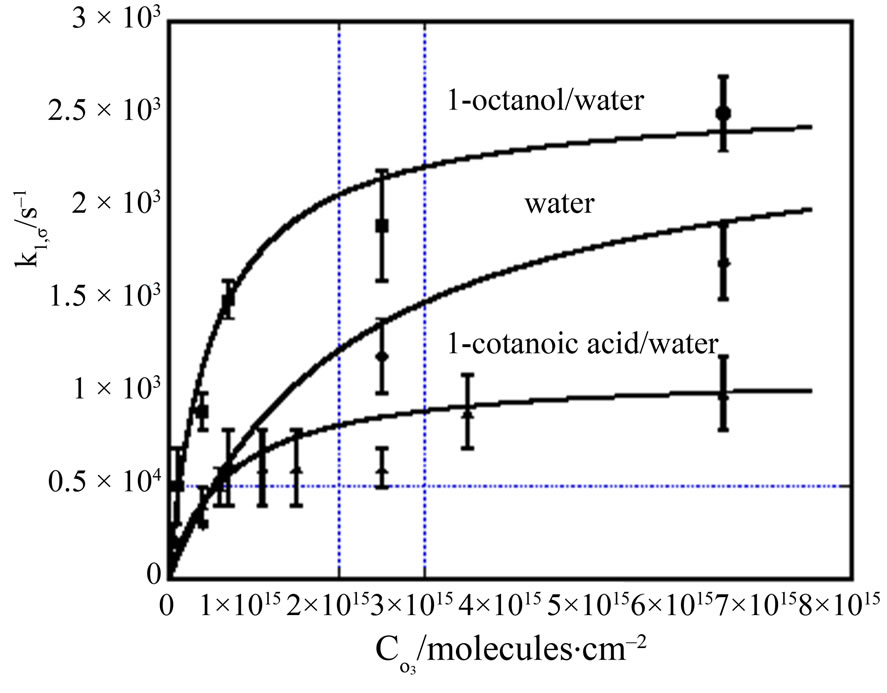
Figure 3. Pseudo first order reaction rate constants for gaseous anthracene reaction with gaseous ozone on different types of aqueous surfaces, viz., 2.5 mM aqueous solution of octanol, pure uncoated water, and 3.8 mM aqueous solution of 1-octanoic acid. Data obtained from Mmereki et al [39]. Note that in each case the rate constant reaches an asymptotic value at high concentrations of gaseous ozone. Values of kmax and Cox,1/2 are given in Table 5.
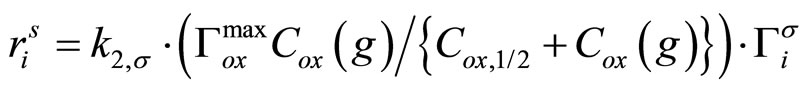 (2)
(2)
Thus, a pseudo first-order reaction rate constant can be defined
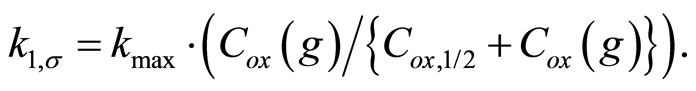 (3)
(3)
The above equation explains the saturation-type behavior for the reaction rate constant with oxidant concentration as shown in Figures 2 and 3. The values of the two constants are given below in Table 5 for different water surfaces. Note that Cox,1/2(g) relates to the binding energy of ozone with the water surface and kmax depends on the different bimolecular reaction rate constants on specific surfaces. Thus, these values change depending on the nature of the water surface [49]. A problem with the above treatment is that molecular O3 has only a small residence time (~10–9 s) on surfaces. The molecular mechanism leading to the L-H surface reaction of ozone with PAH can be reconciled only by invoking the presence of long-lived (>100 s) reactive oxygen intermediates (ROI) on the surface [50].
The overall pseudo-first order reaction rate constant should depend on the surface thickness since there are two parallel channels in which reactions occur in a water film once the organic molecules adsorb (and solvate) on the surface. This is shown in Figure 4. The overall reaction rate is given by the following equation [22]
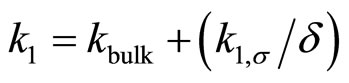 (4)
(4)
where kbulk and k1,σ are respectively the homogenous bulk aqueous phase and heterogeneous surface reaction rate constants. It is evident that as the film thickness becomes smaller, the heterogeneous reaction rate will make a larger contribution towards the overall reaction rate. This fact was demonstrated in our laboratory with regard to the UV photooxidation of two gaseous PAHs (naphthalene and phenanthrene). In both cases the major product concentrations were followed in the aqueous films and the first order rate constants for the degradation of PAH into respective products were obtained [36,51]. For example, the reaction rate constants for phenanthrene conversion to major products are given in Figure 5. It is clear that for all the three major products of oxidation the reaction rate increases as the film thickness decreases. Similar behavior was also observed for naphthalene degradation.

Table 5. Pseudo first order rate constant parameters for the reaction of anthracene with gas phase ozone on various water surfaces.
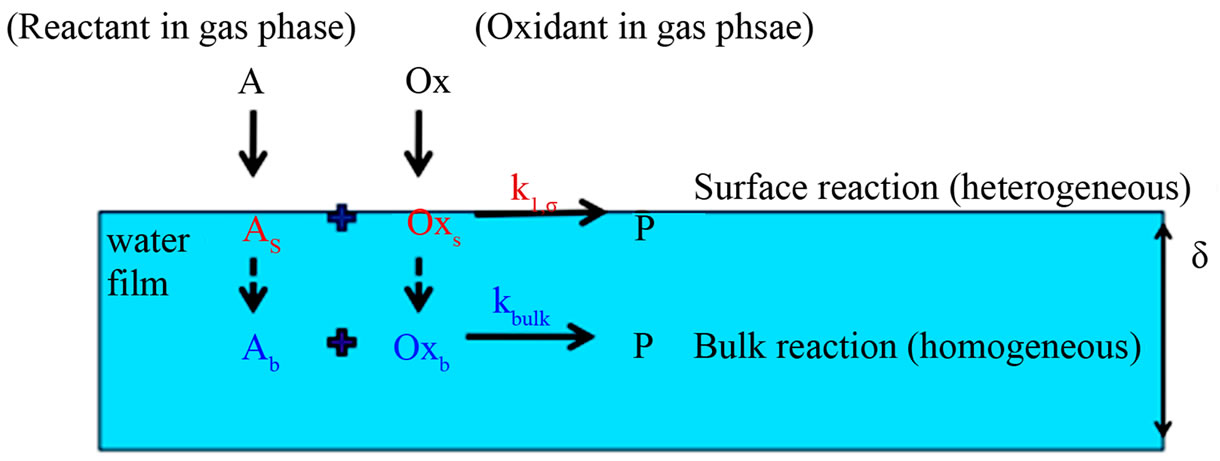
Figure 4. Schematic of the two reaction channels (heterogeneous and homogeneous) for gaseous organic species in water films.
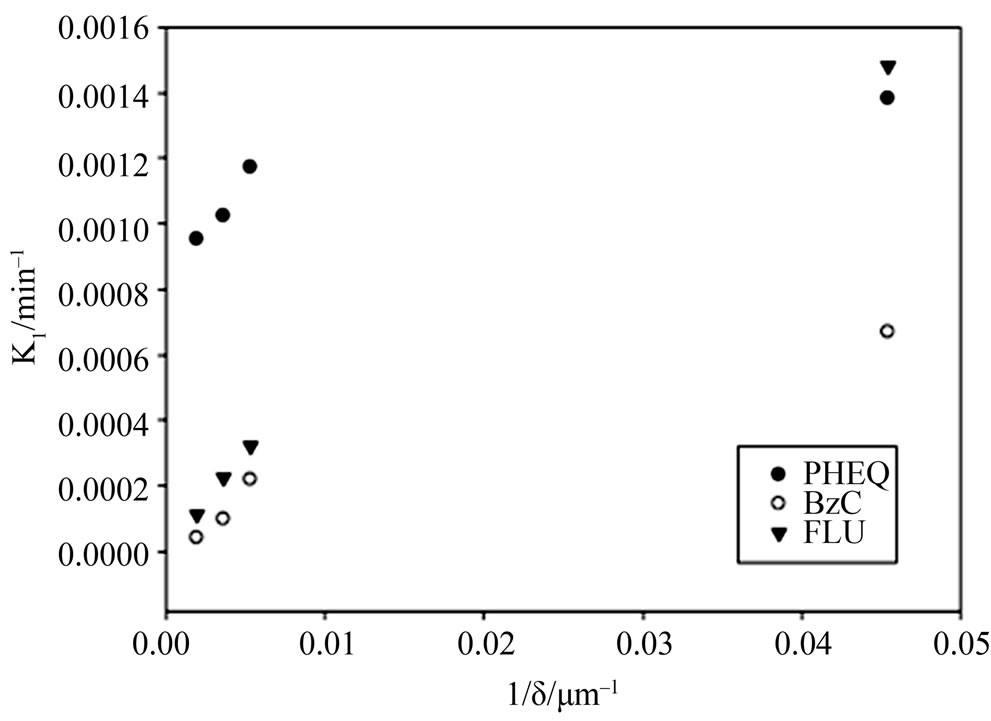
Figure 5. The photooxidation rates of gaseous phenanthrene on water films as a function of film thickness. The formation rate constants for three different compounds (PHEQPhenanthraquinone, BzC-Benzocoumarin, FLU-Fluoranthone) from phenanthrene are given. Data obtained from Chen et al. [36].
Others have also confirmed the surface reactions on water [52]. The reaction of 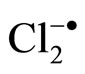 with ethanol in water films was investigated using UV diffuse reflectance laser flash photolysis technique. It was demonstrated that the surface reaction rate is different from the bulk reaction rate. The surface reaction rate varied as the square root of the ethanol concentration and reached an asymptote at high concentrations. However, the bulk reaction rate was much slower and varied linearly with ethanol concentration.
with ethanol in water films was investigated using UV diffuse reflectance laser flash photolysis technique. It was demonstrated that the surface reaction rate is different from the bulk reaction rate. The surface reaction rate varied as the square root of the ethanol concentration and reached an asymptote at high concentrations. However, the bulk reaction rate was much slower and varied linearly with ethanol concentration.
Experimental field observations also lend support to the importance of surface reactions. For example, in analyzing the loss of ozone in coastal regions, it was observed that the surface reaction of ozone on the air-water interface of sea salt particles had to be invoked in models to correctly explain the observations [5]. This observation was confirmed with laboratory work and from molecular dynamics and ab-initio calculations. Fog and cloud droplets can process organic trace gases and particulates via aqueous phase photochemistry. Field observations have shown that fog droplets provide a microreactor environment for the formation of secondary organic aerosols (SOA) by photochemical transformations of organic compounds [6]. Real-time formation of secondary aerosol was detected using on-line aerosol mass spectrometry immediately upon the detection of fog. It was observed that the post-fog atmospheric environment contained particles and gases that were produced by evaporation of water from the fog droplet.
In the organic chemistry literature there is the term “on-water” heterogeneous chemistry that refers to orders of magnitude increase in the rates of catalysis of organic reactions at oil-water interfaces compared to homogeneous reactions in organic solvents [53]. This has been attributed to the breaking of an existing H-bonding network in homogeneous aqueous solution in order to permit catalysis, but not for the “on-water” reactions [54].
In order to explain the preference of surface reactions over bulk reactions for gaseous species on aerosols in the atmosphere, a hypothesis has been proposed [55,56]. For bulk aqueous phase reactions to occur the molecular species that are fully solvated (in a H-bonded water network) have to break their solvation shells. For surface species that are only partly solvated [57] and such an energy penalty does not exist. Diffusion also limits the encounter of the fully solvated species in the case of the bulk solution. There is evidence that the adsorbed molecules at the interface display faster orientational dynamics than in the bulk [58]. Moreover, even when reactions occur within the bulk solution, the diffusion of molecules out of the solvent cage is in competition with recombination reactions. At the surface recombination of reactants is inhibited as shown in photolysis reactions at the surface compared to the bulk [59].
3. Summary
It is evident from principal observations that “surface” water is different from “bulk” water in reactivity. Trace gases (HOC) and atmospheric oxidant species adsorb and react on aqueous surfaces. These processes are of significance for aqueous atmospheric aerosols. Experimental work and molecular simulations support the above conclusion. It is evident that heterogeneous reactions deserve special attention in tropospheric chemistry and is important for thin films of water existing on aqueous aerosols.
4. Acknowledgements
The US National Science Foundation is gratefully acknowledged for support of this work through research grants (AGS 0907261 and AGS 1106569). This material was also presented in part as a Plenary Lecture at the “Recent Advances in Chemical Engineering and Technology” meeting organized by the Indian Institute of Chemical Engineers, Kochi Regional Centre, in Kochi, Kerala, India on 10-12 March 2011.
REFERENCES
- J. H. Seinfeld and S. N. Pandis, “Atmospheric Chemistry and Physics,” John Wiley and Sons, Inc., New York, 2006.
- K. T. Valsaraj, “Elements of Environmental Engineering,” 3rd Edition, Taylor and Francis Publishers, New York, 2009.
- S. Solomon, S. Qin, D. Chen, Z. Marquis, K. B. Averyt, M. Tignor and H. L. Miller (Eds.), “Climate Change 2007: The Physical Basis, Contributions of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change,” IPCC, Cambridge University Press, Cambridge, 2007.
- S. R.-V. Castrillon, N. Giovambattista, I. A. Aksay and P. Debenedetti, “Structure and Energetics of Thin Water Films,” Journal of Physical Chemistry C, Vol. 115, 2011, pp. 4624-4635. doi:10.1021/jp1083967
- E. M. Knipping, M. J. Lakin, K. L. Foster, P. Jungwirth, D. J. Tobias, R. B. Gerber, D. Dabdub and B. J. Finlayson-Pitts, “Experiments and Simulations of Ion-Enhanced Interfacial Chemistry on Aqueous NaCl Aerosols,” Science, Vol. 288, No. 5464, 2000, pp. 301-306. doi:10.1126/science.288.5464.301
- M. Dall’Osto, R. M. Harrison, H. Coe and P. Williams, “Real-Time Secondary Aerosol Formation during a Fog Event in London,” Atmospheric Chemistry and Physics, Vol. 9, No. 7, 2009, pp. 2459-2469. doi:10.5194/acp-9-2459-2009
- S. Raja, R. Ravikrishna, X.-Y. Yu, T. Lee, J. Chen, R. R. Kommalapati, K. Murugesan, X. Shen, K. T. Valsaraj and J. L. Collett, “Fog Chemistry in the Texas-Louisiana Gulf Coast Corridor,” Atmospheric Environment, Vol. 42, No. 9, 2008, pp. 2048-2061. doi:10.1016/j.atmosenv.2007.12.004
- I. J. George and J. P. D. Abbatt, “Heterogeneous Oxidation of Atmospheric Aerosol Particles by Gas-Phase Radicals,” Nature Chemistry, Vol. 2, No. 9, 2010, pp. 713-722. doi:10.1038/nchem.806
- C. J. van Oss, “Interfacial Forces in Aqueous Media,” Taylor and Francis, New York, 2006.
- E. E. Meyer, K. J. Rosenberg and J. N. Israelachvili, Proceedings of the National Academy of Sciences, Vol. 103, 2006, pp. 15739-15746.
- J. N. Israelachvili, “Intermolecular and Surface Forces,” 3rd Edition, Academic Press, New York, 2010.
- J. S. Smith and K. T. Valsaraj, “Solvent Sublation for Industrial Wastewater Treatment,” Chemical Engineering Progress, Vol. 94, No. 5, 1998, pp. 69-76.
- H. F. Rafson (Ed.), “Odor and VOC Control Handbook,” McGraw Hill Pub Co., New York, 1998.
- K. T. Valsaraj, G. J. Thoma, D. D. Reible and L. J. Thibodeaux, “On the Enrichment of Hydrophobic Organic Compounds in Fog Droplets,” Atmospheric Environment, Vol. 27A, No. 2, 1993, pp. 203-210. doi:10.1016/0960-1686(93)90351-X
- J. D. Blando and B. J. Turpin, “Secondary Organic Aerosol Formation in Cloud and Fog Droplets: A Literature Evaluation of Plausibility,” Atmospheric Environment, Vol. 34, No. 10, 2000, pp. 1623-1632. doi:10.1016/S1352-2310(99)00392-1
- D. J. Donaldson and K. T. Valsaraj, “Adsorption and Reaction of Trace Gas-Phase Organic Compounds on Atmospheric Water Film Surfaces: A Critical Review,” Environmental Science & Technology, Vol. 44, No. 3, 2010, pp. 865-873. doi:10.1021/es902720s
- K. T. Valsaraj, “Trace Gas Adsorption Thermodynamics at the Air-Water Interface: Implications in Atmospheric Chemistry,” Pure and Applied Chemistry, Vol. 81, No. 10, 2009, pp. 1889-1901. doi:10.1351/PAC-CON-08-07-06
- D. J. Donaldson and D. Anderson, “Adsorption of Atmospheric Gases at the Air-Water Interface. 2. C1-C4 Alcohols, Acids, and Acetone,” Journal of Physical Chemistry A, Vol. 103, No. 1, 1999, pp. 871-876. doi:10.1021/jp983963h
- R. Braunt and M. J. Conklin, “Dynamic Determination of Vapor/Water Interface Adsorption for Volatile Hydrophobic Organic Compounds (VHOCs) Using Axisymmetric Drop Shape Analysis: Procedure and Analysis of Benzene Adsorption,” Journal of Physical Chemistry B, Vol. 104, No. 47, 2004, pp. 11146-11152. doi:10.1021/jp001140y
- A. Hartkopf and B. L. Karger, “Study of the Interfacial Properties of Water by Gas Chromatography,” Accounts of Chemical Research, Vol. 6, 1973, pp. 209-221. doi:10.1021/ar50066a006
- S. Raja, F. S. Yaccone, R. Ravikrishna and K. T. Valsaraj, “Thermodynamic Parameters for the Adsorption of Aromatic Hydrocarbon Vapors at the Gas-Water Interface,” Journal of Chemical & Engineering Data, Vol. 47, No. 5, 2002, pp. 1213-1219. doi:10.1021/je025520j
- J. Chen, F. S. Ehrenhauser, K. T. Valsaraj and M. J. Wornat, “Uptake and UV-Photooxidation of Gas-Phase PAHs on the Surface of Atmospheric Water Films. 1. Naphthalene,” Journal of Physical Chemistry A, Vol. 110, No. 29, 2006, pp. 161-916. doi:10.1021/jp062560b8
- K. T. Valsaraj, “On the Physico-Chemical Aspects of Partitioning of Non-Polar Hydrophobic Organics at the AirWater Interface,” Chemosphere, Vol. 17, No. 5, 1988, pp. 875-887. doi:10.1016/0045-6535(88)90060-4
- K. T. Valsaraj, “Binding Constants for Non-Polar Hydrophobic Organics at the Air-Water Interface: Comparison of Experimental and Predicted Values,” Chemosphere, Vol. 17, No. 10, 1988, pp. 2049-2061. doi:10.1016/0045-6535(88)90015-X
- K.-U. Goss and R. P. Schwarzenbach, “Linear Free Energy Relationships Used to Evaluate Equilibrium Partitioning of Organic Compounds,” Environmental Science & Technology, Vol. 35, No. 1, 2001, pp. 1-9. doi:10.1021/es000996d
- K.-U. Goss, “Conceptual Model for the Adsorption of Organic Compounds from the Gas Phase to Liquid and Solid Surfaces,” Environmental Science & Technology, Vol. 31, No. 12, 1997, pp. 3600-3605. doi:10.1021/es970361n
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, “Predicting Adsorption Coefficients at Air-Water Interfaces Using Universal Solvation and Surface Area Models,” Journal of Physical Chemistry B, Vol. 108, No. 34, 2004, pp. 12882-12897. doi:10.1021/jp037210t
- K.-U. Goss, “Predicting Adsorption of Organic Chemicals at the Air-Water Interface,” Journal of Physical Chemistry A, Vol. 113, No. 44, 2009, 12256-12259. doi:10.1021/jp907347p
- R. Vacha, P. Jungwirth, J. Chen and K. T. Valsaraj, “Adsorption of Polycyclic Aromatic Hydrocarbons at the Air-Water Interface: Molecular Dynamics Simulations and Experimental Atmospheric Observations,” Physical Chemistry Chemical Physics, Vol. 8, No. 38, 2006, pp. 4461-4467. doi:10.1039/b610253k
- R. Vacha, K. Cwiklik, J. Rezac, P. Hobza, P. Jungwirth, K. Valsaraj, S. Bahr and V. Kempter, “Adsorption of Aromatic Hydrocarbons and Ozone at Environmental Aqueous Surfaces,” Journal of Physical Chemistry A, Vol. 112, No. 22, 2008, pp. 4942-4950. doi:10.1021/jp711813p
- T. Somasundaram, R. M. Lyndon-Bell and C. H. Patterson, “The Passage of Gases through the Liquid Water/ Vapour Interface: A Simulation Study,” Physical Chemistry Chemical Physics, Vol. 1, No. 1, 1999, pp. 143-148. doi:10.1039/a805067h
- C. D. Wick, B. Chen and K. T. Valsaraj, “Computational Investigation of the Influence of Surfactants on the AirWater Interfacial Behavior of Polycylic Aromatic Hydrocarbons,” Journal of Physical Chemistry C, Vol. 114, No. 34, 2010, pp. 14520-14527. doi:10.1021/jp1039578
- J. Chen, F. Ehrenhauser, T. Arachi, F. Hung, K. Valsaraj and M. Wornat, “Adsorption of Gas-Phase Phenanthrene on Atmospheric Water and Ice Films,” Polycyclicarom Atichydrocarbons, Vol. 31, 2011, pp. 1-26. doi:10.1080/10406638.2011.585370
- R. Vacha, P. Slavaicek, M. Mucha, B. Finlayson-Pitts and P. Jungwirth, “Adsorption of Atmospherically Relevant Gases at the Air/Water Interface: Free Energy Profiles of Aqueous Solvation of N2, O2, O3, OH, H2O, HO2, and H2O2,” Journal of Physical Chemistry A, Vol. 108, 2004, pp. 11573-11579.
- R. Vacha, P. Slavicek, M. Mucha, B. J. Finlayson-Pitts and P. Jungwirth, “Adsorption of Atmospherically Relevant Gases at the Air/Water Interface: Free Energy Profiles of Aqueous Solvation of N2, O2, O3, OH, H2O, HO2, and H2O2,” Journal of Physical Chemistry A, Vol. 108, No. 52, 2004, pp. 11573-11579. doi:10.1021/jp046268k
- J. Chen, F. Ehrenhauser, K. T. Valsaraj and M. J. Wornat, “Adsorption and UV Photooxidation of Gas-Phase Phenanthrene on Atmospheric Films,” ACS Symposium Series, Vol. 1005, 2009, pp. 127-146. doi:10.1021/bk-2009-1005.ch009
- I.-F. W. Kuo and C. J. Mundy, “An Ab initio Molecular Dynamics Study of the Aqueous Liquid-Vapor Interface,” Science, Vol. 303, No. 5658, 2004, pp. 658-662. doi:10.1126/science.1092787
- X. D. Zhu, H. Suhr and Y. R. Shen, “Surface Vibrational Spectroscopy by Infrared-Visible Sum Frequency Generation,” Physical Review B, Vol. 35, No. 6, 1987, pp. 3047-3050. doi:10.1103/PhysRevB.35.3047
- B. T. Mmereki, D. J. Donaldson, J. B. Gilman, T. L. Eliason and V. Vaida, “Kinetics and Products of the Reaction of Gas-Phase Ozone with Anthracene Adsorbed at the Air-Aqueous Interface,” Atmospheric Environment, Vol. 38, 2004, pp. 6091-6103. doi:10.1016/j.atmosenv.2004.08.014
- E. S. Enami, M. R. Hoffmann and A. J. Colussi, “Ozonolysis of Uric Acid at the Air/Water Interface,” Journal of Physical Chemistry B, Vol. 112, No. 14, 2008, pp. 4153-4157. doi:10.1021/jp712010k
- R. Vacha, L. Cwiklik, J. Rezac, P. Hobza, P. Jungwirth, K. Valsaraj, S. Bahr and V. Kempter, “Adsorption of Aromatic Hydrocarbons and Ozone at Environmental Aqueous Surfaces,” Journal of Physical Chemistry A, Vol. 112, No. 22, 2008, pp. 4942-4947. doi:10.1021/jp711813p
- S. N. Wren and D. J. Donaldson, “Glancing-Angle Raman Spectroscopic Probe for Reaction Kinetics at Water Surfaces,” Physical Chemistry Chemical Physics, Vol. 12, No. 11, 2010, pp. 2648-2654. doi:10.1039/b922254e
- S. Raja and K. T. Valsaraj, “Heterogeneous Oxidation by Ozone of Naphthalene Adsorbed at the Air-Water Interface of Micron-Size Water Droplets,” Journal of the Air & Waste Management Association, Vol. 55, No. 9, 2005, pp. 1345-1355.
- I. V. Stiopkin, H. D. Jayathilake, A. N. Bordenyuk and A. V. Benderskii, “Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy,” Journal of the American Chemical Society, Vol. 130, No. 7, 2008, pp. 2271-2275. doi:10.1021/ja076708w
- C. Waring, P. A. J. Bagot, M. L. Costen and K. G. McKendrick, “Reactive Scattering as a Chemically Specific Analytical Probe of Liquid Surfaces,” The Journal of Physical Chemistry Letters, Vol. 2, No. 1, 2011, pp. 12-18. doi:10.1021/jz1013032
- Y. R. Shen and V. Ostroverkhov, “Sum-Frequency Vibrational Spectroscopy on Water Interfaces: Polar Orientation of Water Molecules at Interfaces,” Chemical Reviews, Vol. 106, No. 4, 2006, pp. 140-154. doi:10.1021/cr040377d
- K. Harper, B. Minofar, M. R. Sierra-Hernandez, N. N. Casillas-Ituarte, M. Roeselova and H. C. Allen, “Surface Residence and Uptake of Methyl Chloride and Methyl Alcohol at the Air/Water Interface Studied by Vibrational Sum Frequency Spectroscopy and Molecular Dynamics,” Journal of Physical Chemistry A, Vol. 113, No. 10, 2009, pp. 2015-2024. doi:10.1021/jp808630v
- K. B. Eisenthal, “Equilibrium and Dynamic Processes at Interfaces by Second Harmonic and Sum Frequency Generation,” Annual Review of Physical Chemistry, Vol. 43, 1992, pp. 627-661. doi:10.1146/annurev.pc.43.100192.003211
- N.-O. A. Kwamena, M. G. Staikova, D. J. Donaldson, I. J. Goerge and J. P. D. Abbatt, “Role of the Aerosol Substrate in the Heterogeneous Ozonation Reactions of Surface-Bound PAHs,” Journal of Physical Chemistry A, Vol. 111, No. 43, 2007, pp. 11050-11058. doi:10.1021/jp075300i
- M. Shiraiwa, Y. Sosedova, A. Ronviere, H. Yang, Y. Zhang, J. P. D. Abbatt, M. Ammann and U. Poschl, “The Role of Long-Lived Reactive Oxygen Intermediates in the Reaction of Ozone with Aerosol Particles,” Nature Chemistry, Vol. 3, 2011, pp. 291-295. doi:10.1038/NCHEM.988
- J. Chen and K. Valsaraj., “Uptake and UV-Photooxidation of Gas-Phase PAHs on the Surface of Atmospheric Water Films. 1. Naphthalene,” Journal of Physical Chemistry A, Vol. 110, 2006, pp. 9161-9166. doi:10.1021/jp062560b
- R. S. Strekowski, R. Remorov and Ch. George, “Direct Kinetic Study of the Reaction of Cl2 Radical Anions with Ethanol at the Air-Water Interface,” Journal of Physical Chemistry A, Vol. 107, 2003, pp. 2497-2504. doi:10.1021/jp026174f
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, “On Water: Unique Reactivity of Organic Compounds in Aqueous Suspension,” Angewandte Chemie International Edition, Vol. 44, No. 21, 2005, pp. 3275-3279. doi:10.1002/anie.200462883
- Y. Jung and R. A. Marcus, “On the Theory of Organic Catalysis ‘on Water’,” Journal of the American Chemical Society, Vol. 129, No. 17, 2007, pp. 5492-5502. doi:10.1021/ja068120f
- K. T. Valsaraj, “Trace Gas Adsorption Thermodynamics at the Air-Water Interface: Implications in Atmospheric Chemistry,” Pure and Applied Chemistry, Vol. 81, No. 10, 2009, pp. 1889-1901. doi:10.1351/PAC-CON-08-07-06
- P. Nissensson, C. J. X. Knox, B. J. Finlayson-Pitts, L. F. Phillips and D. Dabdub, “Enhanced Photolysis in Aerosols: Evidence for Important Surface Effects,” Physical Chemistry Chemical Physics, Vol. 8, No. 40, 2006, pp. 4700-4710. doi:10.1039/b609219e
- H. Watanabe, S. Yamaguchi, S. Sen, A. Morita and T. Tahara, “‘Half-Hydration’ at the Air/Water Interface Revealed by Heterodyne-Detected Electronic Sum Frequency Generation Spectroscopy, Polarization Second Harmonic Generation, and Molecular Dynamics Simulation,” Journal of Chemical Physics, Vol. 132, No. 14, 2010, Article ID: 144701. doi:10.1063/1.3372620
- M. L. Johnson, C. Rodriguez and I. Benjamin, “Rotational Dynamics of Strongly Adsorbed Solute at the Water Surface,” Journal of Physical Chemistry A, Vol. 113, 2009, pp. 2086-2091. doi:10.1021/jp808842k
- P. Nissenson, D. Dabdub, R. Das, V. Maurino, C. Minero, D. Vione, “Evidence of the Water-Cage Effect on the Photolysis of and FeOH2+. Implications of This Effect and of H2O2 Surface Accumulation on Photochemistry at the Air-Water Interface of Atmospheric Droplets,” tmospheric Environment, Vol. 44, 2010, pp. 4859-4866. doi:10.1016/j.atmosenv.2010.08.035