International Journal of Organic Chemistry
Vol.05 No.02(2015), Article ID:57589,6 pages
10.4236/ijoc.2015.52013
Molecular Structure of Co2(μ-Alkyne) Complex Containing Ph2PC5F6PPh2 Ligand
Makoto Minato*, Yasutada Miyato, Masaki Kakeya
Department of Materials Chemistry, Graduate School of Engineering, Yokohama National University, Yokohama, Japan
Email: minato@ynu.ac.jp
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 1 June 2015; accepted 27 June 2015; published 30 June 2015
ABSTRACT
Oxidative-decarbonylation of Co2(CO)6(μ-PhC≡CH) with Me3NO in the presence of an electron deficient ligand, Ph2PC5F6PPh2(F6FOS), produces Co2(CO)4(μ-PhC≡CH)(F6FOS), (1). The metrical values of 1 have been compared to those of the closely related cobalt carbonyl alkyne compound A containing (Z)-Ph2PCH=CHPPh2 (Z-dppe) ligand. Strikingly anomalous is an alkyne C≡C bond (1.34(1) Å) in 1, which is somewhat elongated compared to A (1.31(1) Å). When taking a strong electron-withdrawing power of fluoride atom into account, F6FOS ligand appeared to reduce the π-back-donation ability of cobalt atom, making this bond shortened in comparison to the same bond in A. Bond lengthening in the alkyne C≡C bond in 1 is attributed to the enhanced electron donor ability of F6FOS compared to Z-dppe and can be understood by examining resonance structures of F6FOS ligand.
Keywords:
Alkyne-Bridged Cobalt Complex, Electron Deficient Ligand, Crystal Structure, Coordinated Alkyne C≡C Bond

1. Introduction
We have become interested in ligands of the type R2PC5F6PR2 (R = Ph, cyclo-C6H11), which are reported by Cullen (Chart 1) [1] .
These ligands seem to display unique electronic properties in comparison to typical diphosphine ligands such as DIPHOS (Ph2PCH2CH2PPh2) because they possess a low-lying π* orbital due to a bridging perfluorocyclopentenyl ring, which is recognized to be a strong electron-withdrawing group [2] . Therefore an organometallic compound with this type of ligand is anticipated to serve as an effective electron reservoir by stabilizing electron
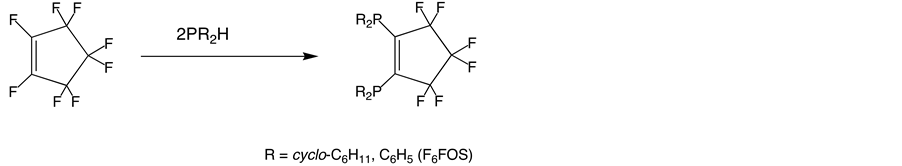
Chart 1. Preparation of diphosphine ligands containing perfluorocyclopentenyl ring.
counts in excess of 18-electrons [3] [4] . To establish the electronic influence of F6FOS (R = Ph, 1,2-bis(diphe- nylphosphino)hexafluorocyclopentene) ligand, we wish to report the synthesis and single-crystal X-ray diffraction study of an alkyne-bridged cobalt complex containing F6FOS.
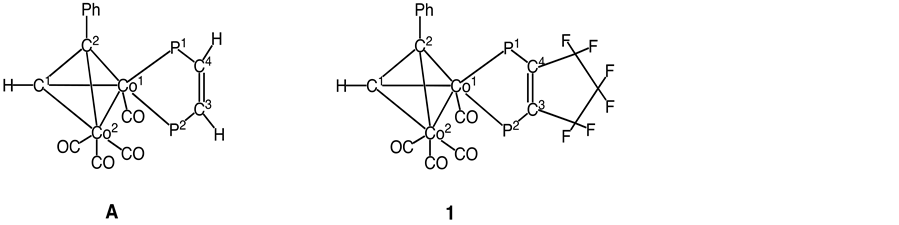
Energetic investigations of cobalt-alkyne compounds involving various bidentate phosphine ligands have been performed [5] -[10] . For example, Bott and coworkers reported that the rigid diphosphine, (Z)-Ph2PCH= CHPPh2 (Z-dppe), reacted with Co2(CO)6(μ-PhC≡CH) to yield the chelating complex Co2(CO)4(μ-PhC≡CH) [(Z)-dppe], (A) [11] . On the basis of Bott’s results, we set out to prepare the alkyne-bridged cobalt complex 1 containing F6FOS ligand that would differ from complex A in the amount of electron density on the metal center but would be sterically very similar with respect to coordination (Scheme 1).
We reckoned that a comparison of the metrical values for 1 and A served to elucidate the electronic effect of F6FOS ligand. In addition, no crystallographic study on F6FOS appears to have been recorded, and consequently data on this ligand are also briefly reported.
2. Experimental Section
2.1. General Considerations
Unless otherwise noted, all reactions were carried out under anaerobic and anhydrous conditions using Ar or N2 and conventional Schlenk techniques using the general methods. NMR spectra were measured on the following instruments, at the frequencies listed, unless stated otherwise: Jeol JMN-AL 400 and Jeol FX270, 1H NMR (400 MHz), 31P (161.7 MHz), 19F NMR (376.05 MHz). Chemical shifts are quoted to the following references: 1H NMR ((CH3)4Si, 0 ppm); 31P NMR (PPh3, 0 ppm); 19F NMR (CF3COOH, −78.50 ppm).
F6FOS [1] and Co2(CO)6(μ-PhC≡CH) [12] were prepared by following literature procedures.
2.2. Synthesis of Complex 1
To 0.080 g (0.21 mmol) of Co2(CO)6(μ-PhC≡CH) and 0.11 g (0.20 mmol) of F6FOS in 15 mL of ethanol was added 0.030 mg (0.40 mmol) of Me3NO2. The solution was stirred for 24 h at ambient temperature. The dark- redprecipitate was formed and the supernatant was removed. The resulting solid was washed with hexane and pumped dry to yield 1 (0.11 g, 62%). Crystals suitable for an X-ray structural analysis were obtained by recrystallization from dichloromethane/n-heptane. Complex 1: 1H NMR (CDCl3, 25˚C, 400 MHz): δ 4.9 - 5.0 (br m, 1H, ≡CH), 7.0 - 7.8 (m, 25H, Ph protons); 31P{1H} NMR (CDCl3, 25˚C, 160 MHz): δ 69.6 (s, 1P), 80.2 (s, 1P); 19F NMR (CDCl3, 25˚C, 376.05 MHz) δ −132.61 (dt, J = 11, 236 Hz, 1F), −131.29 (dt, J = 11, 236 Hz, 1F), −111.34 (d, J = 267, 2F), −109.60 (d, J = 271, 1F), −109.53 (d, J = 267, 1F); IR (KBr, ν (C≡O, cm−1): 1929, 1960, 2012, 2056.
2.3. Single Crystal X-Ray Diffraction
Selected structural details for the structures of F6FOS ligand and complex 1 are included in Table 1 and full crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 1052995 and 1052994, respectively. Copies of these data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
3. Results and Discussion
The ligand, F6FOS, was synthesized from HPPh2 and octafluorocyclopentene. Complex 1 was synthesized in a
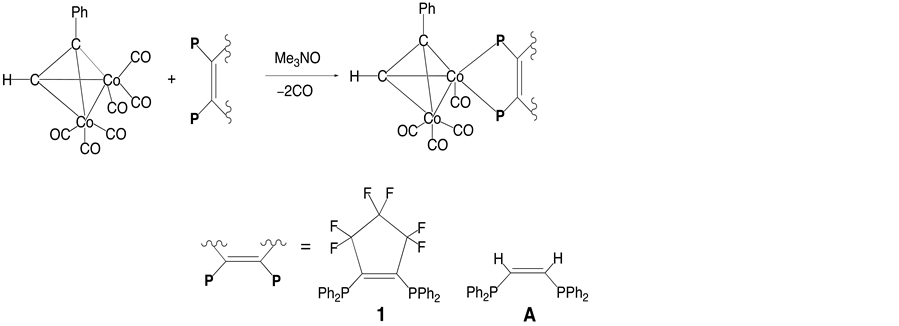
Scheme 1. The reactions of the alkyne-bridged binuclear Co complex with F6FOS and Z-dppe.

Table 1. Experimental details.
Data were collected and processed using CrystalClear (Rigaku).
straightforward fashion from Co2(CO)6(μ-PhC≡CH) and F6FOS in the presence of the oxidative-decarbonyla- tion reagent Me3NO. The IR spectrum of 1 shows three strong CO absorptions in the range of 1900 - 2100 cm−1. In the 1HNMR spectrum of 1, the alkyne proton resonance occurs at δ 5.0 ppm, which is 0.8 ppm upfield approximately from that of the parent complex Co2(CO)6(μ-PhC≡CH). The 31P{1H}NMR spectrum of 1 displays two phosphorus signals at δ 69 ppm and δ 80 ppm, showing a downfield coordination shift (δ −15 ppm in F6FOS). In contrast to F6FOS ligand (giving two sets of fluorine resonances in a 2:1 intensity ratio), 1 shows five types of inequivalent fluorine atoms in the 19F NMR spectrum due to the asymmetry imposed by the formation of the chelate ring. There are two doublets of pseudotriplets at δ −132.61 (J (FF) = 236 and 11 Hz) and −131.29 ppm (J (FF) = 236 and 11 Hz), which may be assigned to the central two fluorine atoms in the C5F6 ring. In addition, the three pseudodoublets at δ −109.53 (J (FF) = 267 Hz), −109.60 (J (FF) = 271 Hz), and −111.34 ppm (J (FF) = 267 Hz) are attributable to rest of the four fluorine atoms in the ring. This spectrum also indicates strong germinal coupling between fluorine atoms above and below the plane of the ring.
The molecular structures of 1 and F6FOS are shown in Figure 1 and Figure 2. Pertinent structural parameters for F6FOS and 1 are compared in Table 2.
Complex 1 crystallizes in the triclinic space P-1 (#2). The alkyne portion of 1 is coordinated through its π-bond to Co1 and Co2, while F6FOS ligand is coordinated to Co1. In the structure, there is a distorted octahedral geometry about each cobalt atom, the two alkyne carbons and two cobalt atoms forming a tetrahedral unit.
It is instructive to compare the geometry about the F6FOS fragment in 1 to that of the parent F6FOS ligand. Upon coordination to cobalt atom, the bond lengths and angles of the ligand itself alter significantly. The mean P-C bond distance in 1 at 1.834 Å is somewhat longer than the corresponding value of 1.824 Å in F6FOS while the C=C bond distance at 1.34(1) Å in the C5F6 ring of 1 is slightly shorter than that observed in F6FOS ligand itself, 1.370(8) Å. This may be explained by a substantial decrease of degree of π-bonding between the phos-

Figure 1. The molecular structure of complex 1.

Figure 2. The molecular structure of ligand F6FOS.
Table 2. Comparison of structural parameters for F6FOS ligand and Complex 1.
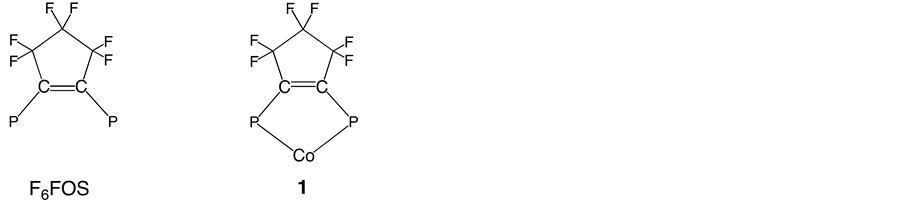
phorus lone-pair orbitals and π-orbital of the C=C bond in 1. For F6FOS ligand, three resonance forms may be drawn as shown in Scheme 2.
The structure I or II makes a considerable contribution to the hybrid because the strong electron-withdrawing effect of fluoride substituents decreases the electron density at the charge-bearing carbons. This structural feature accommodates some degree of P-C multiple bonding. Consequently, this gives the C=C bond of F6FOS a partial single-bond character and lengthens it. Then, coordination of the ligand to cobalt atom would lead to reduce π-donation from the electron pairs on phosphorus atoms. As expected, the mean endocyclic P-C=C bond angle of 119.0˚ in 1 is smaller than that of 123.4˚ in F6FOS ligand.
Our major objective of this study is to establish how the substitution of Z-dppe for F6FOS affects the structure of the rest of the complex. Since complexes 1 and A were expected to be sterically very similar with respect to the polyhedral Co2 core, it could be argued that any observed structural differences would have their origin in the electronic properties inherent to each ancillary diphosphine ligand.
To begin with, we verified the bonding geometry about Co1 atoms in 1 and A. Table 3 compares pertinent distances and bond angles of 1 and A. The P2-Co1-P1 bite angle in 1 is 90.4(1)˚ and, the Co1-P1 bond with a bond length of 2.216(3) and Co1-P2 bond with a bond length of 2.175(3) average at 2.196 Å. These values are analogous to those reported for A (88.39(8)˚ and 2.188 Å, respectively). Furthermore, the Co1-CO bond distance of 1.769(9) Å in 1 is essentially consistent with the value that observed in A (1.769(8) Å). It may be inferred from these facts that A and 1 are sterically very similar with respect to coordination around Co1, and therefore, the steric effects from the substituents (H versus C3F6 frame) of the C3=C4 bonds on the nature of the rest of the molecules are rather weak. As a result, only the electronic effect will be at the origin of any variations in the measured parameters.
Interestingly, the C3=C4 bond distance at 1.34(1) Å in the C5F6 ring of 1 is somewhat longer than the corresponding bond distance in A (1.328(9) Å). This behavior confirms that the C3=C4 bond in 1 still has partial single-bond character, as stated above. One might think the alkyne C1≡C2 bond length is a better criterion for Co- (π*C) interaction due to the bond weakening resulting from back donation of d-electrons on the Co atom into an empty π* orbital. The alkyne C1≡C2 distance of 1 is found to be 1.34(1) Å. Contrary to our expectation, this distance is slightly longer than that in A (1.31(1) Å) and is equal to a value found for the C3=C4 bond in 1. We envisaged that electron-withdrawing C5F6 ring in F6FOS should decrease in the amount of electron density of Co1 atom in 1, which reduces the π*-back-donation ability of the atom, making this bond closer to the triple bond in free PhC≡CH. As a consequence, in the case of 1 the alkyne C1≡C2 bond was expected to be shortened in comparison to the same bond in A. However, the lengthening of the alkyne C1≡C2 distance in 1 relative to A is observed. The result provides evidence that the Co1 atom in 1 may be acting as a better donor of electron density than the corresponding cobalt atom in A. As described above, the C3=C4 bond distance in the C5F6 ring of 1 has partial single-bond character, suggesting that the C3 and C4 atoms have sp3 character to some extend. It is well known that electronegativity values vary with the bond order [13] . So the phosphorus atoms in 1 may have a greater σ-donating ability than those in A because carbon atoms with sp2 bonding generally have a greater electron-withdrawing power than those with sp3 bonding. Hence, it can be concluded that by substitution of Z-dppe by F6FOS the electron density on the cobalt atom has not indeed decreased, but rather resulting in more
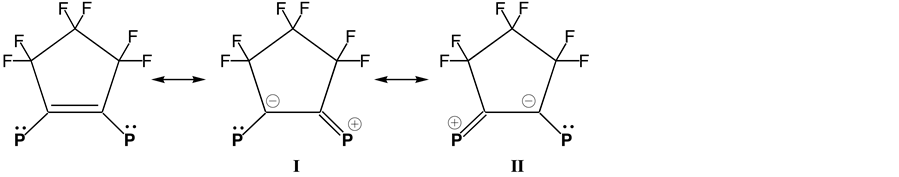
Scheme 2. The resonance forms of F6FOS.
Table 3. Comparison of structural parameters for A and Complex 1.
a Reference [11] .
π-back-donation to the alkyne ligand.
This lengthened the alkyne C1≡C2 distance in 1 is counter-balanced by the short Co1-Co2 distance of 2.471(2) Å, somewhat shorter than the corresponding bond length of 2.495(2) Å found in A. In addition, the Co―C (alkyne) distances in 1 are meaningfully longer than the same bond distances in A (1.92(1) versus 1.920(8), 1.962(8) versus 1.929(7), 2.000(8) versus 1.983(8), 2.004(7) versus 1.991(9) Å, respectively). In comparison to A, it thus appears that in 1 the alkyne ligand remains directed away from Co1 atom, whereas the Co2(CO)3 fragment is located more close to Co1. The Co2-CO bond lengthsin 1 average to 1.773 Å, which is close to the value reported in (A) (1.765Å).
4. Concluding Remarks
The bidentate ligand F6FOS displaces two carbonyl groups from Co2(CO)6(μ-PhC≡CH) to give Co2(CO)4 (μ-PhC≡CH) (F6FOS) (1), the structure of which is compared with that of a closely related cobalt carbonyl alkyne compound (A). The crystal structure of 1 confirms that the electron-withdrawing C5F6 group in F6FOS makes the alkyne C≡C lengthened in comparison to the same bond in A.
Acknowledgements
We wish to express our thanks to Pro. K. Osakada and Dr. Mikio Yamasaki for generous support. We also thank ZEON for valuable gifts of C5F8.
References
- Cullen, W.R., Harbourne, D.A., Liengme, B.W. and Sams, J.R. (1969) The Character of (CH3)2AsC=CAs (CH3)2CF2CF2Fe2 (CO)6 and Related Complexes. Inorganic Chemistry, 8, 95-100. http://dx.doi.org/10.1021/ic50071a023
- Yamada, S., Konno, T., Ishihara, T. and Yamanaka, H. (2005) Reaction of Octafluorocyclopentene with Various Carbon Nucleophiles. Journal of Fluorine Chemistry, 126, 125-133. http://dx.doi.org/10.1016/j.jfluchem.2004.10.047
- Mao, F., Tyler, D.R. and Keszler, D. (1989) Mechanism of the Substitution Reactions of the Nineteen-Electron Co(CO)3L2 Complex [L2 = 2,3-Bis (dipheny1phosphino)maleic An Hydride]. Journal of American Chemical Society, 111, 130-134. http://dx.doi.org/10.1021/ja00183a023
- Mao, F., Philbin, C.E., Weakly, T.J.R. and Tyler, D.R. (1990) Generation of the 19-Electron (18 + δ) Adducts CpMo(CO)3(L2-P) and CpMo(CO)2(L2-P,P') (Cp = η5-CH3C5H4, η5-C5Ph4H, η5-C5Ph5; L2 = 2,3-bis(diphenylphosphino) maleic Anhydride). Crystal structure of the (η5-C5Ph4H)Mo(CO)2L2 Complex. Organometallics, 9, 1510-1516. http://dx.doi.org/10.1021/om00119a024
- Bird, P.H., Franser, A.R. and Hall, D.N. (1977) Reactions of μ-Diphenylethyne-hexacarbonyldicobalt with Bis(diphe- ny1phosphino)methane and Bis(dipheny1arsino)methane. Crystal Structures of (tolan)(dpm)Co2(CO)4 and (tolan) (dam)2Co2(CO)2・C2H4C12. Inorganic Chemistry, 16, 1923-1931. http://dx.doi.org/10.1021/ic50174a018
- Yang, K., Bott, S.G. and Richmond, M. (1994) Reversible Chelate-to-Bridge Ligand Exchange in Co2(CO)(μ-PhC≡CPh) (bma) and Alkyne-Diphosphine Ligand Coupling. Synthesis, Reactivity, and Molecular Structures of Co2(CO)4 (μ-PhC≡CPh) (bma), Co2(CO) (μ-PhC≡CPh) ((Z)-Ph2PCH=CHPPh2), and Co2(CO)4{η2,η2,η1,η1-(Z)-Ph2PC (Ph)=(Ph) CC=C (PPh2)C(O)OC(O)}. Organometallics, 13, 3788-3799. http://dx.doi.org/10.1021/om00022a013
- Yang, K., Bott, S.G. and Richmond, M. (1996) Regioselective Phosphine Attack on the Coordinated Alkyne in Co2 (μ-Alkyne) Complexes Reactivity Studies and X-Ray Diffraction Structures of Co2(CO)4 (bma) (μ-HC≡CtBu) and the Zwitterionichydrocarbyl Complexes Co2(CO)4{η2,η2,η1,η1-(Z)-Ph2PC (Ph)=(Ph)CC=C (PPh2)C(O)OC(O)}. Journal of Organometallic Chemistry, 516, 65-80. http://dx.doi.org/10.1016/0022-328X(95)06090-J
- Chia, L.S., Cullen, W.R., Franklin, M. and Manning, A.R. (1975) Reactions of (RC≡CR')Co2(CO)6 Complexes with Mono- and Bidentate Group 5 Ligands. Inorganic Chemistry, 14, 2521-2526. http://dx.doi.org/10.1021/ic50152a045
- Bianchini, C., Dapporto, P. and Meli, A. (1979) Synthesis and X-Ray Structure of the Dinuclearcobalt (0) Complex (PhC≡CPh)Co2(CO)4(triphos). Journal of Organometallic Chemistry, 174, 205-212. http://dx.doi.org/10.1016/S0022-328X(00)91506-7
- Xia, C.-G., Bott, S.G., Wu, G. and Richmond, M.G. (2004) Reaction of the Dicobalt Alkyne Compound Co2(CO)6 (μ-dmad) with the Dipjosphine Ligand 4,5-Bis(diphenylphosphino)-4-cyclopenten-1,3-dione (bpcd). Spectroscopic Properties, X-Ray Diffraction Data, and Thermal Reactivity of the Chelating Isomer of Co2(CO)4 (bpcd) (μ-dmad). Journal of Chemical Crystallography, 34, 513-521. http://dx.doi.org/10.1023/B:JOCC.0000042019.54785.9a
- Bott, S.G., Yang, K. and Richmond, M.G. (2000) X-Ray Diffraction Structure of Co2(CO)4 (μ-PhC≡CH) [(Z)-Ph2PCH =CHPPh2]: Proof for Diphosphine Ligand Chelation. Journal of Chemical Crystallography, 30, 627-632. http://dx.doi.org/10.1023/A:1011940014273
- Greenfield, H., Sternberg, H.W., Friedel, R.A., Wotiz, J.H., Markby, R. and Wender, I. (1956) Acetylenic Dicobalt Hexacarbonyls. Organometallic Compounds Derived from Alkynes and Dicobalt Octacarbonyl. Journal of American Chemical Society, 78, 120-124. http://dx.doi.org/10.1021/ja01582a036
- Housecroft, C.E. and Sharpe, A.G. (2012) Inorganic Chemistry. 4th Edition, Pearson, Harlow.