International Journal of Organic Chemistry
Vol.4 No.1(2014), Article ID:43848,13 pages DOI:10.4236/ijoc.2014.41008
Enantioselective Aldol Reactions and Michael Additions Using Proline Derivatives as Organocatalysts
Mathieu Wagner, Yohan Contie, Clotilde Ferroud, Gilbert Revial*
Conservatoire National des Arts et Métiers, Laboratoire de Transformations Chimiques et Pharmaceutiques, Paris, France
Email: *gilbert.revial@cnam.fr
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 7 February 2014; revised 10 March 2014; accepted 17 March 2014
ABSTRACT
Six compounds including five proline derivatives have been prepared and tested as chiral organocatalysts for enantioselective aldol reactions and Michael additions. The enantiomeric excesses, which are highly dependent on the molecular structure of catalysts as well as experimental conditions, have reached over 98%.
Keywords:Aldol Reactions; Michael Additions; Enantioselective; Organocatalysts; Proline Derivatives

1. Introduction
The aldol reaction and the Michael addition are very precious tools of the synthetic organic chemist. Indeed, these reactions are widely applied to generate carbon-carbon bonds allowing to link building blocks to generate large and complex molecules. The steric control of chiral centers created during these reactions was initially attained using chiral auxiliaries as is the case, for instance, of chiral imines in Michael additions [1] [2] . For over a decade, many studies have been undertaken worldwide to extend the enantioselectivity in catalytic manner using small organic chiral molecules. These metal-free catalysts, highly efficient, more environmentally friendly, and often stable under both aerobic and aqueous reaction conditions, are always extensively investigated [3] -[8] .
The synthesis of azapyridinomacrocycles N-oxides and their use as organocatalysts for enantioselective allylations of 4-nitrobenzaldehyde with allyltrichlorosilane were reported from our laboratory [9] . The described asymmetric inductions were interesting and promising but too low to afford synthetic applications. Therefore we thought to experiment some of these chiral intermediates themselves as organocatalysts in aldol reactions and Michael additions. This kind of reactions has been extensively described as model to establish the efficiency of small molecules regarding the enantioselectivity. Herein we wish to report the results using some new derivatives 1 - 6 (figure 1) as chiral catalysts in both enantioselective aldol and Michael reactions [10] -[17] .
2. Results and Discussion
The preparation of new proline derivatives 2, 3, 5 and 6 is displayed in Scheme 1. Compound 2 was synthesized starting from amine 1 [9] with N-Boc-(S)-proline to give 7 followed by elimination of the Boc protecting group. In the case of 3, the amidation was carried out through the same pathway starting with the corresponding epimer of 1 derived from (1S,2S)-1,2-diaminocyclohexane. The proline derivative 9 was produced by condensation of commercially available 2-nitrobenzenesulfonamide with N-Boc-(S)-proline while 10 and 11 [18] resulted from the reaction of N-Boc-prolinamine [19] with respectively 2- and 4-nitrobenzenesulfonyl chloride. After acid deprotection, catalysts 4, 5 and 6 were obtained in very good yields. It should be noted that the 4-nitrophenyl regioisomer of 4 was already described [20] .
2.1. Aldol Reaction
With the new derivatives 1 - 6 in hand, we began to evaluate their catalytic behavior in the classical aldol reaction between acetone and 4-nitrobenzaldehyde [21] , and the results are displayed in Table 1. As can be seen, the reaction proceeded smoothly at room temperature under solvent free conditions and was almost total only beyond several dozen hours, except for catalyst 2 (entry 2). If the chemical yields were acceptable (catalysts 2 and 3), the enantiomeric excesses were very low except for catalyst 2. Indeed in that case, a very interesting enantioselectivity of the aldol product was obtained. Regarding the results, we observed the following facts: firstly, the catalyst 2 having proline moieties exhibited higher catalytic efficiencies compared to 1 (entries 1, 2); then, we could notice a strong mismatch effect related to the absolute stereochemistry of the cyclohexane moiety (entries 2, 3). After these initial investigations, catalyst 2, giving the best result, was selected to carry out the optimization of the reaction conditions. We first investigated the solvent effect with or without some additives and the results are displayed in Table 1. While no reaction occurred in aprotic polar solvents (entries 4, 5), apolar solvents (entries 6, 7) induced a significant decrease in catalytic efficiency. Moreover, a catalytic amount of TfOH (entry 9) induced a decrease both rate and yield without significantly affecting the enantiomeric excess. On the other hand, catalytic quantities of AcOH (entry 8) induced an increase of the yield, but involved a slightly decreasing of the enantiomeric excess. With water as additive (entries 10, 11) no significant variations were observed in the reaction rate and enantioselectivity. The reactions, carried out at lower temperatures, have shown as expected a positive effect on the catalytic efficiency and thus the best results were obtained at −20˚C but requiring prolonged reaction time (entries 12, 15). Regarding the loading of the catalyst, the reactions carried out with only 10 mol% of 2 (entries 13 - 15) provided the aldol product with almost the same enantioselectivity but with an expected decreasing of the reaction rate. In order to exclude a quite possible retro-aldol process that could lead to partial racemization, the enantiomeric excess of the reaction was measured several times throughout the reaction, showing no significant variation in value.
To explain the experimental results and especially the different behavior between catalysts 2 and 3, we proposed transition states displayed in Figure 2 for the aldol reaction. Thus, the enamine resulting from the condensation of the catalyst (S,R,R)-2 with acetone adding to the aromatic aldehyde forms a pseudo-cycle stabilized by strong binding interactions between the carbonyl oxygen and two acidic hydrogen atoms. In this cycle, the largest substituent “Ar” is located at the equatorial position. This compact and rigid transition state can explain the relative high enantiomeric excess (78%) promoting the R-isomer of the aldol product. With the isomer (S,S,S)-3 this binding interaction can arise with only one hydrogen, the second one being pushed to the rear of the figure induced by the reverse stereochemistry of the cyclohexane moiety. These very different behaviors between 2 and 3 allowed us to highlight one main characteristic required for catalysts, namely the ability to form a compact cyclic transition state induced by the presence of acidic hydrogen atoms [22] . Following these observations, we thought design proline derivatives 4, 5 and 6 having acidic hydrogen of a sulfonamide function. In addition, one could be expected a significant difference between the catalysts, given the presence in the catalyst 4 of an additional carbonyl group increasing the hydrogen acidity [23] . The experimental results of aldol reactions are displayed in Table 1. The low solubility of catalyst 4 in apolar solvents has limited the range of sol-
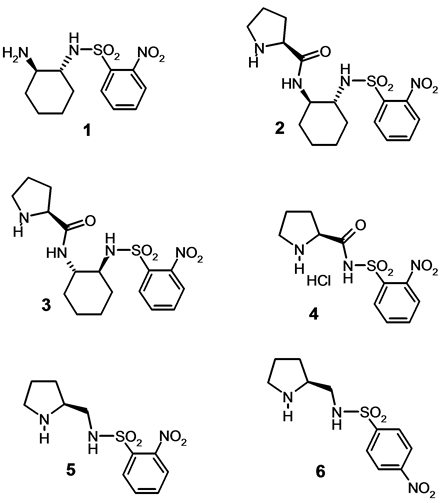
Figure 1. Organocatalysts derived from (R,R)- or (S,S)-1, 2-diaminocyclohexane and/or (S)-proline.
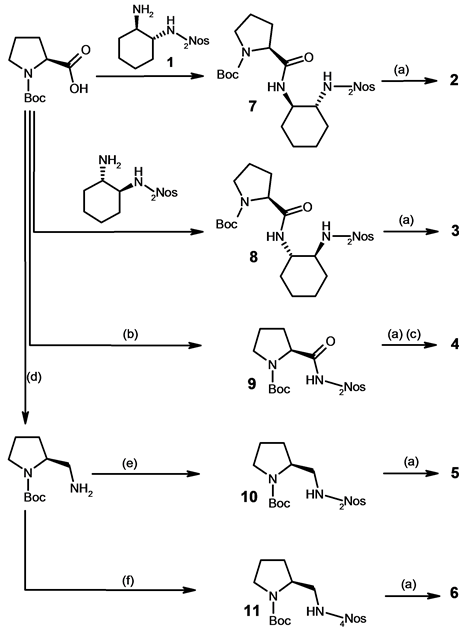
Scheme 1. Reaction conditions: (a) CF3CO2H, CH2Cl2, rt, 2 h; (b) 2Nos-NH2, DMAP, EDC.HCl, THF, rt, 8 h; (c) HCl/ MeOH, Et2O; (d) Reference [18]; (e) 2NosCl, NEt3, CH2Cl2, rt, 4 h; (f) 4NosCl, NEt3, CH2Cl2, rt, 3 h.
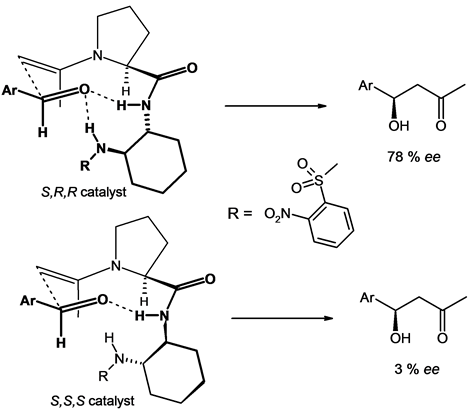
Figure 2. Proposed transition state for the aldol reaction.

Table 1. Catalyst screening and aldol reaction optimizationa.
aReaction conditions: aldehyde (0.33 mmol), acetone (850 μL, 11.5 mmol) using 20 mol% of catalyst and additives (if any); bYield of the isolated product after chromatography on silica gel; cMeasured by chiral HPLC, absolute stereochemistry of the reaction products were assigned according to the literature [21] ; d10 mol%.
vents. Indeed, while no reaction took place in aprotic solvents, excellent results in solvent-free medium were observed (entries 16 - 19, ee up to 96%). In contrast with catalyst 5 in the same conditions, the reaction provided aldol products in only 31% ee (entry 27). Again, with a lower catalyst loading (10%), the reaction occurred (entries 18, 19) and provided the corresponding aldol product with the same high enantioselectivity (96% ee). Finally, the scope and limitation of this process with various aromatic aldehydes under optimized conditions were explored; the results are displayed in Table 1. In all cases, the enantiomeric excesses were of the same order of magnitude as those previously observed. However, some significant differences between aldehydes bearing withdrawing groups and those bearing donor groups can be noted. Indeed, the aldol reaction rate with compound 12h (entry 26, two donor groups) is lower than that observed with other aldehydes; these two donor groups reducing the electrophilicity of the aldehyde function. To conclude this chapter regarding aldol reactions, promising results were obtained showing, once again, that proline derivatives gave the best results.
2.2. Michael Addition
Following these encouraging results, we proposed to test some of these organocatalysts in Michael additions. Organocatalyst activation of this type of reaction has already been intensively studied but still remains a great challenge for the synthetic chemists [13] -[17] . The tests were performed with 2, 4, 5, and 6 to establish their catalytic activities by achieving the reaction model: cyclohexanone/acetone-trans-β-nitrostyrene [24] [25] . The experimental results are displayed in Table 2. As can be seen, the reactions took place smoothly with an extensive reaction time. A surprising result was first obtained with catalyst 2 (entry 1): the enantiomeric excess is near zero while this catalyst was very efficient for the aldol reaction (Table 1, entry 2). With catalyst 4 in acetonitrile, the enantiomeric excess was low at 25˚C (10% ee) with a good diastereomeric ratio (entry 2). However, catalyst 5 (entry 5) proved to be very promising since the enantiomeric excess reached almost 80% in the first experiment. Therefore we choose this catalyst as well as its regioisomer 6 to undertake optimization experiments in order to increase the enantiomeric excess.
The influence of solvent and temperature as well as presence or absence of some additives on the steric course of the reaction was examined and the results are shown in Table 2. Before looking to the results in detail, we can observe that the enantiomeric excesses were generally good to excellent (entries 5 - 19). In acetonitrile solution, addition of a catalytic amount of water or acetic acid (entries 7, 8) significantly increased the reaction rate and, more interestingly, the enantiomeric excesses without altering the diastereomeric ratios. On the other hand, addition of a strong acid (entry 9) induced an important decrease of the enantiomeric excess and reaction rate. We have also tested protic and aprotic solvents, iPrOH with acetic acid at room temperature giving almost the same results (entries 10, 11) and toluene with or without additives yielding better enantiomeric excesses (entries 12 - 15). In the absence of solvent other than the ketone itself, the reaction provided adducts with enantiomeric excesses slightly higher than in acetonitrile (entries 5, 16 and 7, 17). Again a decrease in temperature led to better enantiomeric excesses, but slowed down the reaction rates (entries 11, 15, 18 vs. 10, 14, 17). It should be noted that a spectacular result was observed with catalyst 4 (entries 2 - 4) with which a lowering of the temperature (from 25˚C to - 15˚C) caused an important increase of the enantiomeric excess (from 10% to 60% ee). Similarly to the case of aldol reaction, using a lower catalyst loading (10 mol%, entry 6), the Michael adducts were obtained with a comparable enantiomeric excess but a large reduction of the reaction rate (220 h vs. 120 h). Some improvements of the enantiomeric excesses were observed with catalyst 6 involving the nitro group at the para-position. Enantiomeric excesses were occasionally higher than those obtained with catalyst 5 (entries 28, 15 and 25, 7). We observed a very excellent result (entry 28: 98% ee, 96:4 dr) with a catalytic amount of water in the toluene/cyclohexanone solution at 5˚C.
Opposite results were achieved with acetone and trans-β-nitrostyrene without solvent in presence of catalytic amounts of water (entries 29, 30). The enantiomeric excess (25% at 25˚C and 29% at 5˚C) were the worst results of all examples of Table 2 [26] . One reason that could explain this lower efficiency may be related to the lower size of acetone compared to cyclohexanone which leads to greater degrees of freedom in the transition state. Figure 3 displays the probable catalytic circle [27] of the Michael addition: the reactive enamine from the condensation between chiral secondary amine and cyclohexanone reacts with nitroolefin through the transition state stabilized by some hydrogen bonds including some water molecules [28] [29] . This transition state also explains the increasing of enantioselectivity by the presence of catalytic amounts of water.
Table 2. Catalyst screening and Michael addition optimizationa.
aReaction conditions: trans-β-nitrostyrene (0.60 mmol), cyclohexanone (300 μL, 2.0 mmol) in 900 μL of solvent using 20 mol% of catalyst and additive (if any); bYield of the isolated product; cMeasured by GC/MS analysis; dMeasured by chiral HPLC, absolute stereochemistry of the reaction products were assigned according to the literature [13]; e10 mol%.
3. Conclusion
Among the catalysts we have developed in this study, two of them have proved to be very efficient; catalyst 4 for the aldol reaction and catalyst 6 for the Michael addition. It should be noted that, once again, these two catalysts 4 and 6 both derived from a proline structure [30] . Additional studies are currently in progress in our laboratory to investigate further applications of these new catalysts.
4. Experimental
4.1. General
The reactions were monitored by thin-layer chromatography (TLC) on aluminum plates (0.20 mm, 60 F254) us-
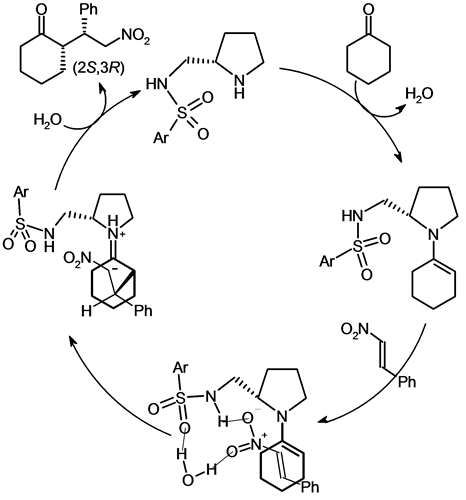
Figure 3. Proposed catalytic cycle of the Michael addition.
ing EtOAc/cyclohexane mixture as eluent. The reaction compounds were first visualized under UV light and then by treatment with iodine vapor. Silica gel (40 - 63 μm) was used for flash chromatographies with EtOAc/ cyclohexane mixtures as eluent. Gas chromatography-mass spectroscopy (GC-MS) was performed with an Agilent 6890N (equipped with a 12 m × 0.20 mm dimethylpolysiloxane capillary column) linked to a Model 5973N (ionization energy: 70 eV). Enantiomeric ratios were determined by chiral HPLC analysis on Daicel Chiralcel AS-H and ADH. Melting points were achieved on a Kofler block. Optical rotations were measured on a Perkin-Elmer model 241. 1H and 13C NMR spectra were recorded in CDCl3 (except specified) respectively at 400 MHz and 100 MHz (Bruker 400). Reagents and materials were obtained from commercial suppliers and were used without further purification.
4.2. Preparation of Organocatalysts
4.2.1. (S)-tert-Butyl 2-[(1R,2R)-2-(2-nitrophenylsulfonamido) cyclohexylcarbamoyl]pyrrolidine-1-carboxylate (7)
To 1.00 g (3.34 mmol) of N-[(1R,2R)-2-aminocyclohexyl]-2-nitrophenylsulfonamide (1) [9] stirred in 33 mL of THF, 740 mg (3.44 mmol) of N-Boc-(S)-proline, and 420 mg (3.44 mmol) of DMAP were successively added. At 0˚C, 660 mg (3.44 mmol) of EDC∙HCl was then added. After 8 h at room temperature the reaction medium was diluted with 70 mL of CH2Cl2 and successively washed with 1N HCl, saturated NaHCO3 and dried over MgSO4. After filtration and concentration of the organic layer under reduced pressure, a flash chromatography of the residue (40% EtOAc in cyclohexane) gave 1.47 g (89% yield) of 7 as a white solid.
7: Rf = 0.5 (40% EtOAc in cyclohexane); mp 160˚C; MS (ES+) m/z 497.20 [M+H]+, 519.17 [M+Na]+, 535.15 [M + K]+, 993.38 [2M + H]+; 1H NMR δ 1.13 - 1.40 (m, 4H), 1.48 (s, 9H), 1.60 - 1.77 (m, 3H), 1.78 - 1.88 (m, 1H), 1.89 - 2.13 (m, 3H), 2.15 - 2.33 (m, 1H), 3.07 - 3.64 (m, 3H), 3.67 - 3.89 (m, 1H), 4.18 - 4.40 (m, 1H), 5.57 - 6.03 (m, 1H), 7.72 - 7.77 (m, 2H), 7.81 - 7.84 (m, 1H), 8.12 (d, J = 7.0 Hz, 1H); 13C NMR δ 24.3 (CH2), 24.7 (CH2), 28.4 (3CH3), 32.9 (CH2), 42.1 (CH), 47.1 (CH2), 52.3 (CH), 80.3 (C), 125.1 (CH), 130.1 (CH), 132.7 (CH), 133.3 (CH), 147.7 (C), 174.2 (C).
4.2.2. (S)-N-[(1R,2R)-2-(2-Nitrophenylsulfonamido)-cyclohexyl]pyrrolidine-2-carboxamide (2)
728 mg (1.47 mmol) of 7 in 15 mL of CF3CO2H/CH2Cl2 (1/4) was stirred 1 h at room temperature. 5 mL of 1N HCl was then added to the reaction medium until pH 1. At 0˚C, the aqueous layer, basified by an excess of NaOH pellets, was extracted with CH2Cl2 and the combined organic layers were dried (MgSO4), filtered and evaporated to give 580 mg of 2 (quantitative yield) as yellow solid.
To 100 mg (0.25 mmol) of 2, dissolved in minimum of HCl/MeOH solution, 25 mL of Et2O was added. After filtration the precipitate was washed with ether to give 103 mg (95% yield) of 2-HCl as yellow solid.
2,HCl: Rf = 0.69 (Al2O3, 5% MeOH in CH2Cl2); mp 94˚C; MS (ES+) m/z 396.95 [M + H]+, 419.09 [M + Na]+; 1H NMR (D2O) δ 0.99 - 1.30 (m, 4H), 1.46 - 1.49 (m, 2H), 1.57 - 1.60 (m, 1H), 1.79 - 1.82 (m, 1H), 1.89 - 2.05 (m, 3H), 2.23 - 2.32 (m, 1H), 3.11 - 3.17 (m, 1H), 3.25 - 3.39 (m, 2H), 3.52 - 3.68 (m, 1H), 4.17 - 4.29 (m, 1H), 7.70 - 7.78 (m, 2H), 7.80 - 7.91 (m, 1H), 7.98 - 8.06 (m, 1H); 13C NMR (D2O) δ 23.6 (CH2), 23.9 (CH2), 24.1 (CH2), 29.8 (CH2), 31.5 (CH2), 31.9 (CH2), 46.4 (CH2), 52.8 (CH), 57.6 (CH), 59.9 (CH), 125.2 (CH), 130.0 (CH), 133.3 (CH), 133.4 (C), 134.5 (CH), 146.9 (C), 169.0 (C).
4.2.3. (S)-tert-Butyl 2-[(1S,2S)-2-(2-nitrophenylsulfonamido)-cyclohexylcarbamoyl]- pyrrolidine-1-carboxylate (8)
Starting from the trans-(S,S)-diaminocyclohexane and using the same protocol as above, 8 was obtained in 91% yields as a white solids.
8: Rf = 0.52 (40% EtOAc in cyclohexane); mp 184˚C; MS (ES+) m/z 497.20 [M + H]+, 519.16 [M + Na]+, 535.15 [M + K]+; 1H NMR δ 1.13 - 1.40 (m, 4H), 1.48 (s, 9H), 1.60 - 1.77 (m, 3H), 1.78 - 1.88 (m, 1H), 1.89 - 2.13 (m, 3H), 2.15 - 2.33 (m, 1H), 3.07 - 3.64 (m, 3H), 3.67 - 3.86 (m, 1H), 4.18 - 4.40 (m, 1H), 5.57 - 6.03 (m, 1H), 7.72 - 7.77 (m, 2H), 7.81 - 7.84 (m, 1H), 8.12 (d, J = 7.0 Hz, 1H); 13C NMR δ 24.3 (CH2), 24.6 (CH2), 27.4 (CH2), 28.4 (3CH3), 31.9 (CH2), 33.6 (CH2), 47.0 (CH2), 51.8 (CH), 59.3 (CH), 59.4 (CH), 80.3 (C), 125.0 (CH), 130.4 (CH), 132.7 (CH), 133.2 (CH), 134.5 (C), 147.7 (C), 174.2 (C).
4.2.4. (S)-N-[(1S,2S)-2-(2-Nitrophenylsulfonamido)-cyclohexyl]pyrrolidine-2-carboxamide (3)
1.00 g (2.0 mmol) of 8 in 20 mL of CF3CO2H/CH2Cl2 (1/4) was stirred 1 h at room temperature. 5 mL of 1N HCl was then added until pH 1. At 0˚C, the separated aqueous layer, basified by an excess of solid NaOH to pH > 10, was extracted with CH2Cl2 and the combined organic layers were dried (MgSO4) and evaporated to give 790 mg of 3 (quantitative yield) as white solid.
3: Rf = 0.58 (15% MeOH in CH2Cl2); mp 103˚C - 105˚C; MS (ES+) m/z 397.25 [M + H]+, 419.26 [M + Na]+, 793.53 [2M + H]+; 1H NMR δ 0.96 - 1.40 (m, 4H), 1.44 - 1.72 (m, 4H), 1.73 - 1.97 (m, 3H), 2.04 - 2.20 (m, 1H), 2.85 - 2.96 (m, 2H), 3.12 - 3.27 (m, 1H), 3.58 - 3.73 (m, 1H), 3.74 - 3.86 (m, 1H), 4.85 (brs, 2H), 7.58 - 7.75 (m, 3H), 7.75 - 7.78 (m, 1H), 8.05 - 8.07 (m, 1H); 13C NMR δ 24.5 (CH2), 24.6 (CH2), 24.1 (CH2), 30.6 (CH2), 32.3 (CH2), 33.2 (CH2), 47.0 (CH2), 52.1 (CH), 59.0 (CH), 60.2 (CH), 124.8 (CH), 130.3 (CH), 132.7 (CH), 133.2 (C), 135.4 (C), 147.7 (C), 173.9 (C).
4.2.5. (S)-tert-Butyl 2-(2-Nitrophenylsulfonylcarbamoyl)-1-carboxylate (9)
To 600 mg (2.80 mmol) of N-Boc-proline stirred at room temperature in 30 mL of THF, 354 mg (2.90 mmol) of DMAP and 506 mg (2.48 mmol) of 2-nitrobenzenesulfonamide were added. At 0˚C, 556 mg (2.90 mmol) of EDC∙HCl was then added. After 8 h at room temperature the reaction medium was diluted with 60 mL of CH2Cl2 and successively washed with 1N HCl, saturated NaCl, and dried over MgSO4. After filtration the organic layer was concentrated over reduced pressure to give 904 mg (91% yield) of 9 as white solid.
9: mp 74˚C; MS (ES+) m/z 422.10 [M + Na]+, 438.08 [M + K]+, 821.22 [2M + Na]+, 837.19 [2M + K]+; 1H NMR δ 1.51 (s, 9H), 1.74 - 2.05 (m, 3H), 2.29 - 2.49 (m, 1H), 3.27 - 3.62 (m, 2H), 4.15 - 4.53 (m, 1H), 7.66 - 7.90 (m, 3H), 8.35 - 8.48 (m, 1H); 13C NMR δ 24.3 (CH2), 28.3 (3CH3), 30.3 (CH2), 47.3 (CH2), 60.7 (CH), 82.1 (C), 124.7 (CH), 132.2 (C), 132.3 (CH), 133.5 (CH), 148.2 (CH), 148.2 (C), 156.8 (C), 171.4 (C).
4.2.6. (S)-N-(2-Nitrophenylsulfonyl-pyrrolidinium)-2-carboxamide (4)
618 mg (1.55 mmol) of 9 in 8 mL of CF3CO2H/CH2Cl2 (1/4) was stirred 1 h at room temperature and then concentrated under reduced pressure. 10 mL of H2O/CH2Cl2 mixture was then added to the residue and the separated aqueous layer, basified by an excess of solid NaOH to pH > 10, was extracted with CH2Cl2. The combined organic layers were dried (MgSO4) and evaporated. The residue was taken up in a minimum of 2.5 N HCl/ MeOH solution and then crystallized by addition of small amounts of ether. After filtration and washing with ether, 477 mg of compound 4-HCl was obtained (92% yield).
4-HCl: mp 192 - 194˚C; MS (ES+) m/z 299.91 [M + H]+, MS (ES–) m/z 297.87 [M – H]+, 1H NMR (D2O) δ 1.80 - 2.13 (m, 3H), 2.26 - 2.45 (m, 1H), 3.23 - 3.67 (m, 3H), 4.22 - 4.26 (dd, J = 9.0, 6.5 Hz, 1H), 7.72 - 7.85 (m, 3H), 8.04 - 8.14 (m, 1H); 13C NMR (D2O) δ 23.6 (CH2), 29.3 (CH2), 46.4 (CH2), 62.0 (CH), 124.5 (CH), 130.1 (CH), 132.7 (CH), 133.3 (C), 134.2 (CH), 147.3 (C), 174.6 (C).
4.2.7. (S)-tert-Butyl 2-[(2-nitrophenylsulfonamido)-methyl]-pyrrolidine-1-carboxylate (10)
To 80 mg (0.40 mmol) of N-Boc-prolinamine stirred at room temperature in 5 mL of CH2Cl2, 70 μL (0.50 mmol) of NEt3 and 88.6 mg (0.40 mmol) of 2-nitrobenzenesulfonyl chloride were added. After 4 h, the reaction medium, diluted with 5 mL of CH2Cl2, was successively washed with 1N HCl, saturated NaCl, and dried over MgSO4. After filtration the concentration of the organic layer over reduced pressure gave 140 mg (91% yield) of 10 as a yellow oil.
10: 1H NMR δ 1.37 (s, 9H), 1.67 - 1.84 (m, 3H), 1.88 - 1.97 (m, 1H), 2.95 - 3.41 (m, 4H), 3.75 - 3.93 (m, 1H), 6.39 (brs, 1H), 7.53 - 7.84 (m, 3H), 8.02 - 8.04 (m, 1H); 13C NMR δ 23.9 (CH2), 28.4 (3CH3), 29.3 (CH2), 46.7 (CH2), 47.4 (CH2), 56.6 (CH), 80.2 (C), 125.1 (CH), 130.9 (CH), 132.5 (CH), 133.4 (CH), 133.8 (C), 148.1 (C), 155.1 (C).
4.2.8. (S)-2-Nitro-N-(pyrrolidin-2-ylmethyl)-benzenesulfonamide (5)
120 mg (0.30 mmol) of 10 in 3 mL of CF3CO2H/CH2Cl2 (1/4) was stirred 5 h at room temperature. 1 mL of 1N HCl was then added to the reaction medium. The aqueous layer, washed by CH2Cl2, was then basified to pH > 10 (NaOH pellets) and extracted with CH2Cl2. The combined organic layers, dried over MgSO4 were evaporated to give 81 mg (92% yield) of 5 as a yellow oil.
5: MS (ES+) m/z 286.1 [M + H]+, 1H NMR δ 1.29 - 1.47 (m, 1H), 1.61 - 1.88 (m, 3H), 2.81 - 2.95 (m, 3H), 3.08 - 3.12 (m, 1H), 3.33 - 3.40 (m, 1H), 4.59 (brs, 2H), 7.62 - 7.68 (m, 2H), 7.73 - 7.75 (m, 1H), 7.99 - 8.09 (m, 1H); 13C NMR δ 25.5 (CH2), 28.8 (CH2), 46.2 (CH2), 47.4 (CH2), 57.8 (CH), 125.1 (CH), 130.9 (CH), 132.6 (CH), 133.3 (CH), 133.9 (C), 148.2 (C).
4.2.9. (S)-4-Nitro-N-(pyrrolidin-2-ylmethyl)-benzenesulfonamide (6)
Starting from 4-nitrobenzenesulfonyl chloride and using the same procedure as above, 11 and 6 were obtained in respectively 92% and 74% yields as yellow oils.
11: 1H NMR δ 1.37 (s, 9H), 1.51 - 1.79 (m, 3H), 1.84 - 2.01 (m, 1H), 2.79 - 2.95 (m, 1H), 2.99 - 3.18 (m, 2H), 3.19 - 3.35 (m, 1H), 3.74 - 3.90 (m, 1H), 7.05 (brs, 1H), 7.97 (d, J = 8.7 Hz, 2H), 8.26 (d, J = 8.7 Hz, 2H); 13C NMR δ 23.7 (CH2), 28.3 (3CH3), 29.5 (CH2), 47.3 (CH2), 48.5 (CH2), 56.6 (CH), 80.6 (C), 124.3 (2CH), 128.2 (2CH), 146.2 (C), 149.8 (C), 156.2 (C).
6: MS (ES+) m/z 286.1 [M + H]+; 1H NMR δ 1.29 - 1.38 (m, 1H), 1.60 - 1.87 (m, 3H), 2.74 (dd, J = 12.6, 8.5 Hz, 1H), 2.74 - 3.82 (m, 1H), 2.84 - 2.92 (m, 1H), 2.99 (dd, J = 12.6, 4.1 Hz, 1H), 4.02 - 4.07 (m, 1H), 4.44 (brs, 2H), 7.97 (d, J = 8.7 Hz, 2H), 8.28 (d, J = 8.7 Hz, 2H); 13C NMR δ 25.7 (CH2), 28.9 (CH2), 46.2 (CH2), 46.9 (CH2), 57.7 (CH), 124.4 (2CH), 128.2 (2CH), 146.2 (C), 149.9 (C).
4.3. Aldol Reactions
4.3.1. General Procedure
To 0.33 mmol of aldehyde 12 in 850 μL (11.5 mmol) of acetone, 66 μmol of organocatalyst 4 was added and the mixture was stirred for appropriate time and temperature (see Table 1). The reaction medium was concentrated under reduced pressure and the residue was purified through column chromatography (10% EtOAc in cyclohexane) to afford aldol 13.
4.3.2. (R)-4-Hydroxy-4-(4'-nitrophenyl)-butan-2-one (13a)
From 4-nitrobenzaldehyde (12a), (R)-4-hydroxy-4-(4’-nitro-phenyl)-butan-2-one (13a) was obtained in 72% yield as pale yellow crystals.
13a: Rf = 0.4 (40% EtOAc in cyclohexane); mp 58˚C - 60˚C (EtOAc/cyclohexane); [α]D20 + 62 (c 0.7, CHCl3); 1H NMR δ 2.17 (s, 3H), 2.78 (d, J = 4 Hz, 2H), 3.0 - 3.5 (m, 1H), 5.19 (t, J = 4.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 2H), 8.12 (d, J = 8.0 Hz, 2H); 13C NMR δ 30.7 (CH3), 51.5 (CH2), 68.9 (CH), 123.8 (2CH), 126.4 (2CH), 147.3 (C), 149.9 (C), 208.5 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 80:20 v/v, 1.0 mL/min, UV 268 nm): tr(major, R) = 9.65 min, tr(minor, S) = 11.43 min, 94% ee.
4.3.3. (R)-4-Hydroxy-4-(2'-nitrophenyl)-butan-2-one (13b)
From 2-nitrobenzaldehyde (12b), (R)-4-hydroxy-4-(2’-nitrophenyl)-butan-2-one (13b) was obtained in 58% yield as a brown solid.
13b: Rf = 0.6 (40% EtOAc in cyclohexane); mp 60˚C - 62˚C (EtOAc/cyclohexane); [α]20D +95 (c 0.62, CHCl3); 1H NMR δ 2.17 (s, 3H), 2.65 (dd, J = 17.8, 9.3 Hz, 1H), 3.07 (dd, J = 17.8, 2.0 Hz, 1H), 5.61 (dd, J = 9.3, 2.0 Hz, 1H), 7.33 - 7.40 (m, 1H), 7.54 - 7.65 (m, 1H), 7.79 - 7.86 (m, 1H), 7.87 - 7.92 (m, 1H); 13C NMR δ 30.5 (CH3), 51.0 (CH2), 65.7 (CH), 124.5 (CH), 128.2 (CH), 128.3 (C), 133.8 (CH), 133.8 (CH), 138.4 (C), 208.9 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 40:60 v/v, 1.0 mL/min, UV 255 nm): tr(major, R) = 5.24 min, tr(minor, S) = 6.47 min, 95% ee.
4.3.4. (R)-4-(4'-Bromophenyl)-4-hydroxy-butan-2-one (13c)
From 4-bromobenzaldehyde (12c), (R)-4-(4’-bromophenyl)-4-hydroxy-butan-2-one (13c) was obtained in 70% yield as a white solid.
13c: Rf = 0.45 (40% EtOAc in cyclohexane); mp 65˚C - 67˚C (EtOAc/cyclohexane); [α]20D +50 (c 0.43, CHCl3); 1H NMR δ 2.10 (s, 3H), 2.69 (dd, J = 17.6, 3.8 Hz, 1H), 2.81 (dd, J = 17.8, 8.5 Hz, 1H), 3.43 (brs, 1H), 5.12 (dd, J = 8.5, 3.3 Hz, 1H), 7.13 - 7.15 (m, 2H), 7.37 - 7.39 (m, 2H); 13C NMR δ 30.8 (CH3), 51.8 (CH2), 69.2 (CH), 121.5 (C), 127.4 (2CH), 131.6 (2CH), 141.8 (C), 208.8 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 85:15 v/v, 1.0 mL/min, UV 221 nm): tr(major, R) = 9.85 min, tr(minor, S) = 12.67 min, 88% ee.
4.3.5. (R)-4-(4'-Chlorophenyl)-4-hydroxy-butan-2-one (13d)
From 4-chlorobenzaldehyde (12d), (R)-4-(4’-chlorophenyl)-4-hydroxy-butan-2-one (13d) was obtained in 61% yield as a white solid.
13d: Rf = 0.45 (40% EtOAc in cyclohexane); mp 60˚C - 62˚C (EtOAc/cyclohexane); [α]20D +59.9 (c 0.85, CHCl3); 1H NMR δ 2.12 (s, 3H), 2.79 (dd, J = 17.6, 4.0 Hz, 1H), 2.85 (dd, J = 17.8, 8.3 Hz, 1H), 3.25 (brs, 1H), 5.12 (dd, J = 8.3, 4.0 Hz, 1H), 7.25 - 7.35 (m, 4H); 13C NMR 30.7 δ (CH3), 51.8 (CH2), 69.2 (CH), 127.0 (2CH), 128.7 (2CH), 133.4 (C), 141.2 (C), 208.8 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 82:18 v/v, 1.0 mL/min, UV 221 nm): tr(major, R) = 11.80 min, tr(minor, S) = 13.80 min, 85% ee.
4.3.6. (R)-4-Hydroxy-4-phenyl-butan-2-one (13e)
From benzaldehyde (12e), (R)-4-hydroxy-4-phenyl-butan-2-one (13e) was obtained in 61% yield as colorless liquid.
13e: Rf = 0.45 (40% EtOAc in cyclohexane); [α]20D +68 (c 0.52, CHCl3); 1H NMR δ 2.19 (s, 3H), 2.74 (dd, J = 17.6, 3.5 Hz, 1H), 2.82 (dd, J = 17.6, 9.0 Hz, 1H), 3.15 (d, J = 3.5 Hz, 1H), 5.15 (ddd, J = 9.0, 3.5, 3.5 Hz, 1H), 7.26 - 7.36 (m, 5H); 13C NMR δ 30.8 (CH3), 52.0 (CH2), 69.8 (CH), 125.6 (2CH), 128.3 (CH), 128.5 (2CH), 142.9 (C), 209.0 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 80:20 v/v, 1.0 mL/min, UV 225 nm): tr(major, R) = 9.98 min, tr(minor, S) = 10.98 min, 90% ee.
4.3.7. (R)-4-Hydroxy-4-(naphthalen-2-yl)-butan-2-one (13f)
From naphthalene-2-carbaldehyde (12f), (R)-4-hydroxy-4-(naphthalen-2-yl)-butan-2-one (13f) was obtained in 68% yield as a colorless oil.
13f: Rf = 0.55 (40% EtOAc in cyclohexane); [α]20D +49 (c 0.72, CHCl3); 1H NMR δ 2.23 (s, 3H), 2.99 (d, J = 5.5 Hz, 1H), 3.0 - 3.5 (m, 1H), 5.96 (t, J = 5.5 Hz, 2H), 7.47 - 7.54 (m, 3H), 7.69 (d, J = 7.3 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.87 (dd, J = 7.5, 7.5 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H); 13C NMR δ 30.8 (CH3), 51.4 (CH2), 66.7 (CH), 122.7 (CH), 123.0 (CH), 125.6 (CH), 125.6 (CH), 126.2 (CH), 128.1 (CH), 129.1 (CH), 129.9 (C), 133.8 (C), 138.2 (C), 209.2 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 78:22 v/v, 1.0 mL/min, UV 217 nm): tr(major, R) = 8.98 min, tr(minor, S) = 9.81 min, 81% ee.
4.3.8. (R)-4-(5-Chloro-2-nitrophenyl)-4-hydroxy-butan-2-one (13g)
From 5-chloro-2-nitrobenzaldehyde (12g), (R)-4-(5-chloro-2-nitrophenyl)-4-hydroxy-butan-2-one (13 g) was obtained in 72% yield as a brown oil.
13g: Rf = 0.65 (40% EtOAc in cyclohexane); 1H NMR δ 2.17 (s, 3H), 2.63 (dd, J = 17.8, 9.5 Hz, 1H), 3.03 (dd, J = 17.8, 2.3 Hz, 1H), 3.5 - 4.0 (m, 1H), 5.63 (dd, J = 9.5, 2.3 Hz, 1H), 7.33 (dd, J = 8.3, 2.5 Hz, 1H), 7.84 (d, J = 2.5 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H); 13C NMR δ 30.4 (CH3), 50.9 (CH2), 63.4 (CH), 126.1 (CH), 128.4 (CH), 128.6 (CH), 140.7 (C), 140.8 (C), 145.1 (C), 208.5 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 60:40 v/v, 1.0 mL/min, UV 225 nm): tr(major, R) = 7.35 min, tr(minor, S) = 8.26 min, 92% ee.
4.3.9. (R)-4-(2,4-Dimethoxyphenyl)-4-hydroxy-butan-2-one (13h)
From 2,4-dimethoxybenzaldehyde (12h), (R)-4-(2,4-dimethoxyphenyl)-4-hydroxy-butan-2-one (13h) was obtained in 29% yield as yellow oil.
13h: Rf = 0.45 (40% EtOAc in cyclohexane); 1H NMR δ 2.10 (s, 3H), 2.73 (dd, J = 17.1, 8.8 Hz, 1H), 2.81 (dd, J = 17.1, 3.5 Hz, 1H), 3.21 (d, J = 4.5 Hz, 1H), 3.72 (s, 3H), 3.74 (s, 3H), 5.23 - 5.27 (m, 1H), 6.37 (d, J = 2.5 Hz, 1H), 6.42 (dd, J = 8.5, 2.3 Hz, 1H), 7.24 (d, J = 8.3 Hz, 1H); 13C NMR δ 30.6 (CH3), 50.6 (CH2), 55.3 (CH3), 55.4 (CH3), 65.5 (CH), 98.6 (CH), 104.2 (CH), 123.5 (C), 127.2 (CH), 157.0 (C), 160.2 (C), 209.3 (C); HPLC (Chiralcel AS-H, n-hexane/iPrOH: 73:27 v/v, 1.0 mL/min, UV 225 nm): tr(major, R) = 12.80 min, tr(minor, S) = 13.90 min, 72% ee.
4.4. Michael Additions
4.4.1. (2S)-2-[(1R)-2-Nitro-1-phenylethyl)-cyclohexanone (14a)
From 90 mg (0.60 mmol) of trans-β-nitrostyrene in 200 μL (2.0 mmol) of cyclohexanone and 900 μL of toluene with a catalytic amount of H2O, 34 mg (0.12 mmol) of 6 was added at room temperature. The mixture was stirred 68 h at room temperature. After evaporation under reduced pressure the residue was purified by flash chromatography (10% EtOAc in cyclohexane) to afford 120 mg (81% yield) of 14a as white crystals.
14a: Rf = 0.5 (10% EtOAc in cyclohexane); mp 134˚C; [α]20D –36 (c 0.28, CHCl3); EIMS m/z 200 (M+-47, 61%), 183 (22), 171 (76), 157 (13), 141 (14), 129 (26), 115 (25), 104 (32), 91 (60); 1H NMR δ 1.12 - 1.19 (m, 1H), 1.48 - 1.65 (m, 4H), 1.82 - 2.02 (m, 1H), 2.24 - 2.42 (m, 2H), 2.57 - 2.64 (m, 1H), 3.68 (td, J = 12.0, 12.0, 4.0 Hz, 1H), 4.54 (dd, J = 12.0, 9.6 Hz, 1H), 4.86 (dd, J = 12.0, 4.0 Hz, 1H), 7.08 - 7.25 (m, 5H); 13C NMR δ 25.0 (CH2), 28.5 (CH2), 33.2 (CH2), 42.7 (CH2), 44.0 (CH), 52.5 (CH), 78.93 (CH2), 127.5 (CH), 128.2 (2CH), 128.9 (2CH), 137.8 (C), 212.0 (C); HPLC (Chiralcel AD7 column, n-hexane/iPrOH: 80:20 v/v, flow rate 0.8 mL/min, wavelength = 260 nm): tR = 8.1 min (2S,3R), tR = 9.2 (2R,3S), tR = 10.5 min (major, 2S,3R), 98% ee.
4.4.2. (−)-(R)-5-Nitro-4-phenyl-pentan-2-one (14b)
From 90 mg (0.60 mmol) of trans-β-nitrostyrene and 150 μL (2.0 mmol) of acetone, and 2.0 mg (0.11 mmol) of water, 35 mg (0.12 mmol) of organocatalyst 6 was added at room temperature. After 48 h at 5˚C, 56 mg (45% yield) of 14b was obtained as white crystals.
14b: Rf = 0.45 (10% EtOAc in cyclohexane); mp 111˚C; [α]20D levorotatory (CHCl3); EIMS m/z 160 (M+-47, 61%), 145 (46), 115 (21), 104 (44), 91 (18), 77 (12); 1H NMR δ 2.11 (s, 3H), 2.91 (d, J = 7.2 Hz, 2H), 3.95 - 4.05 (m, 1H), 4.59 (dd, J = 12.4, 7.6 Hz, 1H), 4.70 (dd, J = 12.5, 6.8 Hz, 1H), 7.2 - 7.35 (m, 5H); 13C NMR δ 30.4 (CH3), 39.0 (CH2), 46.1 (CH2), 79.4 (CH), 127.3 (2CH), 127.9 (CH), 129.1 (2CH), 138.8 (C), 205.4 (C); HPLC (Chiralpak IC, n-hexane/tert-BuOMe: 35:65 v/v, flow rate = 0.8 mL/min, wavelength 220 nm): tR = 12.18 min (major, R), tR = 17.53 min (minor, S), 25% ee.
References
- Pfau, M., Revial, G., Guingant, A. and d’Angelo, J. (1985) Enantioselective Synthesis of Quaternary Carbon Centers through Michael-Type Alkylation of Chiral Imines. Journal of the American Chemical Society, 107, 273-274. http://dx.doi.org/10.1021/ja00287a061
- Pfau, M., Tomas, A., Lim, S. and Revial, G. (1995) Diastereo Selectivity in the Michael-Type Addition of Imines Reacting as their Secondary Enamine Tautomers. Journal of Organic Chemistry, 60, 1143-1147. http://dx.doi.org/10.1021/jo00110a015
- List, B. (2006) The Ying and Yang of Asymmetric Amino-Catalysis. Chemical Communications, 2006, 819-824. http://dx.doi.org/10.1039/b514296m
- Taylor, M.S. and Jacobsen, E.N. (2006) Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angewandte Chemie International Edition, 45, 1520-1543. http://dx.doi.org/10.1002/anie.200503132
- Pellissier, H. (2007) Asymmetric Organocatalysis. Tetrahedron, 63, 9267-9331. http://dx.doi.org/10.1016/j.tet.2007.06.024
- Enders, D., Grondal, C. and Hüttl, M.R.M. (2007) Asymmetric Organocatalytic Domino Reactions. Angewandte Chemie International Edition, 46, 1570-1581. http://dx.doi.org/10.1002/anie.200603129
- Chai, Z. and Zhao, G. (2012) Efficient Organocatalysts Derived from Simple Chiral Acyclic Amino Acids in Asymmetric Catalysis. Catalysis Science & Technology, 2, 29-41. http://dx.doi.org/10.1039/c1cy00347j
- Pellissier, H. (2012) Asymmetric Organocatalytic Cycloadditions. Tetrahedron, 68, 2197-2232. http://dx.doi.org/10.1016/j.tet.2011.10.103
- Sylla-Iyarreta Veitia, M., Joudat, M., Wagner, M., Falguière, A., Guy, A. and Ferroud, C. (2011) Ready Available Chiral Azapyridinomacrocycles n-Oxides; First Results as Lewis Base Catalysts in Asymmetric Allylation of 4-Nitro- benzaldehyde. Heterocycles, 83, 2011-2030.
- Geary, L.M. and Hultin, P.G. (2009) The State of the Art in Asymmetric Induction: The Aldol Reaction as a Case Study. Tetrahedron: Asymmetry, 20, 131-173. http://dx.doi.org/10.1016/j.tetasy.2008.12.030
- Trost, B.M. and Brindle, C.S. (2010) The Direct Catalytic Asymmetric Aldol Reaction. Chemical Society Reviews, 39, 1600-1632. http://dx.doi.org/10.1039/b923537j
- Liu, Y.X., Yang, L., Ma, Z.W., Wang, C.C. and Tao, J.C. (2011) Research Progress in Supported Proline and Proline Derivatives as Recyclable Organocatalysts for Asymmetric C-C Bond Formation Reactions. Chinese Journal of Catalysis, 32, 1295-1311.
- Roca-Lopez, D., Sadaba, D., Delso, I., Herrera, R.P., Tejero, T. and Merino, P. (2010) Asymmetric Organocatalytic Synthesis of γ-Nitrocarbonyl Compounds through Michael and Domino Reactions. Tetrahedron: Asymmetry, 21, 2561-2601.
- Quintard, A., Belot, S., Marchal, E. and Alexakis, A. (2010) Aminal-Pyrrolidine Organocatalysts—Highly Efficient and Modular Catalysts for α-Functionalization of Carbonyl Compounds. European Journal of Organic Chemistry, 2010, 927-936. http://dx.doi.org/10.1002/ejoc.200901283
- Tsakos, M. and Kokotos, C.G. (2012) Organocatalytic “Difficult” Michael Reaction of Ketones with Nitrodienes Utilizing a Primary Amine-Thiourea Based on Di-tert-Butyl Aspartate. European Journal of Organic Chemistry, 2012, 576-580. http://dx.doi.org/10.1002/ejoc.201101402
- Sun, Z.W., Peng, F.Z., Li, Z.Q., Zou, L.W., Zhang, S.X., Li, X. and Shao, Z.H. (2012) Enantioselective Conjugate Addition of both Aromatic Ketones and Acetone to Nitroolefins Catalyzed by Chiral Primary Amines Bearing Multiple Hydrogen-Bonding Donors. Journal of Organic Chemistry, 77, 4103-4110. http://dx.doi.org/10.1021/jo300011x
- Wang, L., Zang, X. and Ma, D. (2012) Organocatalytic Michael Addition of Aldehydes to Trisubstituted Nitroolefins. Tetrahedron, 68, 7675-7679.
- Dahlin, N., Bøgevig, A. and Adolfsson, H. (2004) N-Arene-Sulfonyl-2-Aminomethylpyrrolidines. Novel Modular Ligands and Organocatalysts for Asymmetric Catalysis. Advanced Synthesis & Catalysis, 346, 1101-1105. http://dx.doi.org/10.1002/adsc.200404098
- Veverková, E., Štrasserová, J., Šebesta, R. and Toma, Š. (2010) Asymmetric Mannich Reaction Catalyzed by N-Arylsulfonyl-L-proline Amides. Tetrahedron: Asymmetry, 21, 58-61. http://dx.doi.org/10.1016/j.tetasy.2009.12.013
- List, B., Lerner, R.A. and Barbas III, C.F. (2000) Proline-Catalyzed Direct Asymmetric Aldol Reactions. Journal of the American Chemical Society, 122, 2395-2396. http://dx.doi.org/10.1021/ja994280y
- Bisai, V., Bisai, A. and Singh, V.K. (2012) Enantioselective Organocatalytic Aldol Reaction Using Small Organic Molecules. Tetrahedron, 68, 4541-4580. http://dx.doi.org/10.1016/j.tet.2012.03.099
- Huang, X.-Y., Wang, H.-J. and Shi, J. (2010) Theoretical Study on Acidities of (S)-Proline Amide Derivatives in DMSO and Its Implications for Organocatalysis. The Journal of Physical Chemistry A, 114, 1068-1081. http://dx.doi.org/10.1021/jp909043a
- Lao, J.-H., Zhang, X.-J., Wang, J.-J., Li, X.-M., Yan, M. and Luo, H.-B. (2009) The Effect of Hydrogen Bond Donors in Asymmetric Organocatalytic Conjugate Additions. Tetrahedron: Asymmetry, 20, 2818-2822. http://dx.doi.org/10.1016/j.tetasy.2009.11.029
- List, B., Pojarliev, P. and Martin, H.J. (2001) Efficient Proline-Catalyzed Michael Additions of Unmodified Ketones to Nitro-Olefins. Organic Letters, 3, 2423-2425. http://dx.doi.org/10.1021/ol015799d
- Betancort, J.M. and Barbas III, C.F. (2001) Catalytic Direct Asymmetric Michael Reactions: Taming Naked Aldehyde Donors. Organic Letters, 3, 3737-3740. http://dx.doi.org/10.1021/ol0167006
- Lu, A., Liu, T., Wu, R., Wang, Y., Wu, G., Zhou, Z., Fang, J. and Tang, C.A. (2011) Recyclable Organocatalyst for Asymmetric Michael Addition of Acetone to Nitroolefins. Journal of Organic Chemistry, 76, 3872-3879.
- Singh, K.N., Singh, P., Singh, P., Lal, N. and Sharma, S.K. (2012) Pyrrolidine Based Chiral Organocatalyst for Efficient Asymmetric Michael Addition of Cyclic ketones to β-Nitrostyrenes. Bioorganic & Medicinal Chemistry Letters, 22, 4225-4228.
- Sevin, A., Tortajada, J. and Pfau, M. (1986) Toward a Transition-State Model in the Asymmetric Alkylation of Chiral Ketone Secondary Enamines by Electron-Deficient Alkenes. A Theoretical MO Study. Journal of Organic Chemistry, 51, 2671-2675. http://dx.doi.org/10.1021/jo00364a011
- Sevin, A., Masure, D., Giessnerprettre, C. and Pfau, M. (1990) A Theoretical Investigation of Enantioselectivity— Michael Reaction of Secondary Enamines with Enones. Helvetica Chimica Acta, 73, 552-573. http://dx.doi.org/10.1002/hlca.19900730303
NOTES

*Corresponding author.