 Pharmacology & Pharmacy, 2011, 2, 238-247 doi:10.4236/pp.2011.24031 Published Online October 2011 (http://www.SciRP.org/journal/pp) Copyright © 2011 SciRes. PP Effects of Two Anti-TNF-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on Secreted and Cell-Associated TNF-α in Vitro Ken J. Grattendick, James M. Nakashima, Shri N. Giri Solanan, Inc., Dallas, USA. Email: sgiri@solananinc.com Received June 2nd, 2011; revised July 28th, 2011; accepted August 5th, 2011. ABSTRACT Tumor necrosis factor-alpha (TNF-α) is a potent inflammatory cytokine and its exaggerated production has been im- plicated in acute, chronic and autoimmune inflammatory diseases. Proteinaceous and non-proteinaceous anti-TNF-α agents have been developed to reduce its circulating levels either by neutralizing, binding or inhibiting the de novo synthesis with the aim of achieving desirable therapeutic effects. In the present study, we compared the effects of a pro- tein-based anti-TNF-α drug, etanercept, and a non-protein-based anti-TNF-α small molecule, 5-ethyl-1-phenyl-2-(1H) pyridone (5-EPP), on the LPS-stimulated secretion of TNF-α in the medium and TNF-α associated with the THP-1 cells in vitro. Both drugs had marked concentration-dependent inhibitory effects on the LPS-stimulated secretion of TNF-α. However, their effects on the LPS-stimulated cell-associated TNF-α were diametrically opposed to each other. For in- stance, etanercept further increased the level by up to 12-fold, whereas 5-EPP inhibited the level in a dose dependent manner. In addition, 5- EPP caused a significant reduction in the elevated level of cell associated TNF-α caused by LPS + etanercept. The differences in the levels of cell-associated TNF-α as reported in the present study may partly explain the adverse effects of some protein-based anti-TNF-α drugs including etanercept as opposed to a non-protein-based anti-TNF-α drug such as pirfenidone, a structural analogue of 5-EPP, for treatment of some TNF-α mediated diseases. It was concluded from the findings of this study that drugs which elevate the levels of cell associated-TNF-α will poten- tially have more adverse events even after reducing the secreted levels of TNF-α than the drugs which reduce both the secreted and cell-associated TNF-α levels. Keywords: Tumor Necrosis Factor-Alpha, 5-Ethyl-1-phenyl-2-(1H)-pyridone, Etanercept, Cytokines 1. Introduction The cytokine tumor necrosis factor-alpha (TNF-α) is a key regulator of systemic inflammation and the acute phase response. A precursor form of this cytokine is ex- pressed as a 26 kD type II polypeptide transmembrane protein (mTNF) on the surface of activated macrophages, lymphocytes and other cell types (endothelium). The soluble form of TNF-α, a 17 kD polypeptide, is released when mTNF is cleaved by the metalloproteinase TNF-α conver ting enzyme (T ACE) [1]. Bo th mTNF-α and solu- ble TNF-α readily form biologically-active homotrimeric structures. TNF-α elicits its pathophysiological responses by in- teracting with two structurally distinct transmembrane TNF-α receptors: type I (TNFR1) and type II (TNFR2). Although both receptors are glycoproteins, contain mul- tiple cysteine-rich repeats and share considerable struc- tural and functional homology within their extracellular domains, the intracellular domains of TNFR1 and TNFR2 differ considerably. Signal transduction occurs via both overlapping and discrete pathways. Secreted TNF-α binds to both receptors, while mTNF-α binds mainly to TNFR2 [2-5]. Like TNF-α, these receptors exist both in a soluble form and in a membrane-anchored form. The soluble receptors bind and neutralize the bio- logical activities of TNF-α, whereas the membrane-an- chored form of the receptors mediates the pleiotropic pathophysiological effects of this cytokine [6-8]. The central role of TNF-α in the pathogenesis of chronic inflammatory diseases has led to the develop- ment and widespread clinical uses of protein-based anti- TNF-α agents for treatment of rheumatoid arthritis [9,10],  Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 239 Secreted and Cell-Associated TNF-α in Vitro Crohn’s disease [11], psoriasis [12], ankylosing spondy- litis [13,14], and Behcet’s disease [15]. The FDA has approved five protein-based anti-TNF-α drugs and all of them act, in essence, as neutralizing antibodies of se- creted TNF-α, thus preventing its interactions with cell surface receptors. However, none of these protein-based anti-TNF-α agents have any reported effect on the syn- thesis of TNF-α. Lastly, these drugs are only effective when administered via intravenous, intramuscular or subcutaneous injection—a significant therapeutic con- sideration as compared with a non-protein-based anti- TNF-α. While the role of soluble TNF-α in many diseases has been investigated for over three decades, the contribu- tions of mTNF to TNF-α-associated pathophysiology have only been appreciated relatively recently. Early studies found that mTNF mediates various cytotoxic and inflammatory functions of leucocytes via direct cell-cell contact and binding to TNFRs on target cells [16-18]. More recently, the binding of mTNF to TNFRs was shown to initiate reverse signaling (i.e., receptor-medi- ated ligand signal transduction) which altered the physi- ology of the mTNF-expressing cells. Binding of anti- TNF-α antibodies or soluble TNFRs to mTNF phos- phorylates an intracellular signaling domain that triggers changes in the level of intracellular calcium [18,19], ini- tiates synthesis and release of various cytokines [18,20, 21], increases expression of the E-selectin adhesion mole- cule [22], and alters the cellular response to inflammatory stimuli [23]. Pirfenidone, a low molecular weight pyridone, has been shown to inhibit TNF-α release in vitro, block en- dotoxin-induced and staphylococcus aureus enterotoxin B-induced endotoxic shock in animal models and inhibit TNF-α synthesis at the translational level [24-27]. The pirfenidone analogs, fluorofenidone and 5-ethyl-1-phenyl- 2-(1H) pyridone (5-EPP), also offer a similar degree of protection against endotoxin, lipopolysaccharide/D-ga- lactosamine (LPS/D-GalN) and cecal ligation and punc- ture (CLP) m odel s of se pt i c sh ock [28,29]. The ability of orally-effective pirfenidone to reduce the production and secretion of TNF-α in animal models has led to clinical investigations in diseases where this cytokine has been implicated in the underlying patho- physiology such as secondary progressive multiple scle- rosis (SPMS) and idiopathic pulmonary fibrosis (IPF). For instance, three clinical trials have shown efficacy of pirfenidone treatment for SPMS [30-32], a crippling dis- ease which may be a TNF-α-driven process [33-35] and IPF [36,37]. Recently, Noble et al. published the results of two randomized clinical trials of the CAPACITY study and suggested that pirfenidone offers an appropri- ate treatment option for patients with IPF [38]. Based in part on these clinical results, pirfenidone has been ap- proved for the treatment of IPF in both Japan and the European Union. Protein-based anti-TNF-α drugs and pyridone com- pounds such as pirfenidone have one thing in common— both significantly reduce the elevated levels of biologi- cally active soluble TNF-α, albeit by different mecha- nisms. Protein-based anti-TNF-α drugs bind to TNF-α and thereby block further biological interactions, whereas pyridone-based compounds inhibit the enhanced synthe- sis of TNF-α intrinsic to inflammatory events. This dif- ference in mechanism of action may explain why pir- fenidone appeared to arrest the progression of SPMS and stabilized the condition in clinical studies [31], while infliximab exacerbated the progression of SPMS and forced the discontinuation of the trials [39]. In order to better delineate these differences in mecha- nism of action, we compared the effects of the pro- tein-based anti-TNF-α drug, etanercept, with that of the novel pyridone, 5-EPP, on the regulation of soluble and cell-associated TNF-α in LPS-stimulated THP-1 cells in vitro. While little is presently known about 5-EPP, its chemical structure would imply that it likely possesses pharmacological and toxicological properties similar to those of pirfenidone and fluorofenidone. In this study, we report that both etanercept (ET) and 5-EPP limited the bioavailable TNF-α in the medium following LPS-stimulation. In marked contrast, however, the levels of cell associated TNF-α in etanercept-treated cells increased several-fold, while these levels were sig- nificantly reduced in 5-EPP-treated cells. These results may have important ramifications with respect to adverse events between the use of some protein-based anti-TNF- α drugs and drugs from the pyridone family in the man- agement of TNF-α-drive n di seases. 2. Materials and Methods 2.1. Reagents All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Etanercept (Enbrel™) was purchased from a pharmaceutical supplier in 1 cc syringes at 50 mg/ml and was used in both native carrier and dialyzed (in PBS) forms with no differences in effi- cacy or toxicity between the two. Heating (95˚C × 10 minutes) of etanercept stock solution completely inacti- vated all TNF-α-neutralizing activity as assessed by TNF-α bioassay (data not shown). Five-EPP was synthesized at the Solanan Research Laboratory according to the procedure described earlier [40]. Briefly, the starting compound 5-ethyl-2-pyridone Copyright © 2011 SciRes. PP  Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 240 Secreted and Cell-Associated TNF-α in Vitro (J & W Pharmlab LLC, Levittown, PA) was reacted with bromobenzene in the presence of a Cu-Zn catalyst under a blanket of argon to produce 5-EPP. After extraction, purification and re-crystallization, the material was found to contain >99.8% 5-EPP as determined by reverse phase HPLC and NMR analysis with a sharp melting point of 58.5˚C. All drugs and reagents used in our experiments were diluted in RPMI-1640 with 2% FBS and supple- mented as describe d bel o w. 2.2. In Vitro Cell Treatment THP-1 cells (ATCC, Manassas, VA), a mononuclear cell line which can be differentiated into macrophage-like cells [41], were collected from culture media by cen- trifugation at 250 × g an d resuspended at 1 × 106 cells/ml in RPMI-1640 supplemented with 10% FBS (Hyclone, Logan, UT), 50 mg/ml gentamicin, 25 mM HEPES, 1.25 g/L sodium bicarbonate, 100 µM 2-mercaptoethanol and 50 ng/ml phorbol myristate acetate (PMA; to facilitate differentiation and cell adherence to the culture plates). Five hundred µl of cell suspension were added to each well of a 24-well Costar plate (Thomas Scientific, Swe- desboro, NJ) and allowed to incubate for 18 hours at 37˚C under 5% CO2. After incubation, cells were washed 2X and fresh media (not containing PMA) were added. Cells were allowed to incubate for an additiona l 24 hours to abate the effects of PMA on cell activation. Media were then removed and cells treated with LPS, 5-EPP, or etanercept (ET) dissolved in RPMI for the time described for each experiment. Following incubation, culture media and cell lysates (to be discussed later) were collected and analyzed for TNF-α by bioassay or ELISA, respectively. 2.3. TNF-α Bioassay TNF-α was measured in culture media using a modifica- tion of the TNF-α bioassay utilizing Wehi-164 var13 cells (ATCC) as described previously [42]. The modifi- cation of the protocol involved replacing neutral red staining with the more sensitive and reproducible so luble formazan dye-based viability assay to determine TNF-α mediated cytotoxicity. Briefly, Wehi-164 cells were plated at 1.5 × 104 per well in Falcon Primaria 96-well plates (Thomas Scientific) and allowed to adh ere. After 6 hours, culture media from drug-treated THP-1 cells (as described previously) were centrifuged to remove cell debris and added to the Wehi-164 cells. Serial 1:2 dilu- tions were performed in-well for all samples. Serial dilu- tions of recombinant human TNF-α standard (3.9 - 500 pg/ml; BD Biosciences, San Jose, CA) were also in- cluded to determine the quantity of TNF-α in the experi- mental samples. Finally, 2 µg/ml actinomycin-D were added to each well and the cells were incubated over- night. After 20 hours, media were removed and cell vi- ability was determined by MTS assay (Promega, Madi- son, WI) using manufacturer’s instructions. Absorbance at 492 nm was measured with a Thermo Multiskan EX (Thermo Fisher Scientific, Inc, Waltham, MA). Levels of TNF-α in the samples were calculated based on the for- mula derived from the standard curve for the recombi- nant TNF-α standard. 2.4. Cell Lysate Analysis for TNF-α by ELISA After media were removed from THP-1 cells as de- scribed in the previous section, the adherent cells were washed 3X in PBS and then incubated at 4˚C for 15 min- utes with lysate buffer (10 mM HEPES, 1mM EDTA, 60 mM KCl, 0.5% NP-40, 1 mM DTT, 1 mM PMSF) con- taining protease inhibitor cocktail. After 2 freeze-thaws, lysate mixtures were collected and centrifuged at 10,000 × g at 4˚C and supernatants collected and stored at –80˚C until analyzed for TNF-α by ELISA. Immediately prior to TNF-α quantification, supernatants from each sample group were normalized for protein content as determined by BCA kit (Promega). The TNF-α OptEIA ELISA kit was purchased from BD BioSciences and the protocol used in this study was per manufacturer’s instructions. TMB conversion was measured at 450 nm and 540 nm on a Thermo Multiskan EX plate reader. It was necessary to measure secreted TNF-α and cell- associated TNF-α by different assays. The secreted TNF-α in the present study was measured using bioassay be- cause etanercept adsorbs TNF-α and does not remove it from the assay tubes. The presence of the etanercept- bound TNF- α in the tubes could still bind to the an tibod- ies utilized in the ELISA and this wou ld have resulted in aberrantly high values. This is consistent with reports by Scallon, et al. [43] on the binding properties of TNF-α antagonists. The bioassay measures biologically active available TNF-α only and is better suited for measuring cytokines in the presence of neutralizing agents. Al- though attempts were made to measure TNF-α in the cell lysate by bioassay, it was not possible to separate the resulting cytotoxicity caused by TNF-α from that of the residual lysis buffer present in the assay tubes. The di- alysis of lysis buffer against isotonic saline introduced more variables that further confounded the results. 2.5. Apoptosis Assay THP-1 cells were incubated in 6-well plates with RPMI supplemented with PMA at 1 × 106 cells/ml for 12 h. Fresh media not containing PMA were then added and the cells incubated for 24 hours. Next, cells were incu- bated for 18 h with control media; 1 ng/ml LPS; 1 ng/ml LPS + 200 µg/ml 5-EPP; 1 ng/ml LPS + 0.1 µg/ml eta- Copyright © 2011 SciRes. PP 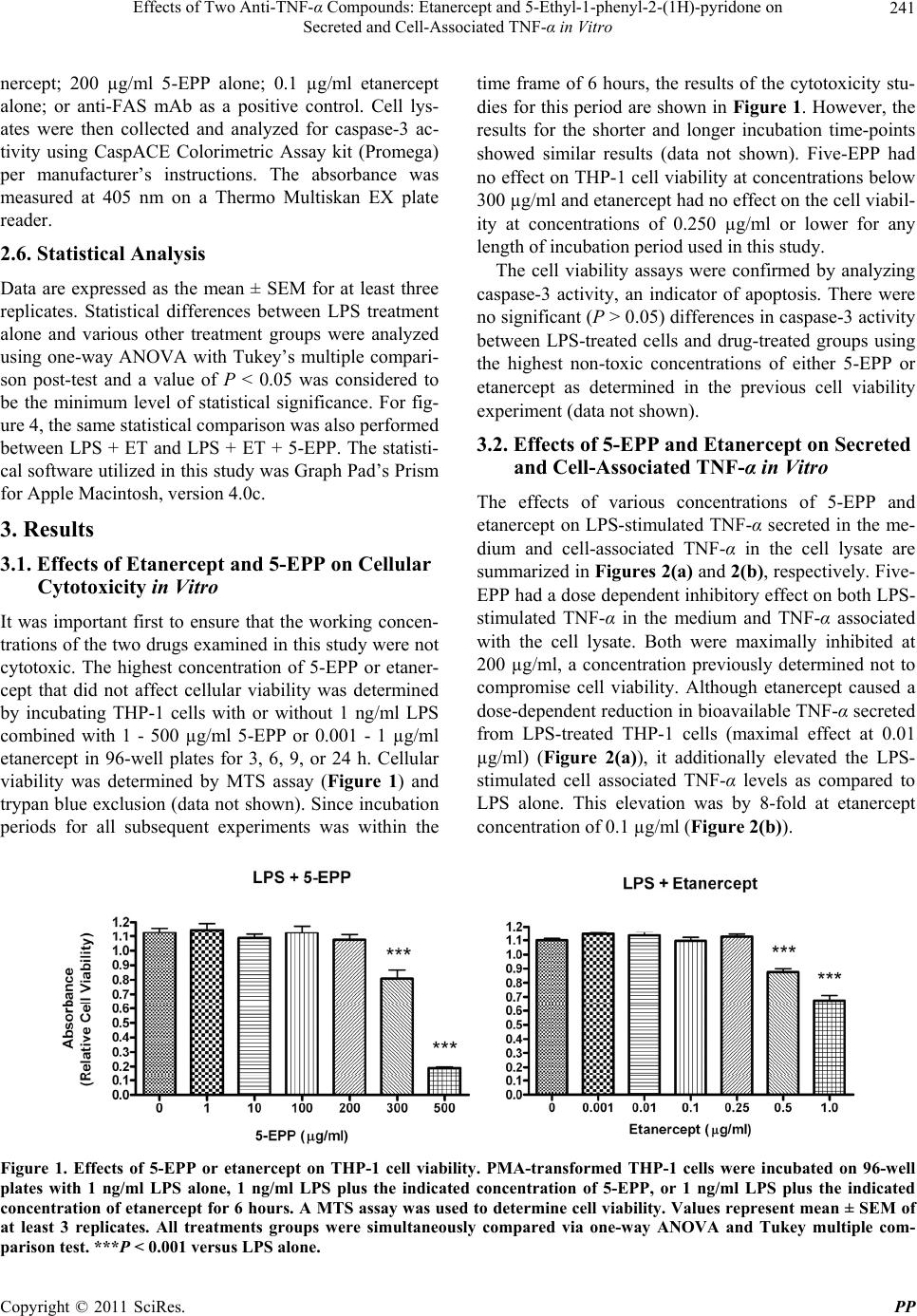 Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on Secreted and Cell-Associated TNF-α in Vitro Copyright © 2011 SciRes. PP 241 nercept; 200 µg/ml 5-EPP alone; 0.1 µg/ml etanercept alone; or anti-FAS mAb as a positive control. Cell lys- ates were then collected and analyzed for caspase-3 ac- tivity using CaspACE Colorimetric Assay kit (Promega) per manufacturer’s instructions. The absorbance was measured at 405 nm on a Thermo Multiskan EX plate reader. time frame of 6 hours, the results of the cytotoxicity stu- dies for this period are shown in Figure 1. However, the results for the shorter and longer incubation time-points showed similar results (data not shown). Five-EPP had no effect on THP-1 cell viability at conc entrations below 300 µg/ml and etanercept had no effect on the cell viabil- ity at concentrations of 0.250 µg/ml or lower for any length of incubation period used in this study. 2.6. Statistical Analysis The cell viability assays were confirmed by analyzing caspase-3 activity, an indicator of apoptosis. There were no significant (P > 0.05) differences in caspase-3 activity between LPS-treated cells and drug-treated groups using the highest non-toxic concentrations of either 5-EPP or etanercept as determined in the previous cell viability experiment (data not shown). Data are expressed as the mean ± SEM for at least three replicates. Statistical differences between LPS treatment alone and various other treatment groups were analyzed using one-way ANOVA with Tukey’s multiple compari- son post-test and a value of P < 0.05 was considered to be the minimum level of statistical significance. For fig- ure 4, the same statistical comparison was also performed between LPS + ET and LPS + ET + 5-EPP. The statisti- cal software utilized in this study was Graph Pad’s Prism for Apple Mac i ntosh, versio n 4. 0c. 3.2. Effects of 5-EPP and Etanercept on Secreted and Cell-Associated TNF-α in Vitro The effects of various concentrations of 5-EPP and etanercept on LPS-stimulated TNF-α secreted in the me- dium and cell-associated TNF-α in the cell lysate are summarized in Figures 2(a) and 2(b), respectively. Five- EPP had a dose dependent inhibitory effect on both LPS- stimulated TNF-α in the medium and TNF-α associated with the cell lysate. Both were maximally inhibited at 200 µg/ml, a concentration previously determined not to compromise cell viability. Although etanercept caused a dose-dependent reduction in bioavailable TNF-α secreted from LPS-treated THP-1 cells (maximal effect at 0.01 µg/ml) (Figure 2(a)), it additionally elevated the LPS- stimulated cell associated TNF-α levels as compared to LPS alone. This elevation was by 8-fold at etanercept concentration of 0.1 µg/ml (Figure 2(b)). 3. Results 3.1. Effects of Etanercept and 5-EPP on Cellular Cytotoxicity in Vitro It was important first to ensure that the working concen- trations of the two drugs examined in this study were not cytotoxic. The highest concentration of 5-EPP or etaner- cept that did not affect cellular viability was determined by incubating THP-1 cells with or without 1 ng/ml LPS combined with 1 - 500 µg/ml 5-EPP or 0.001 - 1 µg/ml etanercept in 96-well plates for 3, 6, 9, or 24 h. Cellular viability was determined by MTS assay (Figure 1) and trypan blue exclusion (data not shown). Since incubation periods for all subsequent experiments was within the Figure 1. Effects of 5-EPP or etanercept on THP-1 cell viability. PMA-transformed THP-1 cells were incubated on 96-well plates with 1 ng/ml LPS alone, 1 ng/ml LPS plus the indicated concentration of 5-EPP, or 1 ng/ml LPS plus the indicated concentration of etanercept for 6 hours. A MTS assay was used to determine cell viability. Values represent mean ± SEM of at least 3 replicates. All treatments groups were simultaneously compared via one-way ANOVA and Tukey multiple com- arison test. ***P < 0.001 versus LPS alone. p 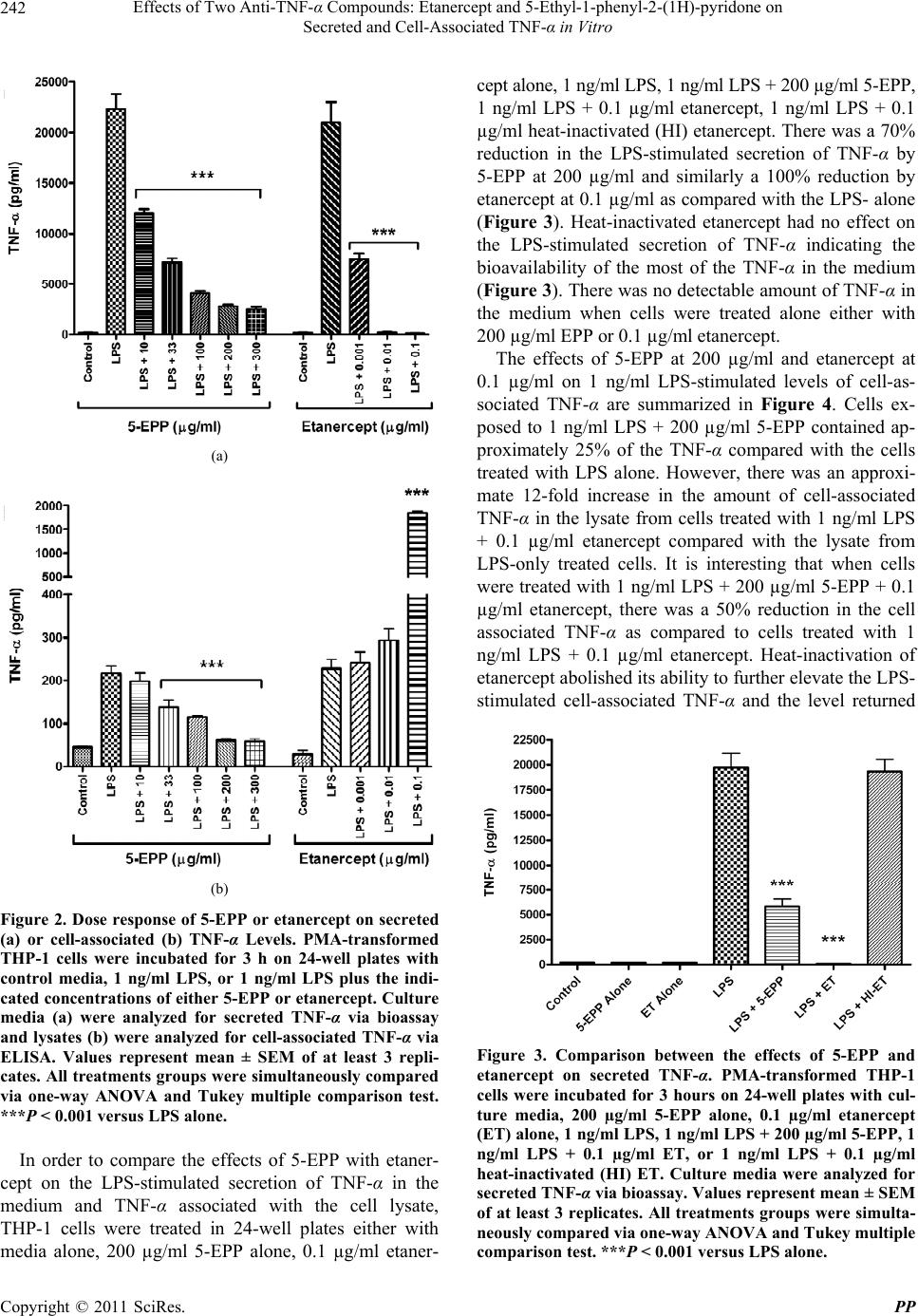 Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 242 Secreted and Cell-Associated TNF-α in Vitro (a) (b) Figure 2. Dose response of 5-EPP or etanercept on secreted (a) or cell-associated (b) TNF-α Levels. PMA-transformed THP-1 cells were incubated for 3 h on 24-well plates with control media, 1 ng/ml LPS, or 1 ng/ml LPS plus the indi- cated concentrations of either 5-EPP or etanercept. Culture media (a) were analyzed for secreted TNF-α via bioassay and lysates (b) were analyzed for cell-associated TNF-α via ELISA. Values represent mean ± SEM of at least 3 repli- cates. All treatments groups were simultaneously compared via one-way ANOVA and Tukey multiple comparison test. ***P < 0.001 versus LPS alone. In order to compare the effects of 5-EPP with etaner- cept on the LPS-stimulated secretion of TNF-α in the medium and TNF-α associated with the cell lysate, THP-1 cells were treated in 24-well plates either with media alone, 200 µg/ml 5-EPP alone, 0.1 µg/ml etaner- cept alone, 1 ng/ml LPS, 1 ng/ml LPS + 200 µg/ml 5-EPP, 1 ng/ml LPS + 0.1 µg/ml etanercept, 1 ng/ml LPS + 0.1 µg/ml heat-inactivated (HI) etanercept. There was a 70% reduction in the LPS-stimulated secretion of TNF-α by 5-EPP at 200 µg/ml and similarly a 100% reduction by etanercept at 0.1 µg/ml as compared with the LPS- alone (Figure 3). Heat-inactivated etanercept had no effect on the LPS-stimulated secretion of TNF-α indicating the bioavailability of the most of the TNF-α in the medium (Figure 3). There was no detectable amount of TNF-α in the medium when cells were treated alone either with 200 µg/ml EPP or 0.1 µg/ml etanercept. The effects of 5-EPP at 200 µg/ml and etanercept at 0.1 µg/ml on 1 ng/ml LPS-stimulated levels of cell-as- sociated TNF-α are summarized in Figure 4. Cells ex- posed to 1 ng/ml LPS + 200 µg/ml 5-EPP contained ap- proximately 25% of the TNF-α compared with the cells treated with LPS alone. However, there was an approxi- mate 12-fold increase in the amount of cell-associated TNF-α in the lysate from cells treated with 1 ng/ml LPS + 0.1 µg/ml etanercept compared with the lysate from LPS-only treated cells. It is interesting that when cells were treated with 1 ng /ml LPS + 200 µg/ml 5-EPP + 0.1 µg/ml etanercept, there was a 50% reduction in the cell associated TNF-α as compared to cells treated with 1 ng/ml LPS + 0.1 µg/ml etanercept. Heat-inactivation of etanercept abolished its ability to further elevate the LPS- stimulated cell-associated TNF-α and the level returned Figure 3. Comparison between the effects of 5-EPP and etanercept on secreted TNF-α. PMA-transformed THP-1 cells were incubated for 3 hours on 24-well plates with cul- ture media, 200 µg/ml 5-EPP alone, 0.1 µg/ml etanercept (ET) alone, 1 ng/ml LPS, 1 ng/ml LPS + 200 µg/ml 5-EPP, 1 ng/ml LPS + 0.1 µg/ml ET, or 1 ng/ml LPS + 0.1 µg/ml heat-inactivated (HI) ET. Culture media were analyzed for secreted TNF-α via bioassay. Values represent mean ± SEM of at least 3 replicates. All treatments groups were simulta- neously compared via one-way ANOVA and Tukey multiple comparison test. ***P < 0.001 versus LPS alone. Copyright © 2011 SciRes. PP 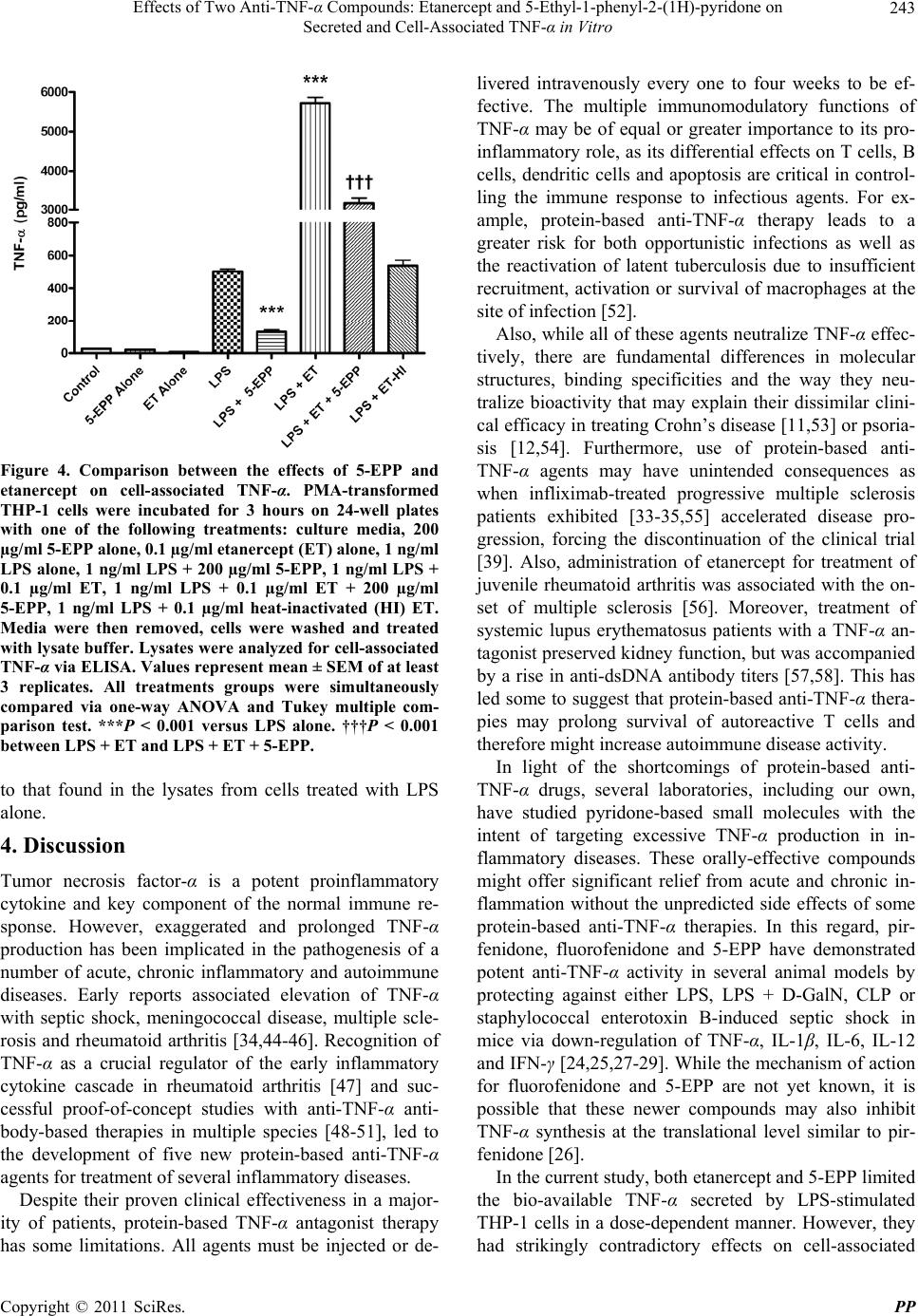 Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 243 Secreted and Cell-Associated TNF-α in Vitro Figure 4. Comparison between the effects of 5-EPP and etanercept on cell-associated TNF-α. PMA-transformed THP-1 cells were incubated for 3 hours on 24-well plates with one of the following treatments: culture media, 200 µg/ml 5-EPP alone, 0.1 µg/ml etanercept (ET) alone, 1 ng/ml LPS alone, 1 ng/ml LPS + 200 µg/ml 5-EPP, 1 ng/ml LPS + 0.1 µg/ml ET, 1 ng/ml LPS + 0.1 µg/ml ET + 200 µg/ml 5-EPP, 1 ng/ml LPS + 0.1 µg/ml heat-inactivated (HI) ET. Media were then removed, cells were washed and treated with lysate buffer. Lysates were analyzed for cell-associated TNF-α via ELISA. Values represent mean ± SEM of at least 3 replicates. All treatments groups were simultaneously compared via one-way ANOVA and Tukey multiple com- parison test. ***P < 0.001 versus LPS alone. †††P < 0.001 between LP S + ET and LPS + ET + 5-EPP. to that found in the lysates from cells treated with LPS alone. 4. Discussion Tumor necrosis factor-α is a potent proinflammatory cytokine and key component of the normal immune re- sponse. However, exaggerated and prolonged TNF-α production has been implicated in the pathogenesis of a number of acute, chronic inflammatory and autoimmune diseases. Early reports associated elevation of TNF-α with septic shock, meningococcal disease, multiple scle- rosis and rheumatoid arthritis [34,44-46]. Recognition of TNF-α as a crucial regulator of the early inflammatory cytokine cascade in rheumatoid arthritis [47] and suc- cessful proof-of-concept studies with anti-TNF-α anti- body-based therapies in multiple species [48-51], led to the development of five new protein-based anti-TNF-α agents for treatment of several inflammatory diseases. Despite their proven clinical effectiveness in a major- ity of patients, protein-based TNF-α antagonist therapy has some limitations. All agents must be injected or de- livered intravenously every one to four weeks to be ef- fective. The multiple immunomodulatory functions of TNF-α may be of equal or greater importance to its pro- inflammatory role, as its differential effects on T cells, B cells, dendritic cells and apoptosis are critical in control- ling the immune response to infectious agents. For ex- ample, protein-based anti-TNF-α therapy leads to a greater risk for both opportunistic infections as well as the reactivation of latent tuberculosis due to insufficient recruitment, activation or survival of macrophages at the site of infection [52]. Also, while all of these agents neutralize TNF-α effec- tively, there are fundamental differences in molecular structures, binding specificities and the way they neu- tralize bioactivity that may explain their dissimilar clini- cal efficacy in treating Crohn’s disease [11,53] or psoria- sis [12,54]. Furthermore, use of protein-based anti- TNF-α agents may have unintended consequences as when infliximab-treated progressive multiple sclerosis patients exhibited [33-35,55] accelerated disease pro- gression, forcing the discontinuation of the clinical trial [39]. Also, administration of etanercept for treatment of juvenile rheumatoid arthritis was associated with the on- set of multiple sclerosis [56]. Moreover, treatment of systemic lupus erythematosus patients with a TNF-α an- tagonist preserved kidney function, but was accompanied by a rise in anti-dsDNA antibody titers [57,58]. This has led some to suggest that protein-based anti-TNF-α thera- pies may prolong survival of autoreactive T cells and therefore might increase autoimmune disease activity. In light of the shortcomings of protein-based anti- TNF-α drugs, several laboratories, including our own, have studied pyridone-based small molecules with the intent of targeting excessive TNF-α production in in- flammatory diseases. These orally-effective compounds might offer significant relief from acute and chronic in- flammation without the unpredicted side effects of some protein-based anti-TNF-α therapies. In this regard, pir- fenidone, fluorofenidone and 5-EPP have demonstrated potent anti-TNF-α activity in several animal models by protecting against either LPS, LPS + D-GalN, CLP or staphylococcal enterotoxin B-induced septic shock in mice via down-regulation of TNF-α, IL-1β, IL-6, IL-12 and IFN-γ [24,25,27-29]. While the mechanism of action for fluorofenidone and 5-EPP are not yet known, it is possible that these newer compounds may also inhibit TNF-α synthesis at the translational level similar to pir- fenidone [26] . In the current study, both etanercept and 5-EPP limited the bio-available TNF-α secreted by LPS-stimulated THP-1 cells in a dose-dependent manner. However, they had strikingly contradictory effects on cell-associated Copyright © 2011 SciRes. PP  Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 244 Secreted and Cell-Associated TNF-α in Vitro TNF-α levels when co-incubated with LPS. Etanercept increased cell associated TNF-α levels by several-fold at non-toxic concentrations, while 5-EPP inhibited TNF-α in a dose-dependent manner as compared with the LPS treatment alone. An intriguing in vivo study by Mohler, et al. reported elevated serum TNF-α levels in mice treated with monomeric sTNFR or low doses of a syn- thetic dimeric sTNFR:Fc (a structure similar to etaner- cept) following LPS treatment as compared to LPS treatment only [59]. Our current findings are consistent with those of Mohler et al., lending credence to our find- ings that etanercept somehow further enhances LPS- stimulated levels of cell-associated TNF-α. The mechanism responsible for enhanced effect of etanercept on cell-associated TNF-α in LPS-treated cells is not clearly understood. It is possible that etanercept- TNF-α complexes are endocytosed making them resistant to repeated washes and resulting in elevated intracellular TNF-α. Alternatively, etanercept only binds trimeric forms of TNF-α and does not maintain a stable complex with either mTNF or soluble TNF-α. Thus the etanercept/ TNF-α complex instability may result in a relatively lar- ger pool of unbound and bio-available TNF-α [43], which is then available to stimulate TNFRs and poten- tially contribute to adverse events. Also, Xin et al. showed that cells previously pre-treated with soluble TNFR (ana- logous to etanercept treatment) became primed for sub- sequent activation by TNF-α through reverse signaling [60]. Thus, in the present study, simultaneous treatment with both etanercept and LPS might allow etanercept- mTN F -α reverse signaling to further prime these cells for additional activation from LPS-stimulated soluble TNF-α release. Lastly, further study is warranted to demonstrate that this in vitro effect seen in the transformed cell line also occurs in human macrophages isolated from blood. The combination of LPS + etanercept that produced elevated levels of cell-associated TNF-α did not have any detectable effects on cell viability as revealed by our cy- totoxicity experiments. However, other cell types may be more sensitive to the cytotoxic effects of mTNF and might exhibit increased adverse effects if exposed to etanercept-treated cells. In situ cells laden with TNF-α on their membranes are capable of altering the physiology of neighboring cells via mTNF-mediated reverse signal- ing [17,18], which is known to trigger a myriad of en- hanced pro-inflammatory responses. For example, re- verse signaling through TNF-α induces the pro-inflam- matory cytokines interleukin-2 and interferon gamma [18] and cell adhesion molecules [22], none of which would be reversed by protein-based TNF-α antagonists. More- over, the near-complete suppression of soluble TNF-α release by TACE inhibition did not prevent hepatocellu- lar necrosis and apoptosis induced by LPS + D-GalN or ConA. In fact, TACE inhibition exacerbated ConA-in- duced liver injury and did not reduce ConA-elevated cell-associated TNF-α [61]. Thus, a protein-based TNF-α antagonist might also cause unexpected adverse effects via up-regulated mTNF-mediated cytotoxicity or cyto- kine release. The elevation of both TNF-α secretion and cell-asso- ciated TNF-α levels by LPS were clearly suppressed by 5-EPP. In addition, 5-EPP was also able to significantly suppress the rise in cell-associated TNF-α induced by a combined LPS + etanercept treatment. The ability of 5-EPP to reduce both LPS-stimulated TNF-α release and cell-associated TNF-α in vitro is entirely consistent with earlier studies with pirfenidone [24,62]. Five-EPP also protects mice from a lethal dose of LPS + D-GalN and completely blocks the simultaneous rise in LPS + D-GalN- stimulated serum TNF-α [28]. Similar protective effects in rodent models have been reported for the related pyri- dones, pirfenidone and fluorofenidone [24,29]. In con- trast to the actions of protein-based TNF-α antagonists or TACE inhibitors, inhibiting the synthesis of this cytokine via pyridone-based compounds would likely reduce both forward and reverse signaling and suppress the multi- factorial inflammatory cascade at its origin. In conclusion, our results confirm that pyridone com- pounds such as 5-EPP can effectively reduce both LPS- induced soluble and cell-associated TNF-α bioactivity in vitro. Etanercept also reduced soluble TNF-α bioactivity in LPS-stimulated cells, but in marked contrast, elevated cell-associated bioactivity. Thus, inhibition of TNF-α production by pyridone compounds such as 5-EPP may provide significant advantages over some of the currently available protein-based anti-TNF-α therapies and may offer a viable therapeutic strategy for the management of TNF-α driven acute and chronic inflammation. However, further research with these and related compounds is required to determine if our find ings can be substantiated for these classes of compounds. REFERENCES [1] R. A. Black, C. T. Rauch, C. J. Kozlosky , J. J. Peschon, J. L. Slack, M. F. Wolfson, et al., “A Metalloproteinase Dis- integrin That Releases Tumour-Necrosis Factor-Alpha from Cells,” Nature, Vol. 385, No. 6618, 1997, pp. 729-733. doi:10.1038/385729a0 [2] E. Decoster, B. Vanhaesebroeck, P. Vandenabeele, J. Grooten and W. Fiers, “Generation and Biological Charac- terization of Membrane-Bound, Uncleavable Murine Tu- mor Necrosis Factor,” The Journal of Biological Chemistry, Vol. 270, No. 31, 1995, pp. 18473-18478. doi:10.1074/jbc.270.31.18473 Copyright © 2011 SciRes. PP  Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 245 Secreted and Cell-Associated TNF-α in Vitro [3] M. Grell, E. Douni, H. Wajant, M. Lohden, M. Clauss, B. Maxeiner, et al., “The Transmembrane Form of Tumor Necrosis Factor Is the Prime Activating Ligand of the 80 kDa Tumor Necrosis Factor Receptor,” Cell, Vol. 83, No. 5, 1995, pp. 793-802. doi:10.1016/0092-8674(95)90192-2 [4] M. Grell, H. Wajant, G. Zimmermann and P. Scheurich, “The Type 1 Receptor (CD120a) Is the High-Affinity Receptor for Soluble Tumor Necrosis Factor,” Proceed- ings of the National Academy of Sciences of the United States of America, Vol. 95, No. 2, 1998, pp. 570-575. doi:10.1073/pnas.95.2.570 [5] D. Wallach, E. E. Varfolomeev, N. L. Malinin, Y. V. Goltsev, A. V. Kovalenko and M. P. Boldin, “Tumor Ne- crosis Factor Receptor and Fas Signaling Mechanisms,” Annual Review of Immunology, Vol. 17, No. 1, 1999, pp. 331-367. doi:10.1146/annurev.immunol.17.1.331 [6] L. J. Old, “Tumor Necrosis Factor (TNF),” Science, Vol. 230, No. 4726, 1985, pp. 630-632. doi:10.1126/science.2413547 [7] P. Vandenabeele, W. Declercq, R. Beyaert and W. Fiers, “Two Tumour Necrosis Factor Receptors: Structure and Function,” Trends in Cell Biology, Vol. 5, No. 10, 1995, pp. 392-399. doi:10.1016/S0962-8924(00)89088-1 [8] P. Vassalli, “The Pathophysiology of Tumor Necrosis Factors,” Annual Review of Immunology, Vol. 10, 1992, pp. 411-452. doi:10.1146/annurev.iy.10.040192.002211 [9] J. M. Bathon, R. W. Martin, R. M. Fleischmann, J. R. Tesser, M. H. Schiff, E. C. Keystone, et al., “A Com- parison of Etanercept and Methotrexate in Patients with Early Rheumatoid Arthritis,” The New England Journal of Medicine, Vol. 343, No. 22, 2000, pp. 1586-1593. doi:10.1056/NEJM200011303432201 [10] P. E. Lipsky, D. M. van der Heijde, E. W. St Clair, D. E. Furst, F. C. Breedveld, J. R. Kalden, et al., “Infliximab and Methotrexate in the Treatment of Rheumatoid Arthri- tis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Ar- thritis with Concomitant Therapy Study Group,” The New England Journal of Medicine, Vol. 343, No. 22, 2000, pp. 1594-1602. doi:10.1056/NEJM200011303432202 [11] S. R. Targan, S. B. Hanauer, S. J. van Deventer, L. Mayer, D. H. Present, T. Braakman, et al., “A Short-Term Study of Chimeric Monoclonal Antibody cA2 to Tumor Necro- sis Factor Alpha for Crohn’s Disease. Crohn’s Disease cA2 Study Group,” The New England Journal of Medi- cine, Vol. 337, No. 15, 1997, pp. 1029-1035. doi:10.1056/NEJM199710093371502 [12] P. J . Mease, B. S. Goffe, J . Metz , A. VanderStoep, B. Finck and D. J. Burge, “Etanercept in the Treatment of Psoriatic Arthritis and Psoriasis: A Randomised Trial,” Lancet, Vol. 356, No. 9227, 2000, pp. 385-390. doi:10.1016/S0140-6736(00)02530-7 [13] J. Braun, J. Brandt, J. Listing, A. Zink, R. Alten, W. Golder, et al., “Treatment of Active Ankylosing Spondy- litis with Infliximab: A Randomised Controlled Multicen- tre Trial,” Lancet, Vol. 359, No. 9313, 2002, pp. 1187- 1193. doi:10.1016/S0140-6736(02)08215-6 [14] J. D. Gorman, K. E. Sack and J. C. Davis Jr., “Treatment of Ankylosing Spondylitis by Inhibition of Tumor Necro- sis Factor Alpha,” The New England Journal of Medicine, Vol. 346, No. 18, 2002, pp. 1349-1356. doi:10.1056/NEJMoa012664 [15] S. Munoz-Fernandez, V. Hidalgo, J. Fernandez-Melon, A. Schlincker and E. Martin-Mola, “Effect of Infliximab on Threatening Panuveitis in Behcet’s Disease,” Lancet, Vol. 358, No. 9293, 2001, p. 1644. [16] G. Caron, Y. Delneste, J. P. Aubry, G. Magistrelli, N. Herbault, A. Blaecke, et al., “Human NK Cells Constitu- tively Express Membrane TNF-Alpha (mTNFalpha) and Present mTNFalpha-De pendent Cytotoxic Activity,” Euro- pean Journal of Immunology, Vol. 29, No. 11, 1999, pp. 3588-3595. doi:10.1002/(SICI)1521-4141(199911)29:11<3588::AID- IMMU3588>3.0.CO;2-O [17] T. Decker, M. L. Lohmann-Matthes and G. E. Gifford, “Cell-Associated Tumor Necrosis Factor (TNF) as a Kill- ing Mechanism of Activated Cytotoxic Macrophages,” Jour- nal of Immunology, Vol. 138, No. 3, 1987, pp. 957-962. [18] M. Higuchi, K. Nagasawa, T. Horiuchi, M. Oike, Y. Ito, M. Yasukawa, et al., “Membrane Tumor Necrosis Factor- Alpha (TNF-Alpha) Expressed on HTLV-I-Infected T Cells Mediates a Costimulatory Signal for B Cell Activa- tion-Characterization of Membrane TNF-Alpha,” Clinical Immunology and Immunopathology, Vol. 82, No. 2, 1997, pp. 133-140. doi:10.1006/clin.1996.4291 [19] A. D. Watts, N. H. Hunt, Y. Wanigasekara, G. Bloom- field, D. Wallach, B. D. Roufogalis, et al., “A Casein Kinase I Motif Present in the Cytoplasmic Domain of Members of the Tumour Necrosis Factor Ligand Family Is Implicated in ‘Reverse Signalling’,” The EMBO Jour- nal, Vol. 18, No. 8, 1999, pp. 2119-2126. doi:10.1093/emboj/18.8.2119 [20] C. Ferran, F. Dautry, S. Merite, K. Sheeha n, R. Schreibe r, G. Grau, et al., “Anti-Tumor Necrosis Factor Modulates Anti-CD3-Triggered T Cell Cytokine Gene Expression in Vivo,” The Journal of Clinical Investigation, Vol. 93, No. 5, 1994, pp. 2189-2196. doi:10.1172/JCI117215 [21] G. H. Waetzig, D. Seegert, P. Rosenstiel, S. Nikolaus and S. Schreiber, “p38 Mitogen-Activated Protein Kinase Is Activated and Linked to TNF-Alpha Signaling in In- flammatory Bowel Disease,” Journal of Immunology, Vol. 168, No. 10, 2002, pp. 5342-5351. [22] S. Harashima, T. Horiuchi, N. Hatta, C. Morita, M. Higuchi, T. Sawabe, et al., “Outside-to-Inside Signal through the Membrane TNF-Alpha Induces E-Selectin (CD62E) Ex- pression on Activated Human CD4+ T Cells,” Journal of Immunology, Vol. 166, No. 1, 2001, pp. 130-136. [23] G. Eissner, S. Kirchner, H. Lindner, W. Kolch, P. Janosch, M. Grell, et al., “Reverse Signaling through Transmem- brane TNF Confers Resistance to Lipopolysaccharide in Human Monocytes and Macrophages,” Journal of Im- munology, Vol. 164, No. 12, 2000, pp. 6193-6198. [24] W. C. Cain, R. W. Stuart, D. L. Lefkowitz, J. D. Starnes, S. Margolin and S. S. Lefkowitz, “Inhibition of Tumor Copyright © 2011 SciRes. PP  Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on 246 Secreted and Cell-Associated TNF-α in Vitro Necrosis Factor and Subsequent Endotoxin Shock by Pir- fenidone,” International Journal of Immunopharmacol- ogy, Vol. 20, No. 12, 1998, pp. 685-695. doi:10.1016/S0192-0561(98)00042-3 [25] M. L. Hale, S. B. Margolin, T. Krakauer, C. J. Roy and B. G. Stiles, “Pirfenidone Blocks the in Vitro and in Vivo Effects of Staphylococcal Enterotoxin B,” Infection and Immunity, Vol. 70, No. 6, 2002, pp. 2989-2994. doi:10.1128/IAI.70.6.2989-2994.2002 [26] H. Nakazato, H. Oku, S. Yamane, Y. Tsuruta and R. Su- zuki, “A Novel Anti-Fibrotic Agent Pirfenidone Sup- presses Tumor Necrosis Factor-Alpha at the Translational Level,” European Journal of Pharmacology, Vol. 446, No. 1-3, 2002, pp. 177-185. doi:10.1016/S0014-2999(02)01758-2 [27] H. Oku, H. Nakazato, T. Horikawa, Y. Tsuruta and R. Suzuki, “Pirfenidone Suppresses Tumor Necrosis Fac- tor-Alpha, Enhances Interleukin-10 and Protects Mice from Endotoxic Shock,” European Journal of Pharma- cology, Vol. 446, No. 1, 2002, pp. 167-176. doi:10.1016/S0014-2999(02)01757-0 [28] K. J. Grattendick, J. M. Nakashima and S. N. Giri, “Ef- fects of 5-Ethyl-1-phenyl-2-(1H) Pyridone on Serum Bio- markers of Multiorgan Dysfunction and Mortality in Li- popolysaccharide/Glactosamine and Cecal Ligation and Puncture Models of Septic Shock in Mice,” Research Communication in Molecular Pathology and Pharma- cology, In Press. [29] Y. Tang, B. Li, N. Wang, Y. Xie, L. Wang, Q. Yuan, et al., “Fluorofenidone Protects Mice from Lethal Endotoxemia through the Inhibition of TNF-Alpha and IL-1beta Re- lease,” International immunopharmacology, Vol. 10, No. 5, 2010, pp. 580-583. doi:10.1016/j.intimp.2010.02.005 [30] J. D. Bowen, K. Maravilla and S. B. Margolin, “Open- Label Study of Pirfenidone in Patients with Progressive Forms of Multiple Sclerosis,” Multiple Sclerosis (Hound- mills, Basingstoke, England), Vol. 9, No. 3, 2003, pp. 280-283. [31] J. E. Walker, S. N. Giri and S. B. Margolin, “A Double- Blind, Randomized, Controlled Study of Oral Pirfenidone for Treatment of Secondary Progressive Multiple Sclero- sis,” Multiple Sclerosis (Houndmills, Basingstoke, Eng- land), Vol. 11, No. 2, 2005, pp. 149-158. [32] J. E. Walker and S. B. Margolin, “Pirfenidone for Chronic Progressive Multiple Sclerosis,” Multiple sclerosis (Hound- mills, Basingstoke, England), Vol. 7, No. 5, 2 001, pp. 305- 312. [33] K. Akassoglou, J. Bauer, G. Kassiotis, H. Lassmann, G. Kollias and L. Probert, “Transgenic Models of TNF In- duced Demyelination,” Advances in Experimental Medi- cine and Biology, Vol. 468, 1999, pp. 245-259. [34] F. M. Hofman, D. R. Hinton, K. Johnson and J. E. Merrill, “Tumor Necrosis Factor Identified in Multiple Sclerosis Brain,” The Journal of Experimental Medicine, Vol. 170, No. 2, 1989, pp. 607-612. doi:10.1084/jem.170.2.607 [35] J. E. Merrill and E. N. Benveniste, “Cytokines in In- flammatory Brain Lesions: Helpful and Harmful,” Trends in Neurosciences, Vol. 19, No. 8, 1996, pp. 331-338. doi:10.1016/0166-2236(96)10047-3 [36] G. Raghu, W. C. Johnson, D. Lockhart and Y. Mageto, “Treatment of Idiopathic Pulmonary Fibrosis with a New Antifibrotic Agent, Pirfenidone: Results of a Prospective, Open-Label Phase II Study,” American Journal of Respi- ratory and Critical Care Medicine, Vol. 159, No. 4, 1999, pp. 1061-1069. [37] H. Taniguchi, M. Ebina, Y. Kondoh, T. Ogura, A. Azuma, M. Suga, et al., “Pirfenidone in Idiopathic Pulmonary Fi- brosis,” The European Respiratory Journal: Official Journal of the European Society for Clinical Respiratory Physiology, Vol. 35, No. 4, 2010, pp. 821-829. [38] P. W. Noble, C. Albera , W. Z. Bradford, U. Costabel, M. K. Glassberg, D. Kardatzke, et al., “Pirfenidone in Pa- tients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials,” Lancet, Vol. 377, No. 9779, 2011, pp. 1760-1769. doi:10.1016/S0140-6736(11)60405-4 [39] B. W. van Oosten, F. Barkhof, L. Truy en, J. B. Boringa, F. W. Bertelsmann, B. M. von Blomberg, et al., “Increased MRI Activity and Immune Activation in Two Multiple Sclerosis Patients Treated with the Monoclonal Anti-Tu- mor Necrosis Factor Antibody cA2,” Ne urology, Vol. 47, No. 6, 1996, pp. 1531-1534. [40] S. B. Margolin, S. N. Giri and W. J. Goux, “Treatment of Sepsis with 5-Ethyl-1-phenyl-2-(1H)-pyridone and Novel Methods for Synthesis,” International Application Pub- lished under Patent Cooperation Treaty, WO 2009/111785 A2, 2009. [41] S. Tsuchiya, M. Yamabe, Y. Yamaguchi, Y. Kobayashi, T. Konno and K. Tada, “Establishment and Characteriza- tion of a Human Acute Monocytic Leukemia Cell Line (THP-1),” International Journal of Cancer, Vol. 26, No. 2, 1980, pp. 171-176. doi:10.1002/ijc.2910260208 [42] C. D. Morgan, K. C. Mills, D. L. Lefkowitz and S. S. Le- fkowitz, “An Improved Colori metric Assay for Tumor Ne- crosis Factor Using WEHI 164 Cells Cultured on Novel Microtiter Plates,” Journal of Immunological Methods, Vol. 145, No. 1-2, 1991, pp. 259-262. doi:10.1016/0022-1759(91)90336-E [43] B. Scallon, A. Cai, N. Solowski, A. Rosenberg, X. Y. Song, D. Shealy, et al., “Binding and Functional Compariso ns of Two Types of Tumor Necrosis Factor Antagonists,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 301, No. 2, 2002, pp. 418-426. doi:10.1124/jpet.301.2.418 [44] T. Calandra, J. D. Baumgartner, G. E. Grau, M. M. Wu, P. H. Lambert, J. Schellekens, et al., “Prognostic Values of Tumor Necrosis Factor/Cachectin, Interleukin-1, Inter- feron-Alpha, and Interferon-Gamma in the Serum of pa- tients with Septic Shock. Swiss-Dutch J5 Immunoglobu- lin Study Group,” The Journal of Infectious Diseases, Vol. 161, No. 5, 1990, pp. 982-987. doi:10.1093/infdis/161.5.982 [45] S. J. Hopkins and A. Meager, “Cytokines in Synovial Copyright © 2011 SciRes. PP  Effects of Two Anti-T N F-α Compounds: Etanercept and 5-Ethyl-1-phenyl-2-(1H)-pyridone on Secreted and Cell-Associated TNF-α in Vitro Copyright © 2011 SciRes. PP 247 Fluid: II. The Presence of Tumour Necrosis Factor and Interferon,” Clinical and Experimental Immunology, Vol. 73, No. 1, 1988, pp. 88-92. [46] A. Waage, A. Halstensen and T. Espevik, “Association between Tumour Necrosis Factor in Serum and Fatal Out- come in Patients with Meningococcal Disease,” Lancet, Vol. 329, No. 8529, 1987, pp. 355-357. doi:10.1016/S0140-6736(87)91728-4 [47] F. M. Brennan, D. Chantry, A. Jackson, R. Maini and M. Feldmann, “Inhibitory Effect of TNF Alpha Antibodies on Synovial Cell Interleukin-1 Production in Rheumatoid Ar- thritis,” Lancet, Vol. 334, No. 8657, 1989, pp. 244-247. doi:10.1016/S0140-6736(89)90430-3 [48] P. F. Piguet, G. E. Grau, C. Vesin, H. Loet scher, R. Gentz and W. Lesslauer, “Evolution of Collagen Arthriti s in Mice Is Arrested by Treatment with Anti-Tumour Necrosis Fac- tor (TNF) Antibody or a Recombinant Soluble TNF Re- ceptor,” Immunology, Vol. 77, No. 4, 1992, pp. 510-514. [49] R. O. Williams, M. Feldmann and R. N. Maini, “Anti- Tumor Necrosis Factor Ameliorates Joint Disease in Mur- ine Collagen-Induced Arthritis,” Proceedings of the Na- tional Academy of Sciences of the United States of America, Vol. 89, No. 20, 1992, pp. 9784-9788. doi:10.1073/pnas.89.20.9784 [50] K. J. Tracey, Y. Fong, D. G. Hesse, K. R. Manogue, A. T. Lee, G. C. Kuo, et al., “Anti-Cachectin/TNF Monoclonal Antibodies Prevent Septic Shock during Lethal Bacte- raemia,” Nature, Vol. 330, No. 6149, 1987, pp. 662-664. doi:10.1038/330662a0 [51] S. M. Opal, A. S. Cross, J. C. Sadoff, H. H. Collins, N. M. Kelly, G. H. Victor, et al., “Efficacy of Antilipopolysac- charide and Anti-Tumor Necrosis Factor Monoclonal An- tibodies in a Neutropenic Rat Model of Pseudomonas sepsis,” The Journal of Clinical Investigation, Vol. 88, No. 3, 1991, pp. 885-890. doi:10.1172/JCI115390 [52] J. J. Gomez-Reino, L. Carmona, V. R. Valverde, E. M. Mola and M. D. Montero, “Treatment of Rheumatoid Ar- thritis with Tumor Necrosis Factor Inhibitors May Predis- pose to Significant Increase in Tuberculosis Risk: A Mul- ticenter Active-Surveillance Report,” Arthritis and Rheu- matism, Vol. 48, No. 8, 2003, pp. 2122-2127. doi:10.1002/art.11137 [53] W. J. Sandborn, S. B. Hanauer, S. Katz, M. Safdi, D. G. Wolf, R. D. Baerg, et al., “Etanercept for Active Crohn’s Disease: A Randomized, Double-Blind, Placebo-Controlled Trial,” Gastroenterology, Vol. 121, No. 5, 2001, pp. 1088- 1094. doi:10.1053/gast.2001.28674 [54] U. Chaudhari, P. Romano, L. D. Mulcahy, L. T. Dooley, D. G. Baker and A. B. Gottlieb, “Efficacy and Safety of In- fliximab Monotherapy for Plaque-Type Psoriasis: A Ran- domised Trial,” Lancet, Vol. 357, No. 9271, 2001, pp. 1842-1847. doi:10.1016/S0140-6736(00)04954-0 [55] B. W. van Oosten, F. Barkhof, P. E. Scholten, B. M. von Blomberg, H. J. Ader and C. H. Polman, “Increased Pro- duction of Tumor Necrosis Factor Alpha, and Not of In- terferon Gamma, Preceding Disease Activity in Patients with Multiple Sclerosis,” Archives of Neurology, Vol. 55, No. 6, 1998, pp. 793-798. doi:10.1001/archneur.55.6.793 [56] N. L. Sicotte and R. R. Voskuhl, “Onset of Multiple Scle- rosis Associated with Anti-TNF Therapy,” Neurology, Vol. 57, No. 10, 2001, pp. 1885-1888. [57] M. Aringer and J. S. Smolen, “SLE—Complex cytokine Effects in a Complex Autoimmune Disease: Tumor Ne- crosis Factor in Systemic Lupus Erythematosus,” Arthri- tis Research & Therapy, Vol. 5, No. 4, 2003, pp. 172-177. doi:10.1186/ar770 [58] R. A. Mageed and D. A. Isenberg, “Tumour Necrosis Factor Alpha in Systemic Lupus Erythematosus and Anti- DNA Autoantibody Production,” Lupus, Vol. 11, No. 2, 2002, pp. 850-855. doi:10.1191/0961203302lu306rr [59] K. M. Mohler, D. S. Torrance, C. A. Smith, R. G. Good- win, K. E. Stremler, V. P. Fung, et al., “Soluble Tumor Necrosis Factor (TNF) Receptors Are Effective Thera- peutic Agents in Lethal Endotoxemia and Function Si- multaneously as Both TNF Carriers and TNF Antago- nists,” Journal of Immunology, Vol. 151, No. 3, 1993, pp. 1548-1561. [60] L. Xin, J. Wang, H. Zhang, W. Shi, M. Yu, Q. Li, et al., “Dual Regulation of Soluble Tumor Necrosis Fac- tor-Alpha Induced Activation of Human Monocytic Cells via Modulating Transmembrane TNF-Alpha-Mediated ‘Reverse Signaling’,” International Journal of Molecular Medicine, Vol. 18, No. 5, 2006, pp. 885-892. [61] C. C. Solorzano, R. Ksontini, J. H. Pruitt, P. J. Hess, P. D. Edwards, A. Kaibara, et al., “Involve ment of 26-kDa Cell- Associated TNF-Alpha in Experimental Hepatitis and Exacerbation of Liver Injury with a Matrix Metallopro- teinase Inhibitor,” Journal of Immunology, Vol. 158, No. 1, 1997, pp. 414-419. [62] K. J. Grattendick, J. M. Nakashima, L. Feng, S. N. Giri and S. B. Margolin, “Effects of Three Anti-TNF-Alpha Drugs: Etanercept, Infliximab and Pirfenidone on Release of TNF-Alpha in Medium and TNF-Alpha Associated with the Cell in Vitro,” International Immunopharma- cology, Vol. 8, No. 5, 2008, pp. 679-687.
|