 Vol.2, No.6, 412-418 (2009) doi:10.4236/jbise.2009.26059 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/JBISE/ JBiSE A MCMC strategy for group-specific 16S rRNA probe design Yi-Bo Wu1*, Li-Rong Yan2*, Hui Liu1, Han-Chang Sun1, Hong-Wei Xie1§ 1Department of Automatic Control, College of Mechatronics and Automation, National University of Defense Technology, Changsha , Hunan, China; 2Department of Information, Wuhan General Hospital of Guangzhou Command, Wuhan, Hubei, China; *These au- thors contributed equally to this work; §Corresponding author. Email: wybbok@hotmail.com Received 17 April 2008; revised 6 July 2009; accepted 17 July 2009. ABSTRACT Revealing biodiversity in microbial communities is essential in metagenomics researches. With thousands of sequenced 16S rRNA gene avail- able, and advancements in oligonucleotide mi- croarray technology, the detection of microor- ganisms in microbial communities consisting of hundreds of species may be possible. Many of the existing strategies developed for oligonu- cleotide probe design are dependent on the re- sult of global multiple sequences alignment, which is a time-consuming task. We present a novel program named OligoSampling that uses MCMC method to design group-specific oli- gonucleotide probes. The probes generated by OligoSampling are group specific with weak cross-hybridization potentials. Furthermore a high coverage of target sequences can be ob- tained. Our method does not need to globally align target sequences. Locally aligning target sequences iteratively based on a Gibbs sam- pling strategy has the same effect as globally aligning sequences in the process of seeking group-specific probes. OligoSampling provides more flexibility and speed than other software programs based on global multiple sequences alignment. Keywords: 16S Rrna; Probe Desig n; MCMC 1. INTRODUCTION Metagenomics is a new field combining molecular biol- ogy and genetics in an attempt to reveal the vast scop e of biodiversity in a wide range of environment, as well as new functional capacities of individual cells and com- munities, and the complex evolutionary relationships between them [1,2,3,4]. Apparently, revealing biodiver- sity in microbial communities is the first step [1,5]. The vast majority of microbial diversity had been missed by cultivation-based methods [2]. The analysis of 16S rRNA gene sequences is the most common approach to determine microbial diversity [6]. Oligonucleotide microarrays now afford an idea tool for identifying sequence variants (even single-base-pair var ian t) in 1 6S rRNA gene copies of diverse micro o rgan- isms simultaneously in a single assay [7,8,9,10]. Many 16S rRNA-based oligonucleotide microarrays have been designed to detect multiple pathogens simultaneously [11, 12,13,14,15]. Such technology has the potential to revolu- tio n iz e cl inical diagnostics [15,16,17,18,19,20,21,22,23]. A critical issue for oligonucleotide microarray design is to find appropriate oligonucleotide probes specific to their target sequences. To improve efficiency in probe design, many softwares or databases have been devel- oped. Kaderali et al. proposed a combination of suffix trees and dynamic programming based alignment algo- rithms to compute melting temperature (T), and pre- sented an efficient algorithm to select probes with high specificity in detecting the target [24]. Loy et al. built a comprehensive database containing more than 700 pub- lished rRNA-targeted oligonucleotide probe sequences with supporting bibliographic and biological annotation [25]. Kumar et al. provided a software package ARB to evaluate sequence alignments and oligonucleotide probes with respect to three-dimensional structure of ribosomal RNA [26]. DeSantis et al. proposed an align- ment compression algorithm, NAST (Nearest Alignment Space Termination), to find Operational Taxonomic Units (OTUs) for automated design of effective probes [27,28]. m Global multiple sequences alignment plays an im- portant role in comprehensive analysis of group-spe- cific oligonucleotide probe for those methods men- tioned above. It is a challenge for personal computer to align a large amount of sequences. Here we present a novel program named OligoSampling that uses MCMC method to design group-specific oligonucleo- 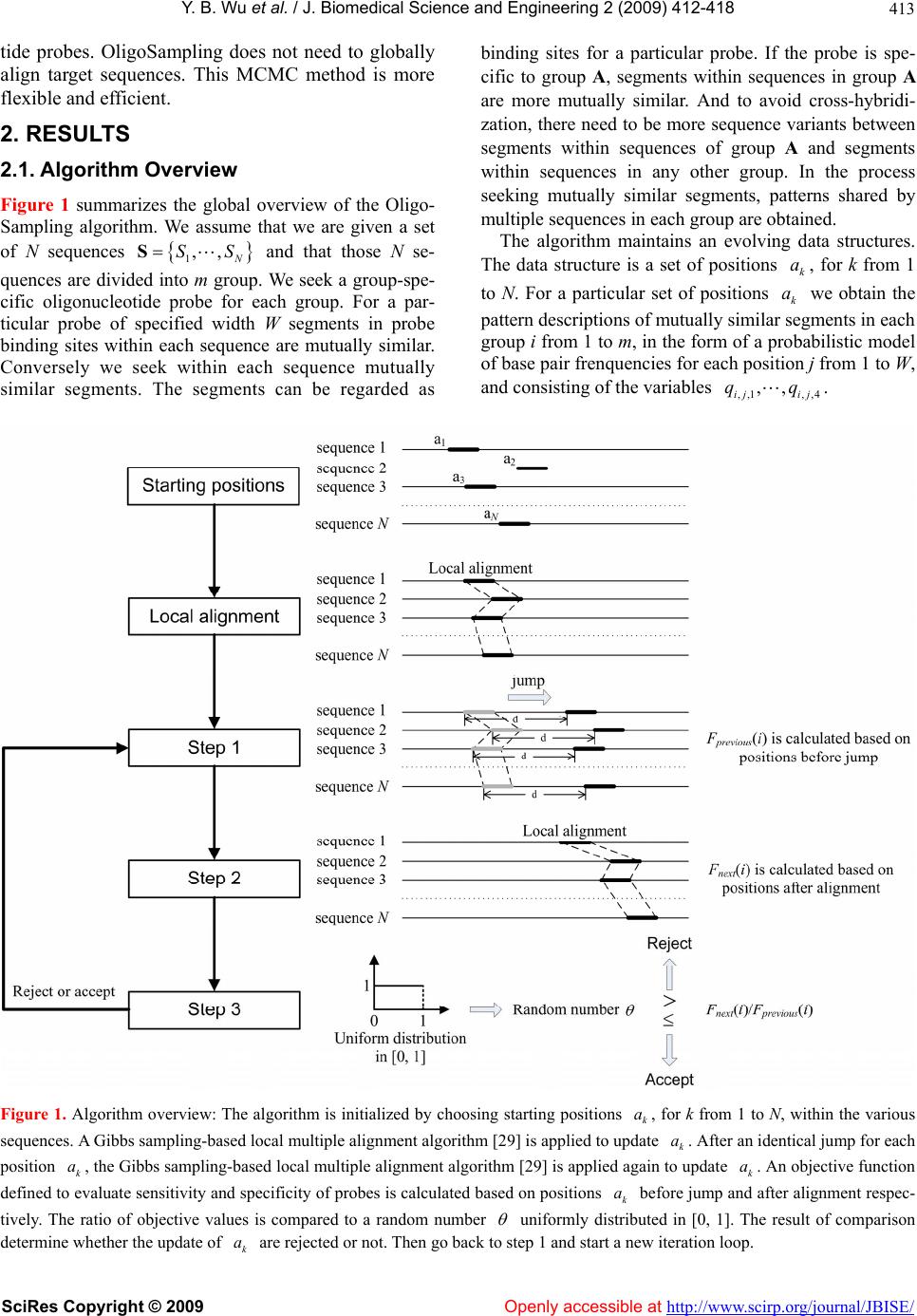 Y. B. Wu et al. / J. Biomedical Science and Engineering 2 (2009) 412-418 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/JBISE/ 413 tide probes. OligoSampling does not need to globally align target sequences. This MCMC method is more flexible and efficient. 2. RESULTS 2.1. Algorithm Overview Figure 1 summarizes the global overview of the Oligo- Sampling algorithm. We assume that we are given a set of N sequences 1,, N SSS and that those N se- quences are divided into m group. We seek a group-spe- cific oligonucleotide probe for each group. For a par- ticular probe of specified width W segments in probe binding sites within each sequence are mutually similar. Conversely we seek within each sequence mutually similar segments. The segments can be regarded as binding sites for a particular probe. If the probe is spe- cific to group A, segments within sequences in group A are more mutually similar. And to avoid cross-hybridi- zation, there need to be more sequence variants between segments within sequences of group A and segments within sequences in any other group. In the process seeking mutually similar segments, patterns shared by multiple sequences in each group are obtained. The algorithm maintains an evolving data structures. The data structure is a set of positions , for k from 1 to N. For a particular set of positions we obtain the pattern descriptions of mutually similar segments in each k a k a group i from 1 to m, in the form of a probabilistic model of base pair frenquencies for each position j from 1 to W, and consisting of the variables . ,,1 ,,4 ,, ij ij qq Figure 1. Algorithm overview: The algorithm is initialized by choosing starting positions , for k from 1 to N, within the various sequences. A Gibbs sampling-based local multiple alignment algorithm [29] is applied to update . After an identical jump for each position , the Gibbs sampling-based local multiple alignment algorithm [29] is applied again to update . An objective function defined to evaluate sensitivity and specificity of probes is calculated based on positions before jump and after alignment respec- tively. The ratio of objective values is compared to a random number k a k k a k ak a a uniformly distributed in [0, 1]. The result of comparison determine whether the update of are rejected or not. Then go back to step 1 and start a new iteration loop. k a 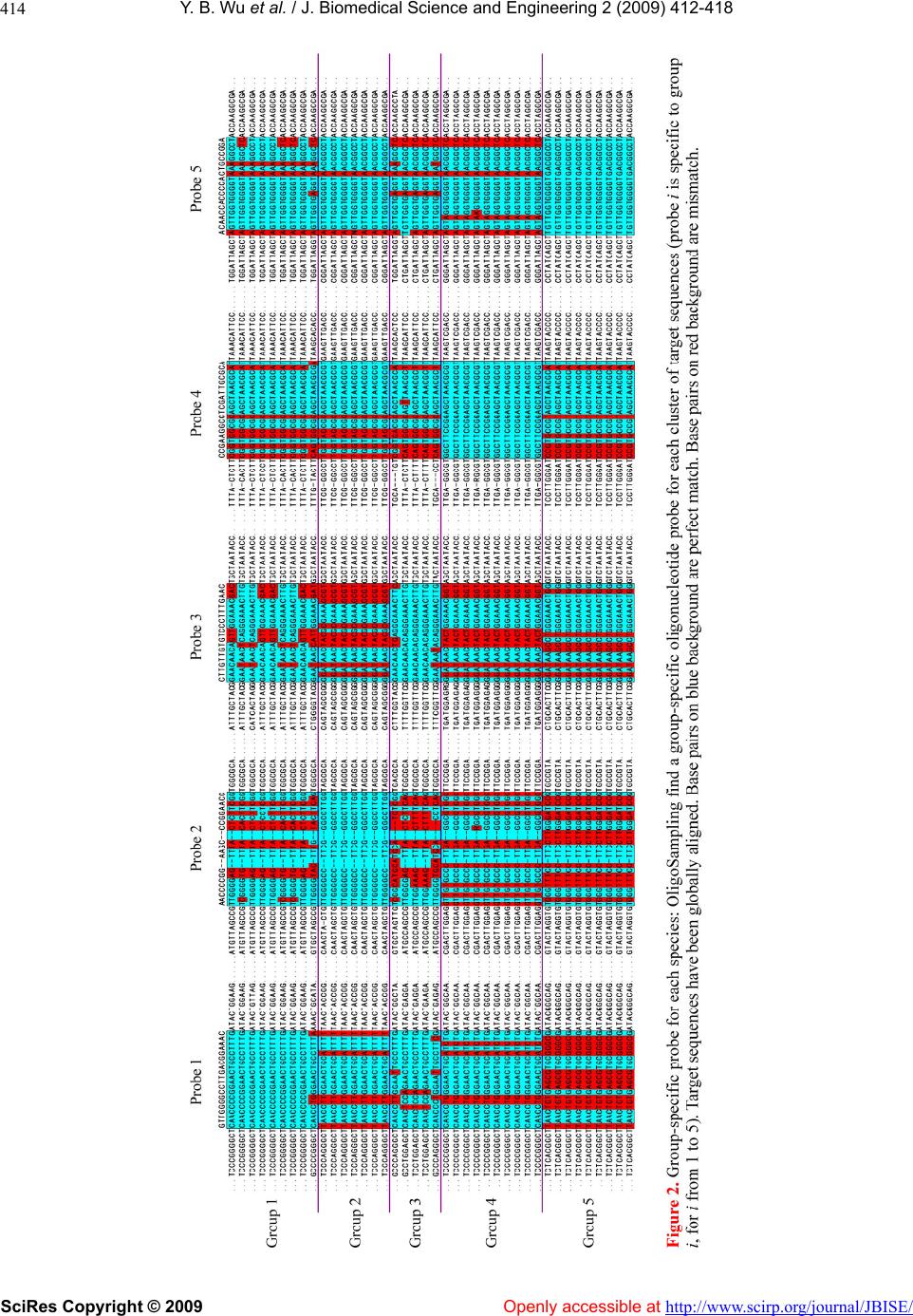 Y. B. Wu et al. / J. Biomedical Science and Engineering 2 (2009) 412-418 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/JBISE/ 414  Y. B. Wu et al. / J. Biomedical Science and Engineering 2 (2009) 412-418 SciRes Copyright © 2009 http://www.scirp.org/journal/JBISE/ 415 In the process of seeking group-specific probe for group i the algorithm is initialized by choosing starting positions 0 k a within the various sequences. A Gibbs sampling-based local multiple alignment algorithm [29] is applied to update 0 k a to 1 k a. After alignment the algorithm then proceeds through many iterations to exe- cute the following three steps: Openly accessible at 1) Jump step. Draw a sample d from a normal distri- bution. Positions 1 k a are updated to 21 kk aad. 2) Local multiple alignment step. Gibbs sampling- based local multiple alignment algorithm is applied re- garding po sitions 2 k a as initial s tarting point. Positio ns 2 k a are updated to 3 k a. Rejection step. We define an objective function (Eq.1). Based on positions 1 k a and 3 k a, the objective func- tion previous i and tnex i are calculated respective- ly. We generate a random number uniformly distrib- uted in [0, 1]. If next previous iF i , we accept 3 k a. Otherwise, positions resume to 1 k a. Then, go back to step 1. 44 ,, ,, ,,,, 1 1111 ,, log minlog WW jk j ijkijkjjk m jkjk ijk i q Fiq qqq (1) In the objective function sensitivity of the probe is measured by entropy, and specificity is measured by Kullback-Leibler divergence. By selecting a set of that maximizes t he objecti ve funct i on, t he algori t hm fi nds the most group-specific oli gonucleoti de probe for group i. k a 2.2. Design Group-Specific Oligonucleotide Probes for 5 Bacterial Species To examine the performance of OligoSampling on group-specific probe design, we select 5 bacteria species (Afipia sp., Bordetella pertussis, Brucella sp., Es- cherichia coli O157:H7, and Mycobacterium tuberculo- sis) with a total of 40 sequences. For OligoSampling, the input dataset is 5 clusters of homolog ous sequences. The objective of this algorithm is to find a group-specific oligonucleotide probe for each cluster. As shown in Fig- ure 2, the probes generated by OligoSamplin g are group specific with weak cross-hybridization potentials. Oligo- Sampling also obtains a high c overage of ta r get sequences. 3. DISCUSSION The secondary structure model of 16S rRNA consists of a conserved core that is interspersed with a number of variable areas. 16S rRNA evolves slowly and is often not very convenient to resolve bacterial strains at the species level. Therefore, multiple copies of 16S rRNA gene in different species share high sequence similarity. In jump step each aligned position is updated by jumping a same distance in the same direction. Convergence will be achieved rapidly in the following alignment due to high sequence similarity between sequences (Figure 3). In the process of updating positions , Gibbs sam- pling-based local multiple alignment algorithm is ap- plied to keep sequences locally aligned. And then, sensi- tivity and specificity of probe is evaluated based on en- tropy and Kullback-Leibler divergence. This process is functionally equivalent to oligonucleotide probe design based on global multiple sequences alignment, but has less calculation amount. k a k a Through the iterative algorithm mentio ned above, dif- ferent initializations sometimes lead to different local optimal solutions. Therefore, searches beginning with multiple initializations have more possibility to achieve global optimal solution. Searches beginning with differ- ent initializations are mutually independent and can be executed in parallel. It is convenient to parallelize this algorithm. Gibbs sampling-based local multiple alignment algo- rithm is sensitive to starting positions k. Appropriate starting positions can quicken convergence speed. a k a Figure 3. Convergence of local alignment: Optimal ob- jective values converge in 10000 iterations under three conditions: after jump, beginning with starting positions proportional to sequence length, and beginning with random starting positions. To make the comparison of convergence speed independent of initialization, ten alignment procedures beginning with different sets of starting positions are executed under each condition. Av- erage optimal objective values in iterations 1 to 10000 were normalized to that in last iteration. 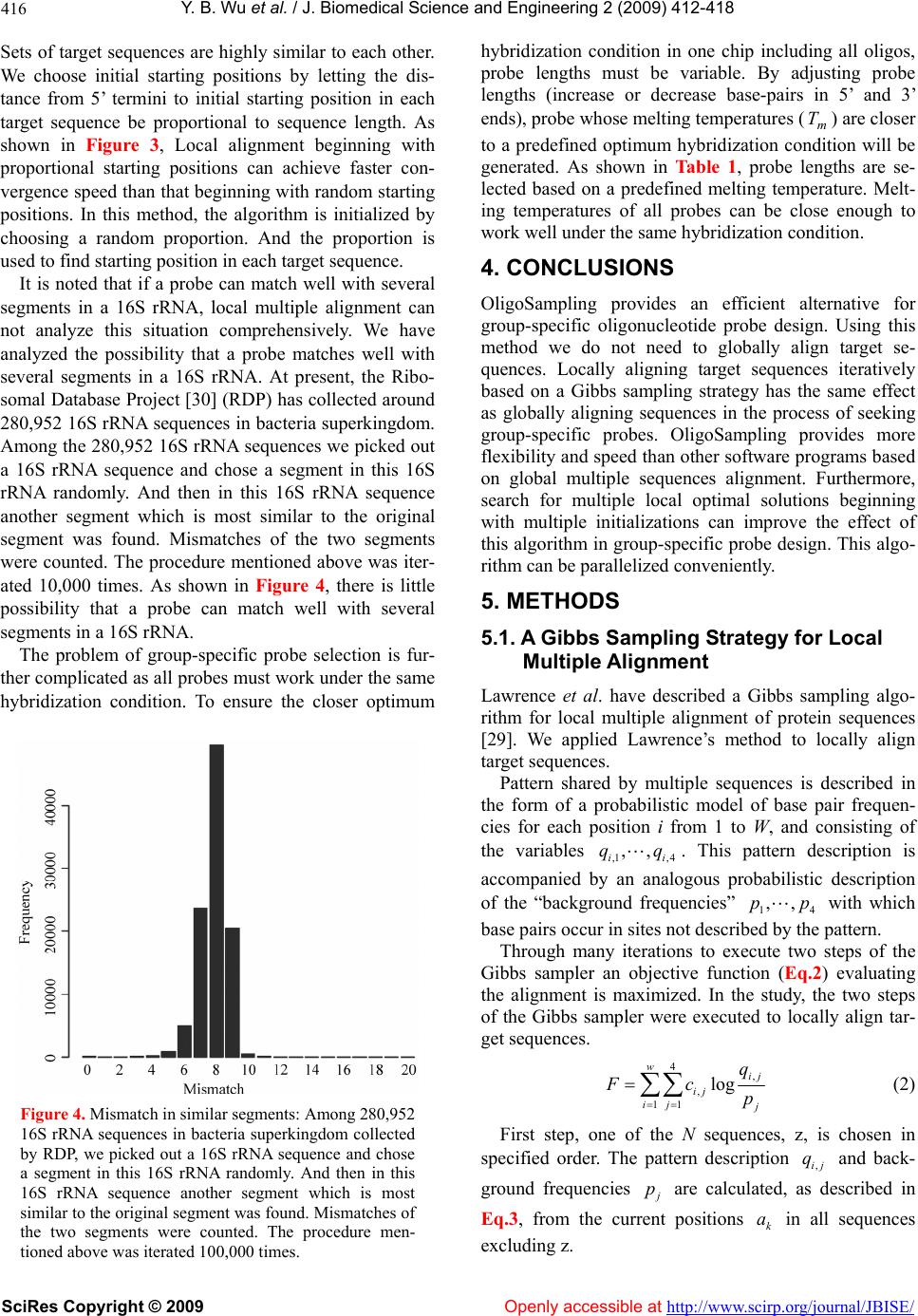 Y. B. Wu et al. / J. Biomedical Science and Engineering 2 (2009) 412-418 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/JBISE/ 416 Sets of target sequences are highly similar to each other. We choose initial starting positions by letting the dis- tance from 5’ termini to initial starting position in each target sequence be proportional to sequence length. As shown in Figure 3, Local alignment beginning with proportional starting positions can achieve faster con- vergence speed than that beginning with random starting positions. In this method, the algorithm is initialized by choosing a random proportion. And the proportion is used to find starting positio n in each target sequence. It is noted that if a probe can match well with several segments in a 16S rRNA, local multiple alignment can not analyze this situation comprehensively. We have analyzed the possibility that a probe matches well with several segments in a 16S rRNA. At present, the Ribo- somal Database Project [30] (RD P) has collected around 280,952 16S rRNA sequences in bacteria superkingdom. Among the 280,952 16S rRNA sequences we picked out a 16S rRNA sequence and chose a segment in this 16S rRNA randomly. And then in this 16S rRNA sequence another segment which is most similar to the original segment was found. Mismatches of the two segments were counted. The procedure mentioned above was iter- ated 10,000 times. As shown in Figure 4, there is little possibility that a probe can match well with several segments in a 16S rRNA. The problem of group-specific probe selection is fur- ther complicated as all probes must work under the same hybridization condition. To ensure the closer optimum Figure 4. Mismatch in similar segments: Among 280,952 16S rRNA sequences in bacteria superkingdom collected by RDP, we picked out a 16S rRNA sequence and chose a segment in this 16S rRNA randomly. And then in this 16S rRNA sequence another segment which is most similar to the original segment was found. Mismatches of the two segments were counted. The procedure men- tioned above was iterated 100,000 times. hybridization condition in one chip including all oligos, probe lengths must be variable. By adjusting probe lengths (increase or decrease base-pairs in 5’ and 3’ ends), probe whose melting temperatures () are closer to a predefined optimum hybridization condition will be generated. As shown in Table 1, probe lengths are se- lected based on a predefined melting temperature. Melt- ing temperatures of all probes can be close enough to work well under the same hybridization conditio n. m T 4. CONCLUSIONS OligoSampling provides an efficient alternative for group-specific oligonucleotide probe design. Using this method we do not need to globally align target se- quences. Locally aligning target sequences iteratively based on a Gibbs sampling strategy has the same effect as globally aligning sequences in the process of seeking group-specific probes. OligoSampling provides more flexibility and speed than o ther software programs based on global multiple sequences alignment. Furthermore, search for multiple local optimal solutions beginning with multiple initializations can improve the effect of this algorithm in group-specific prob e design. This algo- rithm can be parallelized conveniently. 5. METHODS 5.1. A Gibbs Sampling Strategy for Local Multiple Alignment Lawrence et al. have described a Gibbs sampling algo- rithm for local multiple alignment of protein sequences [29]. We applied Lawrence’s method to locally align target sequences. Pattern shared by multiple sequences is described in the form of a probabilistic model of base pair frequen- cies for each position i from 1 to W, and consisting of the variables . This pattern description is accompanied by an analogous probabilistic description of the “background frequencies” with which base pairs occur in sites not described by the pattern. ,1 ,4 ,, i qqi 4 p 1 ,,p Through many iterations to execute two steps of the Gibbs sampler an objective function (Eq.2) evaluating the alignment is maximized. In the study, the two steps of the Gibbs sampler were executed to locally align tar- get sequences. 4, , 11 log wij ij ij q Fc p (2) First step, one of the N sequences, z, is chosen in specified order. The pattern description and back- ground frequencies ,ij q p are calculated, as described in Eq.3, from the current positions in all sequences excluding z. k a  Y. B. Wu et al. / J. Biomedical Science and Engineering 2 (2009) 412-418 SciRes Copyright © 2009 http://www.scirp.org/journal/JBISE/ 417 Table 1. Adjust prob e leng t h s a c c o r d i n g to a predefined optimum hybridization condition (70℃). Original probe m T Adjusted probe m T 3’-GTTGGGGCCTTGACGGAAAC-5’ 74.98 3’-GGGGCCTTGACGGA-5’ 71.07 3’-AACCCCGGAAGCCCGGAACC-5’ 80.38 3’-CCGGAAGCCCGGA-5’ 70.93 3’-CTTGTTGTGTCCCTTTGAAC-5’ 66.40 3’-CCTTGTTGTGTCCCTTTGAAC-5’ 70.05 3’-CCGAAGGCCTCGATTGCGCA-5’ 78.59 3’-GAAGGCCTCGATTGCGC-5’ 70.69 3’-ACAACCACCCCACTGCCGGA-5’ 82.37 3’-ACCACCCCACTGCCG-5’ 71.66 , ,1 ij j ij cb qNB (3) For the ith position of the pattern we have 1N ob- served nucleotides, because sequence z has been ex- cluded; let be the coun t of nucleotide j in this posi- tion. These are supplemented with nucleotide-dep- endent “pseudocounts” to yield pattern probabilities. B is the sum of the ,ij c ,ij c j b b. Openly accessible at Second step, the probability Q of generating seg- ment x in position a according to the current pattern description are calculated, as are the probability ,ij q P of generating this segment by the background prob- abilities . The weight j p xx QP is assigned to current segment x. And then draw a sample d from a normal distribution. Positions a are updated to . In the updated position weight z ad is calculated in the same way. We generate a random number uniformly distributed in [0, 1]. If x A , we accept the update of positions a. Otherwise, we reject the update. Then go back to first step. 5.2. Selection of Weight Set in Sensitivity and Specificity Evaluation Pozhitkov et al. examined the effects of single-base pair mismatch (all possible types and positions) on signal intensities of hybridization through a series of calibra- tion experiments [31]. The results of experiments indi- cated that the most optimal discrimination of mismatch from perfect match probe-target duplexes is provided with the mismatch in the middle of the duplex. To sup- press non-specific binding of probe to target, the posi- tion of the mismatch should be moved from the 5’ or 3’ termini to the center of the probe. Therefore, in evalua- tion of sensitivity and specificity of group-specific oligonucleotide probes the weight coefficients (in Eq.1) should dependent on the signal intensity values of mismatches in different positions. Based on normalized signal intensity values of mismatch duplexes at position 1 to 20 provided by Pozhitkov et al., the weight coeffi- cients are assigned. Position in the middle has the highest weight. 3. ACKNOWLEDGEMENTS This work was supported by a grant from the National Natural Science Foundation of China (No. 30800253). The authors thank anonymous referees and the associate editor for many helpful comments. REFERENCES [1] S. R. Gill, M. Pop, R. T. Deboy, P. B. Eckburg, P. J. Turnbaugh, B. S. Samuel, J. I. Gordon, D. A. Relman, C. M. Fraser-Liggett, and K. E. Nelson, (2006) Metage- nomic analysis of the human distal gut microbiome, Sci- ence, 312, 1355–1359. [2] E. Pennisi, (2007) Metagenomics, massive microbial sequence project proposed, Science, 315, 1781. [3] A. Jurkowski, A. H. Reid, and J. B. Labov, (2007) Me- tagenomics: A call for bringing a new science into the classroom (while it’s still new), CBE Life Sci. Educ., 6, 260–265. [4] P. J. Turnbaugh, R. E. Ley, M. Hamady, C. M. Fra- ser-Liggett, R. Knight, and J. I. Gordon, (2007) The hu- man microbiome project, Nature, 449, 804–810. [5] P. B. Eckburg, E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman, (2005) Diversity of the human intestinal mi- crobial flora, Science, 308, 1635–1638. [6] S. C. Lau and W. T. Liu, (2007) Recent advances in mo- lecular techniques for the detection of phylogenetic markers and functional genes in microbial communities, FEMS Microbiol Lett., 275, 183–190. [7] S. P. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu, and D. Solas, (1991) Light-directed, spatially addressable parallel chemical synthesis, Science, 251, 767–773. [8] S. P. Fodor, R. P. Rava, X. C. Huang, A. C. Pease, C. P. Holmes, and C. L. Adams, (1993) Multiplexed bio- chemical assays with biological chips, Nature, 364, 555– 556. [9] A. C. Pease, D. Solas, E. J. Sullivan, M. T. Cronin, C. P. Holmes, and S. P. Fodor, (1994) Light-generated oli- gonucleotide arrays for rapid DNA sequence analysis, Proc. Natl. Acad. Sci., USA, 91, 5022–5026. [10] R. W. Ye, T. Wang, L. Bedzyk, and K. M. Croker, (2001) Applications of DNA microarrays in microbial systems, J. Microbiol Methods, 47, 257–272. [11] A. E. Warsen, M. J. Krug, S. LaFrentz, D. R. Stanek, F. J.  Y. B. Wu et al. / J. Biomedical Science and Engineering 2 (2009) 412-418 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/JBISE/ 418 Loge, and D. R. Call, (2004) Simultaneous discrimina- tion between 15 fish pathogens by using 16S ribosomal DNA PCR and DNA microarrays, Appl. Environ. Micro- biol., 70, 4216–4221. [12] D. Z. Jin, S. Y. Wen, S. H. Chen, F. Lin, and S. Q. Wang, (2006) Detection and identification of intestinal patho- gens in clinical specimens using DNA microarrays, Mol Cell Probes, 20, 337–347. [13] D. Harmsen, C. Singer, J. Rothganger, T. Tonjum, G. S. de Hoog, H. Shah, J. Albert, and M. Frosch, (2001) Di- agnostics of neisseriaceae and moraxellaceae by ribo- somal DNA sequencing: Ribosomal differentiation of medical microorganisms, J. Clin. Microbiol., 39, 936– 942. [14] C. P. Sun, J. C. Liao, Y. H. Zhang, V. Gau, M. Mastali, J. T. Babbitt, W. S. Grundfest, B. M. Churchill, E. R. McCabe, and D. A. Haake, (2005) Rapid, species-specific detection of uropathogen 16S rDNA and rRNA at am- bient temperature by dot-blot hybridization and an elec- trochemical sensor array, Mol. Genet. Metab., 84, 90– 99. [15] J. K. Loy, F. E. Dewhirst, W. Weber, P. F. Frelier, T. L. Garbar, S. I. Tasca, and J. W. Templeton, (1996) Molecu- lar phylogeny and in situ detection of the etiologic agent of necrotizing hepatopancreatitis in shrimp, Appl. Envi- ron. Microbiol., 62, 3439–3445. [16] B. X. Hong, L. F. Jiang, Y. S. Hu, D.Y. Fang, and H. Y. Guo, (2004) Application of oligonucleotide array tech- nology for the rapid detection of pathogenic bacteria of foodborne infections, J. Microbiol Methods, 58, 403– 411. [17] W. Deng, D. Xi, H. Mao, and M. Wanapat, (2007) The use of molecular techniques based on ribosomal RNA and DNA for rumen microbial ecosystem studies: A re- view, Mol. Biol. Rep. [18] G. Taroncher-Oldenburg and B. B. Ward, (2007) Oli- gonucleotide microarrays for the study of coastal micro- bial communities, Methods Mol. Biol., 353, 301–315. [19] C. R. Kuske, S. M. Barns, C. C. Grow, L. Merrill, and J. Dunbar, (2006) Environmental survey for four patho- genic bacteria and closely related species using phyloge- netic and functional genes, J. Forensic. Sci., 51, 548–558. [20] D. R. Call, (2005) Challenges and opportunities for pathogen detection using DNA microarrays, Crit. Rev. Microbiol., 31, 91–99. [21] T. Mohammadi, P. H. Savelkoul, R. N. Pietersz, and H. W. Reesink, (2006) Applications of real-time PCR in the screening of platelet concentrates for bacterial contami- nation, Expert Rev. Mol. Diagn., 6, 865–872. [22] L. Bodrossy and A. Sessitsch, (2004) Oligonucleotide microarrays in microbial diagnostics, Curr. Opin. Micro- biol., 7, 245–254. [23] K. Lemarchand, L. Masson, and R. Brousseau, (2004) Molecular biology and DNA microarray technology for microbial quality monitoring of water, Crit. Rev. Micro- biol., 30, 145–172. [24] L. Kaderali and A. Schliep, (2002) Selecting signature oligonucleotides to identify organisms using DNA arrays. Bioinformatics, 18, 1340–1349. [25] A. Loy, M. Horn, and M. Wagner, (2003) ProbeBase: An online resource for rRNA-targeted oligonucleotide probes, Nucleic. Acids. Res., 31, 514–516. [26] Y. Kumar, R. Westram, P. Kipfer, H. Meier, and W. Ludwig, (2006) Evaluation of sequence alignments and oligonucleotide probes with respect to three-dimensional structure of ribosomal RNA using ARB software package, BMC Bioinformatics, 7, 240. [27] T. Z. DeSantis, I. Dubosarskiy, S. R. Murray, and G. L. Andersen, (2003) Comprehensive aligned sequence con- struction for automated design of effective probes (CAS- CADE-P) using 16S Rdna, Bioinformatics, 19 , 1461– 1468. [28] T. Z. DeSantis, P. Hugenholtz, K. Keller, E. L. Brodie, N. Larsen , Y. M. Piceno, R. Phan, an d G. L. Andersen, (2006) NAST: A multiple sequence alignment server for com- parative analysis of 16S rRNA genes, Nucleic. Acids. Res., 34, 394–399. [29] C. E. Lawrence, S. F. Altschul, M. S. Boguski, J. S. Liu, A. F. Neuwald, and J. C. Wootton, (1993) Detecting sub- tle sequence signals: A gibbs sampling strategy for mul- tiple alignment, Science, 262, 208–214. [30] B. L. Maidak, J. R. Cole, T. G. Lilburn, C. T. Parker, P. R. Saxman, R. J. Farris, G. M. Garrity, G. J. Olsen, T. M. Schmidt, and J. M. Tiedje, (2001) The RDP-II (Ribo- somal Database Project), Nucleic. Acids. Res., 29, 173– 174 [31] A. Pozhitkov, P. A. Noble, T. Domazet-Loso, A. W. Nolte, R. Sonnenberg, P. Staehler, M. Beier, and D. Tautz, (2006) Tests of rRNA hybridization to microarrays suggest that hybridization characteristics of oligonucleotide probes for species discrimination cannot be predicted, Nucleic. Acids. Res., 34, 66.
|