 Journal of Quantum Informatio n Science, 2011, 1, 96-103 doi:10.4236/jqis.2011.12013 Published Online September 2011 (http://www.SciRP.org/journal/jqis) Copyright © 2011 SciRes. JQIS Time Dependent Wave Packet Study of the H− + H2 Nonreactive Scattering Sinan Akpinar*, Seda Surucu Department of Physics, Faculty of Science, Firat University, Elazig, Turkey E-mail: * sakpinar@firat.edu.tr Received July 4, 2011; revised August 10, 2011; accepted August 22, 2011 Abstract Time dependent wave packet calculations have been performed for the H− + H2 nonreactive scattering, summed of elastic and inelastic probabilities, on the recent reported potential energy surface of the 3 H systems. The total probabilities for total angular momentum J up to 35 have been calculated to get the con- verged integral cross sections over collision energy range of 0.20 - 1.42 eV. Integral cross-sections and rate constants have been calculated from the wave packet transition probabilities for the initial states (υ = 0, j = 0) by means of J-shifting method and uniform J-shifting method for J > 0. Keywords: Reaction Dynamics, Non-Reactive Scattering 1. Introduction The quantum wave packet method is especially useful and transparent for studying the dynamics of elementary chemical processes, because it allows the direct calcula- tion of the observables and shows the possible elemen- tary mechanisms. Over the past years, several wave packet methods were suggested and become very popular, and significant progress in this method has been done in order to solve better the time dependent Schrödinger equation (i.e., more efficient iterative method, the use of better representations) [1-9]. The ion-molecule reactions are of interest in under- standing the collision processes in interstellar media, the plasma, and high-energy physic studies [10-24]. How- ever, there have been a number of experimental and theoretical studies on the reaction cross sections of H− + H2 reactions and its isotopic variants [10-16]. Experi- mentally, Michels and Paulson [15] measured the reac- tion cross sections for the collisions of H− and D− with H2, D2, and HD using the tandem mass spectrometer (TMS). Muller et al. [17] performed crossed beam measurement of rotationally inelastic scattering of H2 from H−. Haufler et al. [18] determined the integral cross sections for the H− + D2 and D− + H2 reactions and a pronounced isotope effect was found in the experiment. Theoretically, an analytical ab initio Potential Energy Surface (PES) for the ground electronic state of 3 was reported by Starck and Meyer (SM) [19]. Gianturco and Kumar [20] calculated the differential and integral cross sections for vibrationally in elastic processes in (H-,H2) collisions over collision energy (107.69-922.4 kcal/mol) on SM PES. Recently, Panda and Sathya- murthy [21] have computed an ab initio PES of 3 systems using coupled cluster singles and doubles with nonperturbative triples method for a wide range of ge- ometries. Subsequently, there are many theoretical stud- ies about the title reactions using this global ab initio potential energy surface (PES) [10,21,22]. The pioneer work by Michels and Paulson [15] reported the biggest theoretical value of the cross section of this system (i.e, 2.5 Angs squared). Panda and Sathyamurthy [21] used the time dependent quantum mechanics method within the centrifugal sudden approximation for computing the integral reaction cross section values for H− + H2 reac- tion (υ = 0, j = 0) and its isotopic variants. Their results were also found to be in good agreement with the ex- perimental results of Muller et al. [17] but larger than those of Haufler et al. [18]. Using SM PES, Morari and Jaquet [14] calculated the excitation function for the ex- change reaction by including coriolis coupling and they found the results were in agreement with experimental results of Muller et al. [17] for all the range of energies studied and not with those of Haufler et al. [18]. Re- cently, using ab initio PES of Ref. 21 and time-depend- ent wave-packet quantum method, Yao et al. [10] calcu- lated the cross sections for both the reaction D− + H2 and the reaction H− + D2 in the collision energy range of 0.2 -  97 S. AKPINAR ET AL. 2.4 eV. Their calculations showed that the Coriolis cou- pled method was more consistent with the experimental ones than the centrifugal sudden approximation, and a pronounced isotopic effect was also observed to compare the two reactive systems in their previous report [10]. Giri et all. have computed the differential and integral cross sections for elastic and two dimensional inelastic H− + H2 (υ = 0, j = 0,1) reactions at four different relative translational energies (Etrans = 1.66, 2.03, 2.40 and 2.79 eV) by a time independent quantum mechanical ap- proach [23]. To our knowledge, there are no experiment- tal rate constant results of H− + H2 nonreactive scattering to compare in all the energy ranges studied in this paper. So, up to now, theoretically, there are not any three-di- mensional studies of H− + H2 nonreactive scattering us- ing time dependent quantum wave packet method to ob- tain cross sections and the reaction rates. Here, we report three dimensional time dependent quantum calculations of + H2(υ, j) + H2(υ', j') nonreactive scattering with υ and j being the vibrational and rotational quantum numbers. We use the 3 po- tential energy surface of Panda and Sathyamurthy for all the calculations. For computational reasons, we restrict our calculations for total angular momentum J = 0 and use the J-shifting approximation [25] and uniform J- shifting approximation [26] to evaluate cross sections and rate constants. The paper is organized as follows. In Section 2 we give a brief description of methodology. The results of our calculations are presented in Section 3. 2. Theory In this work, we employed Jacobi coordinates (R, r, γ), which are ideally suited for the calculation of the nonre- active state-to state and total probabilities. The corre- sponding Hamiltonian operator for J = 0 is expressed as 22 2 2222 11 11 ˆˆ 22 22 ,, Rr Rr j RrRr VRr (1) where r and R, respectively, the diatomic (H-H) and atom-diatom (H–-H2) distances with and r as their reduced masses. γ is the angle between R and r. denotes the diatomic rotational angular momentum, ˆ j ,,VRr is the potential energy function for atom- molecule reaction. Using the Hamiltonian operator in the form given in Equation (1) makes it necessary to use a large number of grid points in both R and r and an imaginary damping potential in the end of each grid. Having added and sub- tracted to the Hamiltonian opera- tor given by Equation (1), we get ,, 180VR r 22 22 22 ˆˆ ˆ,, 22BC RR jURrH r RR (2) where HBC(r) is the Hamiltonian operator for the dia- tomic molecule and U(R,r,γ) = V(R,r,γ) – V(R = ,r, γ = 180). Starting from the initial wave packet at t = 0 which is constructed according to the initial system, the time-de- pendent Schrödinger equation is solved in terms of modified complex Chebichev polynomials [2], min 2 0 0 ,,,2 2 EN iVt nn n Et Rr teJ n (3) where ˆ,,,0 n nnorm CiH Rrt with ψ(R, r, γ, t = 0) is the initial wavefunction, Cn(x) are complex Che- bichev polynomials (CP), Jn(x) is the Bessel functions and ΔE is the magnitude of the entire energy spread of the spectrum of the unnormalized Hamiltonian operator ˆ . The propagation requires the operation of the ˆ H nnorm on ψ. This is performed by using a three- term recursion relation of the Chebichev polynomials Ci 1 ˆ 2 nnorm nn iH 1 (4) where the recurrence is started by setting two initial values as 0,,,0Rr t and 1ˆ,,, 0. norm iHR rt The initial wavepacket is located in the asymptotic re- gion of entrance channel and propagated on the potential energy surface toward the strong interaction region. We wish to compute state-to-state nonreactive scattering probabilities and we have to follow the development of the wavepacket being reflected from the interaction re- gion. The flux that goes into reactive channel is absorbed and not analyzed. In order to extract the cross section and other observable quantities from the wavepacket dynam- ics, the wavepacket is analyzed at each time step by tak- ing cuts through at a fixed value of the scattering coor- dinate R = R∞. 0 ,, , vjkjkkvj k r CtRr tPwrdr (5) where ,, , k Rr t is initial wave function, k P is an angular wave function for a rotational state j, vj is vibrational wave function of H2 molecule, wk are the weights in Gauss quadrature formula. The transi- tion probabilities for the production of specific final vi- brational–rotational states from a specified initial reac- tant level are given by [26-28]. r 2 2 ' 0 ,' ' vj J vj vjvjvj Rr v E PE kk fk (6) Copyright © 2011 SciRes. JQIS  S. AKPINAR ET AL. 98 where Av′j′(E) is the Fourier transform of time-dependent coefficients (Cν′j′(t)). vj and are the wave vectors for initial and final channels. k'vj k v k is the Fourier transform of initial Gaussian. When using a time-dependent quantum method for scattering problems one is always faced with numerical difficulties associated with the reflection of the wave function from the end of the grid. Therefore, in order to avoid such a reflection, an imaginary potential is used to damp the wave packet at the edges of the grid. The ab- sorbing potential parameters are optimized as instructed by Vibok and Balint-Kurti [29]. At present calculations, a negative complex damping potential with a quadratic form has been used at both edges of the grid. Usually, it is not possible to obtain the integral cross sections and the thermal rate constants by nonreactive probabilities calculated for J = 0. Because, all important J values must be used to calculate the cross sections, and for high J values each calculation becomes very difficult. This problem is often handled approximately by the J-shifting method [25]. In general, J-shifting method, which is relies on the identification of “bottleneck” ge- ometry, such as a transition state, works very well when the reaction proceeds through an energy barrier, as is the case for present system. The changes in rotational energy of the system, when fixed at this geometry, provide an energy shift , K hift E, which is used in estimating the non- reactive probabilities and depends on the J and K quan- tum numbers. When K = 0, there is no component on an- gular momentum and the relationship is 0 ,, JJ J vj vjvj vjshift PPEE (7) where is the accurately computed nonreac- tive probability for J = 0, at the total energy E, and , is the estimated nonreactive probability for another value of J. 0 , J vj vj PE E J vj vj P The calculation of total cross sections requires having the transition probabilities for all available J values: 2 0 π21 J vj Colvj J vj EJ k PE (8) where Ecol = E – εvj is the collision energy and εvj is the initial rovibrational energy of the diatomic molecule, is the energy-dependent total reaction probabil- ity for a given initial state J vj PE The state-to-state rate constant can be calculated by Boltzmann averaging of the integral cross section over collision energy 12 / 33 0 8de Col B EkT vjCol ColvjCol RB kTEEE kT (9) where kB is the Boltzmann constant, T is temperature [30]. Another very appealing method for evaluating nonre- active rate constants is the Uniform J-Shifting approach developed by Zhang and Zhang [26]. In this approach, the optimized value of rotational constant (B) at a given temperature (T) for a range (Ji and Ji + 1) of J value is ex- tracted from these accurate probability functions, 1 11 ln1, 2 11 i i J B iJ ii ii kT Q BT i JJ JJQ (10) where i Q and 1i J Q are partition like functions for Ji and Ji + 1 reference angular momentum and can be written in a simple form / ed iicolB JJEkT col Col QT PEE (11) and 11/ ed iicolB JJEkT col col QT PEE (12) In result, the rate constant is given by 1/2 1 0 33 3 2π21e iB TJJkT J B kTQ TJ kT (13) 3. Results and Discussions We have calculated the rovibrational nonreactive prob- abilities at J = 0 for 0,0, 1,2j propagating of initial wave packet in Jacobi coordinates R, r, and , using the parameters of Table 1. The calculation re- quired 40000 iterations steps to converge. The potential energy surface of Ref. [21] has been used in this paper. It has in the following features: 1) Barrier height (0.47 eV) of proposed potential energy surface found to be a mini- mum for the collinear geometry, with the saddle point located at 1.999a0. 2) A van der Waals minimum was also found for the collinear geometry at r = 1.419a0 and R = 5.915a0 (a0 is Bohr radius) with a well of depth 0.0475 eV. 3) This potential has been constructed by fit- ting an analytical function to the ab initio potential en- ergy values computed using coupled cluster singles and doubles. Figure 1 shows nonreactive transition probabilities in 0, 0j at total angular momentum J = 0, 15, 25, 35 obtained from Equation (7) as a function of the colli- sion energy. Transition probabilities show no threshold and decrease with increasing collision energy as ex- pected for a nonreactive scattering with a small barrier to the reactive channel. That is, after barrier height for J = 0, the nonreactive probability decreases slowly in the en- ergy interval considered. Therefore, it can be seen non- reactive probabilities rapidly shift towards higher ener- gies on increasing J values. Probabilities are very small Copyright © 2011 SciRes. JQIS 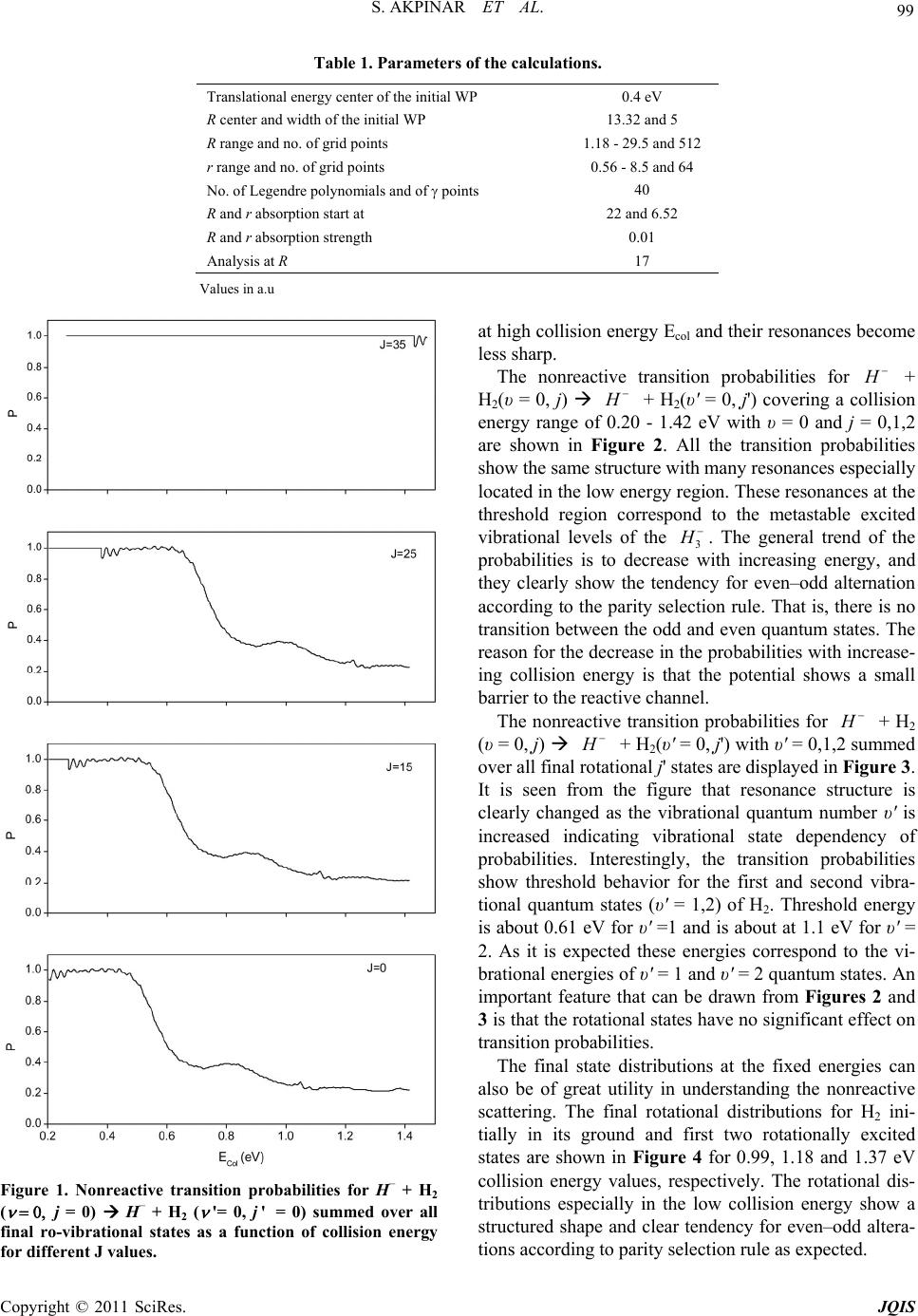 S. AKPINAR ET AL. Copyright © 2011 SciRes. JQIS 99 Table 1. Parameters of the calculations. Translational energy center of the initial WP 0.4 eV R center and width of the initial WP 13.32 and 5 R range and no. of grid points 1.18 - 29.5 and 512 r range and no. of grid points 0.56 - 8.5 and 64 No. of Legendre polynomials and of points 40 R and r absorption start at 22 and 6.52 R and r absorption strength 0.01 Analysis at R 17 Values in a.u at high collision energy Ecol and their resonances become less sharp. The nonreactive transition probabilities for + H2(υ = 0, j) + H2(υ' = 0, j') covering a collision energy range of 0.20 - 1.42 eV with υ = 0 and j = 0,1,2 are shown in Figure 2. All the transition probabilities show the same structure with many resonances especially located in the low energy region. These resonances at the threshold region correspond to the metastable excited vibrational levels of the 3 . The general trend of the probabilities is to decrease with increasing energy, and they clearly show the tendency for even–odd alternation according to the parity selection rule. That is, there is no transition between the odd and even quantum states. The reason for the decrease in the probabilities with increase- ing collision energy is that the potential shows a small barrier to the reactive channel. The nonreactive transition probabilities for + H2 (υ = 0, j) + H2(υ' = 0, j') with υ' = 0,1,2 summed over all final rotational j' states are displayed in Figure 3. It is seen from the figure that resonance structure is clearly changed as the vibrational quantum number υ' is increased indicating vibrational state dependency of probabilities. Interestingly, the transition probabilities show threshold behavior for the first and second vibra- tional quantum states (υ' = 1,2) of H2. Threshold energy is about 0.61 eV for υ' =1 and is about at 1.1 eV for υ' = 2. As it is expected these energies correspond to the vi- brational energies of υ' = 1 and υ' = 2 quantum states. An important feature that can be drawn from Figures 2 and 3 is that the rotational states have no significant effect on transition probabilities. The final state distributions at the fixed energies can also be of great utility in understanding the nonreactive scattering. The final rotational distributions for H2 ini- tially in its ground and first two rotationally excited states are shown in Figure 4 for 0.99, 1.18 and 1.37 eV collision energy values, respectively. The rotational dis- tributions especially in the low collision energy show a structured shape and clear tendency for even–odd altera- tions according to parity selection rule as expected. Figure 1. Nonreactive transition probabilities for H– + H2 ( j = 0) H– + H2 ( '= 0, j' = 0) summed over all final ro-vibrational states as a function of collision energy for different J values. 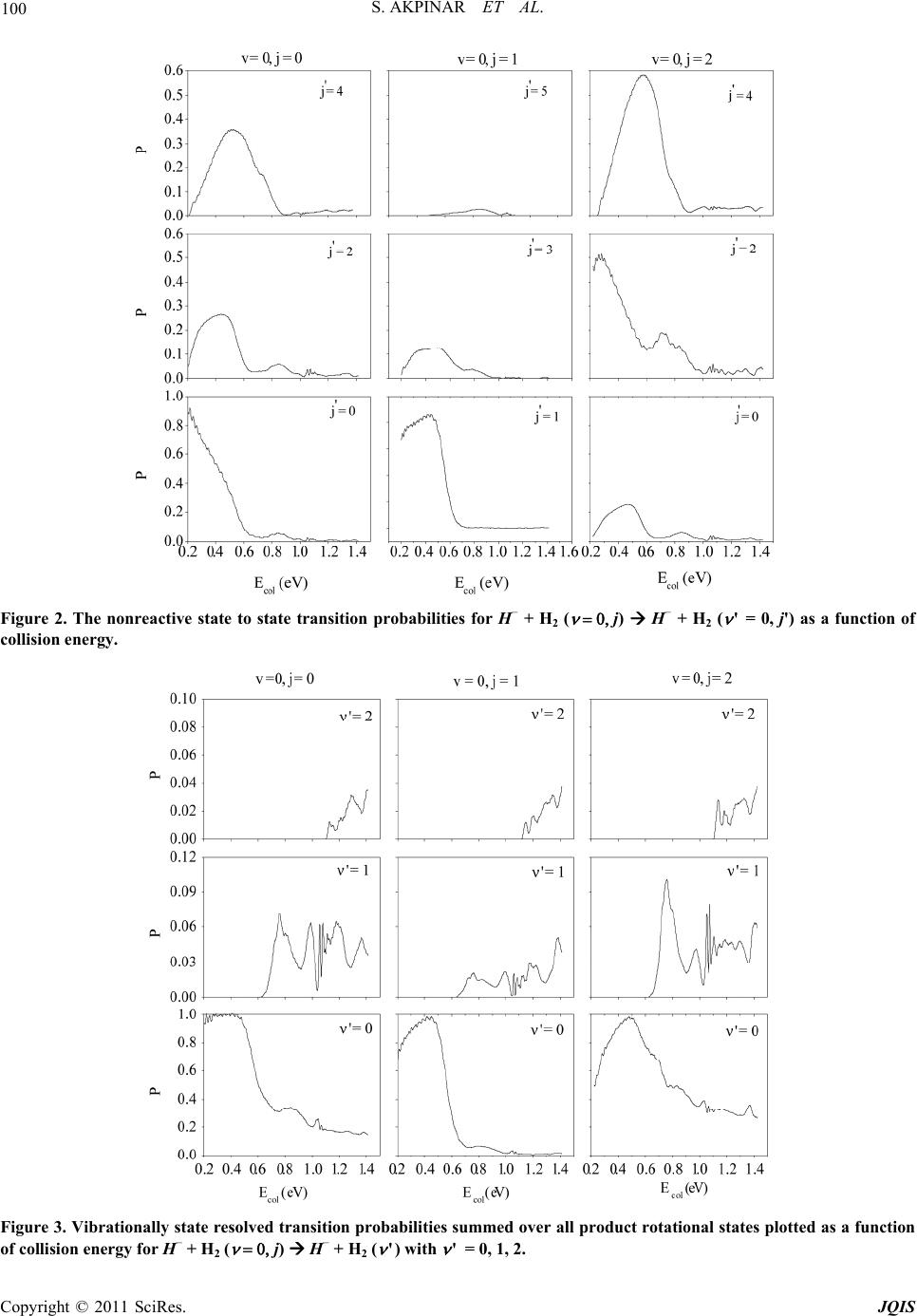 S. AKPINAR ET AL. 100 Figure 2. The nonreactive state to state transition probabilities for H– + H2 ( j) H– + H2 ( ' = 0, j') as a function of collision energy. Figure 3. Vibrationally state resolved transition probabilities summed over all product rotational states plotted as a function of collision energy for H– + H2 ( j) H– + H2 ( ') with ' = 0, 1, 2. Copyright © 2011 SciRes. JQIS 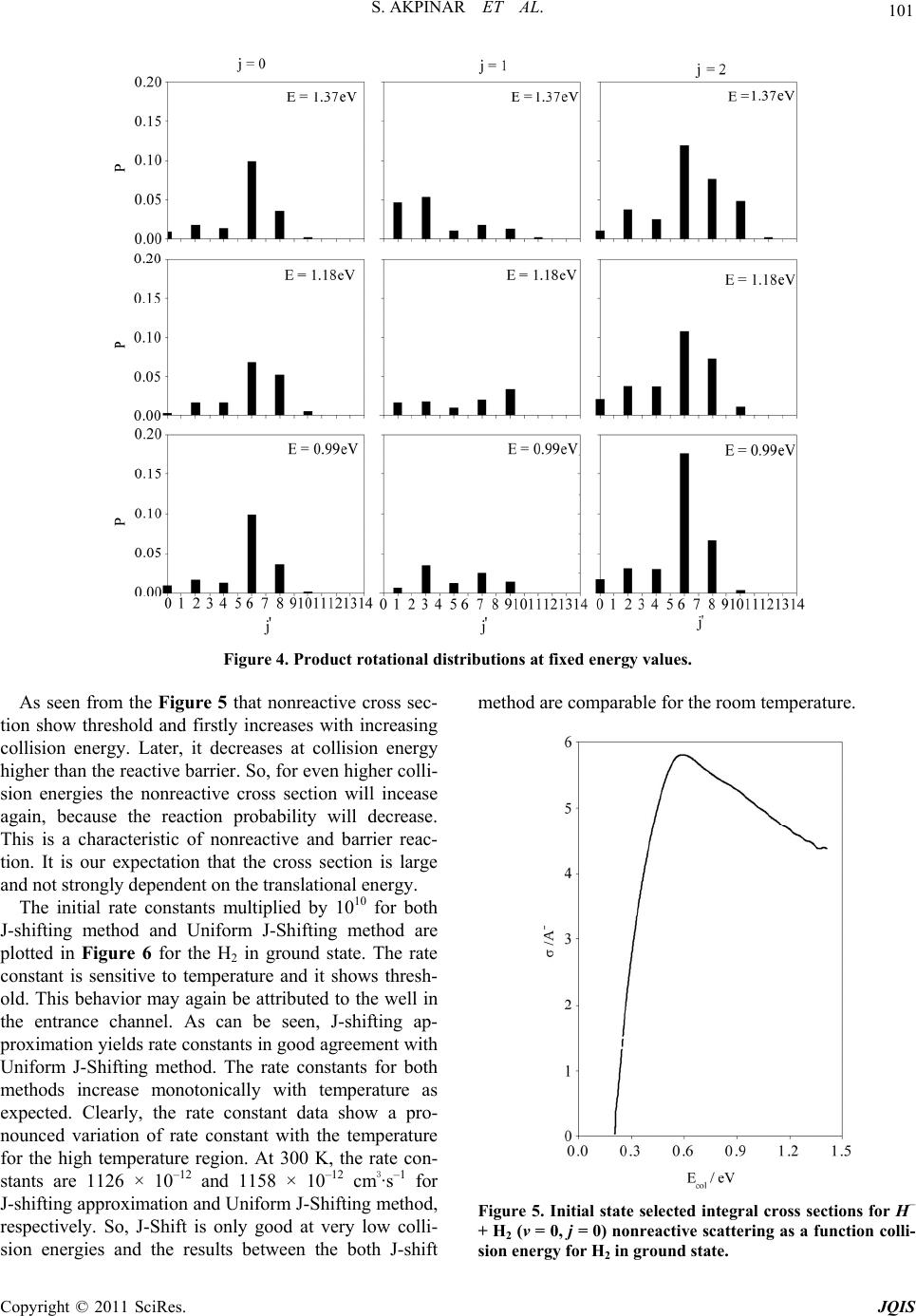 101 S. AKPINAR ET AL. Figure 4. Product rotational distributions at fixed energy values. As seen from the Figure 5 that nonreactive cross sec- tion show threshold and firstly increases with increasing collision energy. Later, it decreases at collision energy higher than the reactive barrier. So, for even higher colli- sion energies the nonreactive cross section will incease again, because the reaction probability will decrease. This is a characteristic of nonreactive and barrier reac- tion. It is our expectation that the cross section is large and not strongly dependent on the translational energy. The initial rate constants multiplied by 1010 for both J-shifting method and Uniform J-Shifting method are plotted in Figure 6 for the H2 in ground state. The rate constant is sensitive to temperature and it shows thresh- old. This behavior may again be attributed to the well in the entrance channel. As can be seen, J-shifting ap- proximation yields rate constants in good agreement with Uniform J-Shifting method. The rate constants for both methods increase monotonically with temperature as expected. Clearly, the rate constant data show a pro- nounced variation of rate constant with the temperature for the high temperature region. At 300 K, the rate con- stants are 1126 × 10–12 and 1158 × 10–12 cm3·s–1 for J-shifting approximation and Uniform J-Shifting method, respectively. So, J-Shift is only good at very low colli- sion energies and the results between the both J-shift method are comparable for the room temperature. Figure 5. Initial state selected integral cross sections for H– + H2 (v = 0, j = 0) nonreactive scattering as a function colli- sion energy for H2 in ground state. Copyright © 2011 SciRes. JQIS  S. AKPINAR ET AL. 102 01000 2000 3000 0 1 2 3 Uniform method J-Shifting method k x 1010 / cm 3 s-1 T / K Figure 6. Nonreactive rate constants depend on the initial quantum numbers for H– + H2(v = 0, j = 0) nonreactive scattering as a function of temperature. 4. Conclusions In this paper, we presented a time-dependent quantum wave packet calculation for the + H2 nonreactive scattering and gave the dynamics information of nonre- active probability, cross section and rate constant. The + H2 nonreactive system has an energy barrier height on potential energy surface. The nonreactive cross section has been obtained by summing up the nonreac- tive probabilities. The probability for J = 0 was calcu- lated while the probabilities for J > 0 was estimated by means of two different J-shifting approximation. The calculations showed that the reaction cross section shows a threshold behavior and the initial state selected rate constant is significantly dependent on the temperature. 5. Acknowledgements We would like to thank Prof. N. Sathyamurthy for his providing of potential energy function. Authors are in- debted to Dr. Paolo Defazio for many stimulating dis- cussions on quantum wave packet theory. Partial finan- cial support from the Firat University Scientific Research Projects Unit (FUBAP) (Project No: FUBAP-1775) is gratefully acknowledged. Therefore, the numerical cal- culations reported in this paper were performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TR-Grid e-Infrastructure) and this work was supported by the Turkish Scientific and Tech- nological Research Council of TURKEY (TUBITAK) (Project No. 109T447). Authors wish to thank TUBI- TAK and FUBAP. 6. References [1] S. C. Althorpe, “Quantum Scattering Studies of Reac- tions,” In: P. V. R. Schleyer, Ed., The Encyclopedia of Computational Chemistry, Wiley, Athens, 2005. [2] H. Tal-Ezer and R. Kosloff, “An Accurate and Efficient Scheme for Propagating the Time Dependent Schrödinger Equation,” Journal of Chemical Physics, Vol. 81, No. 9, 1984, pp. 3967-3971. doi:10.1063/1.448136 [3] T. J. Park and J. C. Light, “Unitary Quantum Time Evo- lution by Iterative Lanczos Reduction,” Journal of Chemical Physics, Vol. 85, No. 10, 1986, p. 5870 doi:10.1063/1.451548 [4] J. Broeckhove and L. Lathouwers (Eds.), “Time-Depen- dent Quantum Molecular Dynamics,” New York, Plenum, 1992. [5] R. E. Wyatt and J. Z. H. Zhang (Eds.), “Dynamics of Molecules and Chemical Reactions,” Dekker, New York, 1996. [6] N. Balakrishnan, C. Kalyanaraman and N. Sathyamurthy, “Time-Dependent Quantum Mechanical Approach to Reactive Scattering and Related Processes,” Physical Report, Vol. 280, No. 2, 1997, pp. 79-144. doi:10.1016/S0370-1573(96)00025-7 [7] S. C. Althorpe and D. C. Clary, “Quantum Scattering Calculations on Chemical Reactions,” Annual Review of Physical Chemistry, Vol. 54, 2003, pp. 493-529. doi:10.1146/annurev.physchem.54.011002.103750 [8] S. K. Gray and G. G. Balint-Kurti, “Quantum Dynamics with Real Wave Packets, including Application to Three- Dimensional (J = 0) D+H2→HD+H Reactive Scattering,” Journal of Chemical Physics, Vol. 108, No. 3, 1998, p. 950 doi:10.1063/1.475495 [9] P. Defazio and C. Petrongolo, “Dynamics of the N(2D)+H2 Reaction on the 2 A Surface, Propagating Real Wave Packets with an Arccos Mapping of the Hamiltonian,” Journal of Theoretical and Computational Chemistry, Vol. 2, No. 4, 2003, p. 547. doi:10.1142/S0219633603000732 [10] L. Yao, L. P. Ju, T. S. Chu and K. L. Han, “Time-De- pendent Wave-Packet Quantum Scattering Study of the Reactions D−+H2→H−+HD and H−+D2→D−+HD,” Physical Review A, Vol. 74, No. 6, 2006, p. 062715. doi:10.1103/PhysRevA.74.062715 [11] S. Mahapatra, N. Sathyamurthy, S. Kumar and F. A. Gi- anturco, “Transition State Resonances in Collinear (H−, H2) Collisions,” Chemical Physics Letters, Vol. 241, No. 3, 1995, p. 223. doi:10.1016/0009-2614(95)00633-F [12] A.N. Panda, K. Giri and N. J. Sathyamurthy, “Three Di- mensional Quantum Dynamics of (H−, H2) and Its Iso- topic Variants,” The Journal of Physical Chemistry A, Vol. 109, No. 10, 2005, pp. 2057-2061. doi:10.1021/jp044953l [13] A.N. Panda and N. Sathyamurthy, “Time-Dependent Quantum Mechanical Wave Packet Study of the He+H2+(v,j)→HeH++H reaction,” Journal of Chemical Physics, Vol. 122, No. 5, 2005, p. 054304. doi:10.1063/1.1839866 [14] C. Morari and R. Jaquet, “Time-Dependent Reactive Scattering for the System H−+D2↔HD+D− and Com- Copyright © 2011 SciRes. JQIS  103 S. AKPINAR ET AL. parison with H-+ H2↔ H2+ H,” The Journal of Physical Chemistry A, Vol. 109, 2005, p. 3396. doi:10.1021/jp0462963 [15] H. H. Michels and J. F. Paulson, In: D. G. Truhlar, Ed., Potential Energy Surface and Dynamics Calculations, Plenum Press, New York, 1981. [16] M. S. Huq, L. D. Doverspike and R. L. Champion, “Elec- tron Detachment for Collisions of H− and D− with Hy- drogen Molecules,” Physical Review A, Vol. 27, No. 6, 1982, pp. 2831-2839. doi:10.1103/PhysRevA.27.2831 [17] H Müller, M Zimmer and F Linder, “State-Resolved Measurements of Rotational Excitation in H−+H2 Colli- sions at Low Energies,” Journal of Physics B-Atomic Molecular and Optical Physics, Vol. 29, No. 18, 1996, p. 4165. [18] E. Haufler, S. Schlemmer and D. Gerlich, “Absolute In- tegral and Differential Cross Sections for the Reactive Scattering of H−+D2 and D−+H2,” The Journal of Physi- cal Chemistry A, Vol. 101, No. 36, 1997, pp. 6441-6447. doi:10.1021/jp9707246 [19] J. Starck and W. Meyer, “Ab-Initio Potential-Energy Surface for the Collisional System H−+H2 and Properties of Its Van-Der-Waals Complex,” Journal of Chemical Physics, Vol. 83, 1993, p. 176. [20] F.A. Gianturco and S. Kumar, “Selective Efficiency of Vibrational Excitations in Ion–Molecule Collisions: A Comparison of Behavior for H+–H2 and H−–H2,” Journal of Chemical Physics, Vol. 103, No. 8, 1995, p. 2940. doi:10.1063/1.470481 [21] A.N. Panda and N. Sathyamurthy, “Dynamics of (H−, H2) Collisions: A Time-Dependent Quantum Mechanical In- vestigation on a New Ab Initio Potential Energy Sur- face,” Journal of Chemical Physics, Vol. 121, No. 19, 2004, p. 9343. doi:10.1063/1.1797711 [22] T. S. Chu, R. F. Lu, K. L. Han, X. N. Tang, H. F. Xu and C. Y. Ng, “A Time-Dependent Wave-Packet Quantum Scattering Study of the Reaction H2 +(v = 0–2,4,6; j = 1)+He→HeH++H,” Journal of Chemical Physics, Vol. 122, No. 24, 2005, p. 4322. doi:10.1063/1.1948380 [23] K. Giri and N. Sathyamurthy, “Rotational Excitation in (H−, H2) Collisions: A Quantum Mechanical Study,” Journal of Physics B: Atomic, Molecular and Optical Physics, Vol. 39, No. 20, 2006, p.4123. doi:10.1088/0953-4075/39/20/010 [24] W. Li, M. Wang, C. Yang, X. Ma, D. Wang and W. Liu, “Quasi-Classical Trajectory Study of the Cross Sections of the Reactions of D−+H2→H−+HD and H−+D2→ D−+HD,” Chemical Physics Letters, Vol. 445, No. 4-6, September 2007, pp. 125-128. doi:10.1016/j.cplett.2007.08.013 [25] J. M. Bowman, “Reduced Dimensionality Theory of Quantum Reactive Scattering,” The Journal of Physical Chemistry, Vol. 95, No. 13, 1991, pp. 4960-4968 doi:10.1021/j100166a014 [26] D. H. Zhang and J. Z. H. Zhang, “Uniform J-Shifting Approach for Calculating Reaction Rate Constant,” Journal of Chemical Physics, Vol. 110, No. 16, 1999, p. 7622. doi:10.1063/1.478802 [27] F. Gögtas, G. G. Balint-Kurti and A. R. Offer, “Quantum Mechanical Three-Dimensional Wavepacket Study of the Li+HF->LiF+H Reaction,” Journal of Chemical Physics, Vol. 104, 1996, p. 7927. [28] G. G. Balint-Kurti, R. N. Dixon and C. C. Marston, “Time-Dependent Quantum Dynamics of Molecular Photofragmentation Processes,” Journal of the Chemical Society-Faraday Transactions, Vol. 86, 1990, p. 1741. [29] A. Vibok and G. G. Balint-Kurti, “Parametrization of Complex Absorbing Potentials for Time-Dependent Quantum Dynamics,” The Journal of Physical Chemistry, Vol. 96, No. 22, 1992, pp. 8712-8719 doi:10.1021/j100201a012 [30] S. Y. Lin and H. Guo, “Quantum Wave Packet Studies of the C(1D)+H2→CH+H Reaction?” Integral Cross Section and Rate Constant, Vol. 108, No. 12, 2004, pp. 2141- 2148. doi:10.1021/jp031184h Copyright © 2011 SciRes. JQIS
|