 Journal of Quantum Informatio n Science, 2011, 1, 87-95 doi:10.4236/jqis.2011.12012 Published Online September 2011 (http://www.SciRP.org/journal/jqis) Copyright © 2011 SciRes. JQIS Modeling of the Chemico-Physical Process of Protonation of Molecules Entailing Some Quantum Chemical Descriptors Sandip K. Rajak, Nazmul Islam, Dulal C. Ghosh* Department of Chemistry, University of Kalyani, Kalyani, India E-mail: *dcghosh1@rediffmail.com Received May 27, 2011; revised July 25, 2011; accepted August 6, 2011 Abstract Relying upon the basic tenets of scientific modeling, an ansatz for the evaluation of proton affinity of mole- cules are evolved in terms of a four component model. The components of the model chosen are global de- scriptors like ionization energies, global softness, electronegativity and electrophilicity index. These akin quantum mechanical descriptors of atoms and molecules are linked with the charge rearrangement and po- larization that occur during the physico-chemical process of protonation of molecules. The suggested ansatz is invoked to compute the protonation energy of as many as 43 compounds of diverse physico-chemical na- ture viz, hydrocarbons, alcohols, carbonyls, carboxylic acids, esters, aliphatic amines and aromatic amines. A detailed comparative study of theoretically evaluated protonation energies of the above mentioned molecules vis-à-vis their corresponding experimental counterparts reveals that there is a close agreement between the theory and experiment. Thus the results strongly suggest that the proposed modeling and the ansatz for computing PA, the proton affinity, of molecules for studying the physico-chemical process of protonation may be valid proposition. Keywords: Physico-Chemical Process of Protonation, Proton Affinity, Conceptual Density Functional Descriptors, Commonality between Density Functional Descriptors and Proton Affinity, Muliti-Linear Regression Model 1. Introduction The protonation reaction or the physico-chemical process of protonation is ubiquitous in almost all the areas of chemistry and biochemistry [1-5]. The majority of che- mical reaction occurs in acid medium. The chemical process of protonation is fundamental first step of many chemical rearrangements, and enzymatic reactions [4]. The resulting protonated molecule is frequently a pivotal intermediate that guides the succeeding steps of the overall process. The knowledge of the intrinsic basicity and the site of protonation of a compound is central for the understanding of the mechanism of chemical reac- tions. The legend proton affinity is defined as the nega- tive of the enthalpy change of a protonation reaction at the standard conditions. The gas-phase proton affinities are a quantitative measure of the intrinsic basicity of a molecule [6]. The study of thermochemistry of the pro- ton transfer reaction in the gas phase is well-known ex- periment of acid-base reaction [7]. Dynamics of proton transfer is also important for ionization processes in mass spectroscopy [8]. Basicity is defined [9] as the tendency of a molecule, B, to accept a proton, H+, in the following Base-Acid (Proton) adduct BH+ formation reaction BHBH PA (1) where PA is the proton affinity of the base B. This concept of basicity was generalized further and freed from reference to a specific acid (H+) by Lewis [10]. During the physico-chemical process of protonation, electronic charge is soaked by the proton from the entire skeleton of the molecule. As a result, all the structural parameters i.e. bond lengths and bond angles, and other charge dependent physical properties like the polarizabil- ity and the dipole etc are affected. A plethora of informa- tion has appeared on the study of this important chemico- physical process [6,7]. Although, experimentally the proton affinity can be  S. K. RAJAK ET AL. 88 determined by several techniques like the measurement of the heats of formation [5] of the species involved in the adduct formation reaction, by mass spectroscopic measurement techniques [7-9,11] and by the measure- ment of the ionization thresholds [7]. The “acid-base adducts” are not always stable and/or does not exist in all cases and also it is well known [12] that the experimental determination of the proton affinities of molecules is not easy always. For this reason, in recent years, much em- phasis has been given to the calculation of proton affini- ties through some quantum mechanical and its young branch, the density functional theoretical models [13-17]. It is now established [18] that the ab initio quantum mechanical approaches and its numerous variants are very successful in providing reliable values of proton affinity and gas phase basicity for small molecules. However, due to the reason of heavy computational cost , application of ab initio methods for the estimation of proton affinities is still impractical for larger molecules [19]. It is also recognized [20] that the popular semi em- pirical methods such as AM1, MNDO and PM3 etc are not that reliable in calculating proton affinities. Although there are some attempts of modeling to compute protona- tion energy for specific groups of compounds [6,21-26], but fact remains that no universal model has been, so far, put forward subsuming the energetic effects necessarily appearing in the physico-chemical process of protonation as a substitute for experimental or theoretical measure- ment of the energy of protonation. Currently the conceptual density functional theory [27-44] of chemical reactivity have introduced many de- scriptors, global and local, like electronegativity, hard- ness, softness, fukui functions, electrophilicity index etc in theoretical chemistry. Such descriptors have made serious inroad in science and opened a new paradigm of chemical thinking, modeling and computation [27-44]. In this work, we have developed a model for the evaluation of proton affinity in terms of some akin con- ceptual reactivity descriptors which can be conceptually linked and associated with the physico-chemical process of protonation. The akin descriptors are the ionization energy (I), the global softness(S), the electronegativity (χ), and the global electrophilicity index (ω). 1.1. The Physico-Chemical Process of Protonation In the terminology classification of chemical reaction according to the reagent and the substrate, proton is an electrophile. In the physico-chemical process of protona- tion, when a proton dynamically approaches towards a nucleophile from a long distance it is attracted by the electron cloud of the molecule. The proton acting as an electrophile soaks the electron density from the entire skeleton of the nucleophile [45]. As a result, the electron cloud of the nucleophile is redistributed and remains under the influence of the electrophile, the proton. In some circumstances, the proton fixes at a site of lone pair, if available, in the molecule. However, if there is no lone pair in the structure of the molecule, the proton remain- ing attached to the sphere of the charge cloud of the molecule. The polarizing power of the proton induces a physical process of structural and energetic changes in the molecule and the effect is expected to be at its maxi- mum at the gas phase of the molecule. Thus, the gas- phase basicity is certainly the ideal revelator of the structural and energetic characteristics of the molecular protonation process. 1.2. The Physico-Chemical Process of Protonation Entailing the Ionization Energy, the Electronegativity, the Chemical Hardness, the Softness, and the Electrophilicity Index In order to suggest a mathematical modeling of comput- ing the protonation energy of molecules involving the above akin theoretical descriptors that may be associated and directly linked with the physico-chemical process of protonation, we depict the glimpses of the role of each descriptor in the process separately viz. 1) The ionization energy (I) This is a fundamental descriptor of the chemical reac- tivity of atoms and molecules. High ionization energy indicates high stability and chemical inertness and small ionization energy indicates high reactivity of the atoms and molecules [46]. Mills et al. [47] discovered a linear relationship between the proton affinity and the additive inverse of the ionization energies of molecules. 2) Electronegativity (χ) Electronegativity though defined in many different ways, the most logical and rational definition of it is the electron holding power of the atoms or molecules. Elec- tronegativity is defined and measured as the power (force) with which the valence electron of an atom is held by its screened nuclear charge. The more electronegative ele- ments hold electrons more tightly and the less electro- negative elements hold less tightly. Lohr [48] has dis- cussed the physico-chemical process of protonation from a deeper insight and discovered the important relation- ship between the protonation and electronegativity. He [48] further went to conclude that there is a protonic counter part of electronegativity as a organizing principle of acidity and basicity. However, the inverse relationship between the electronegativity and protonation process and associated energetic effect is straight forward. Copyright © 2011 SciRes. JQIS  89 S. K. RAJAK ET AL. 3) Global Softness (S) The softness is in fact the inverse concept of hardness, a fundamental descriptor of the stability and reactivity of atoms and molecules. It is apparent that the chemical hardness fundamentally signifies the resistance towards the deformation or polarization of the electron cloud of the atoms, ions or molecules under small perturbation of chemical reaction. The softness is simply the reciprocal of the hardness. Thus the general operational signifi- cance of the hard-soft chemical species may be under- stood in the following statement. If the electron cloud is strongly held by the nucleus, the chemical species is “hard” but if the electron cloud is loosely held by the nucleus the system is “soft” [40,49]. Hence the polariza- tion process associated with protonation should be di- rectly controlled by the softness of the molecule. Or in other words, the energetic effect associated with the pro- tonation should be directly proportional to the softness of the molecule. 4) Electrophilicity Index (ω) In reference to nucleophilic-electrophilic, acid-base or donor-acceptor reaction, the electrophilicity index [50,51] of atoms and molecules seems to be an absolute and fundamental property of such chemical species because it signifies the energy lowering process on soaking elec- trons from the donors. This tendency of charge soaking and energy lowering must emanate and develop from the attraction between the soaked electron density and screened nuclear charge of the atoms and molecules. As the process of electron soaking by proton continues, the accumulated electron density will, however, shield the proton. However, the electrophilicity of the substrate will oppose the charge soaking by the proton during the physico-chemical process of protonation and hence the protonation is hindered by the electrophilicity and hence protonation energy should bear an inverse relationship with electrophilicity index. 2. The Modeling of the Physico-Chemical Process of Protonation and Algorithm for Computing the Proton Affinity of the Molecules The descriptors like the ionization process of atoms and molecules, the physical property like hardness, softness, the electronegativity and the electrophilicity have close relation i.e. akin with each other in their operational sig- nificance and origin. We have tried to posit above that the physico-chemical process of protonation can be linked to the above akin descriptors –the ionization process, the hardness, soft- ness, electronegativity and electrophilicity. Recently, we [52-61] have published good number of papers where we have discussed that the three descriptors, the electro- negativity, the hardness and the electrophilicity index of atoms and molecules are fundamentally qualitative per se and operationally the same. All these three descriptors represent the attraction of screened nuclei towards the electron pair/bond. Thus, we can safely and reasonably conclude that the proton affinity and the three descriptors have inverse relationship. Thus, since the above four parameters have dimension of energy and can be linked to the process of charge re- arrangement and polarization during the physico-che- mical process of protonation, they can be components of a probabilistic scientific modeling of proton affinity. The physico-chemical process of protonation has direct link to the charge polarization and alteration of electron dis- tribution in the molecule. The proton affinity or the ability of donating the lone pair of a Lewis base and the ability for the deformation of electron cloud of a species, the softness, and /or the tendency of the molecule to lose electron, the ionization potential, are fundamentally similar in physical appear- ance stemming from the attraction power of the nuclei of the atoms forming the molecule. The softness, the ioni- zation energy, the electronegativity (chemical potential) and the electrophilicity index have direct link to the process of polarization and transfer of charge from a substrate and hence control the energetic effect—the protonation energy. Considering all the above mentioned fundamental nature of the physico-chemical process of protonation and its probable relationship with the quan- tum mechanical descriptors, we suggest an ansatz for the computation of the proton affinity in terms of these theoretical descriptors. The physico-chemical process and the energetic effect must entail the above stated four parameters. To derive an explicit relation to compute the proton affinity in terms of the above stated descriptors, we suggest explicit inter relationships between the pro- tonation energy and the descriptors relying upon their response towards the protonation. PA I (2) PA S (3) PA 1 (4) PA 1 (5) Combining the above four relations we get, 123 4 PACCICC 1C1S (6) where PA is proton affinity, C, C1, C2, C3 , and C4 are the constants I is ionization energy, S is global softness, χ is the electronegativity and ω is the global electrophilicity index of the molecule. Copyright © 2011 SciRes. JQIS 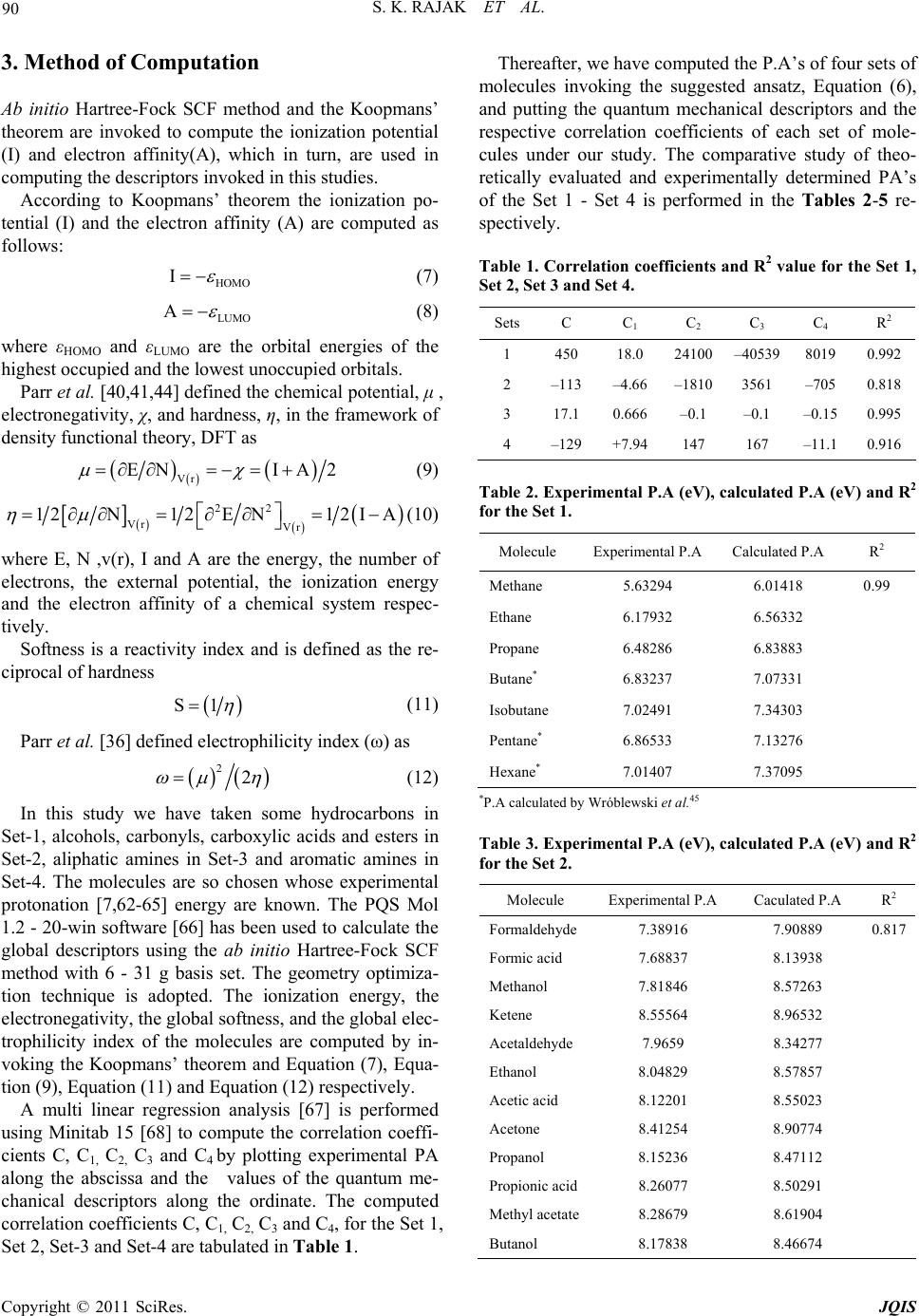 S. K. RAJAK ET AL. 90 3. Method of Computation Ab initio Hartree-Fock SCF method and the Koopmans’ theorem are invoked to compute the ionization potential (I) and electron affinity(A), which in turn, are used in computing the descriptors invoked in this studies. According to Koopmans’ theorem the ionization po- tential (I) and the electron affinity (A) are computed as follows: HOMO I (7) LUMO A (8) where εHOMO and εLUMO are the orbital energies of the highest occupied and the lowest unoccupied orbitals. Parr et al. [40,41,44] defined the chemical potential, μ , electronegativity, χ, and hardness, η, in the framework of density functional theory, DFT as Vr EN IA2 (9) 22 Vr Vr 12N12EN12 IA (10) where E, N ,v(r), I and A are the energy, the number of electrons, the external potential, the ionization energy and the electron affinity of a chemical system respec- tively. Softness is a reactivity index and is defined as the re- ciprocal of hardness S1 (11) Parr et al. [36] defined electrophilicity index (ω) as 22 (12) In this study we have taken some hydrocarbons in Set-1, alcohols, carbonyls, carboxylic acids and esters in Set-2, aliphatic amines in Set-3 and aromatic amines in Set-4. The molecules are so chosen whose experimental protonation [7,62-65] energy are known. The PQS Mol 1.2 - 20-win software [66] has been used to calculate the global descriptors using the ab initio Hartree-Fock SCF method with 6 - 31 g basis set. The geometry optimiza- tion technique is adopted. The ionization energy, the electronegativity, the global softness, and the global elec- trophilicity index of the molecules are computed by in- voking the Koopmans’ theorem and Equation (7), Equa- tion (9), Equation (11) and Equation (12) respectively. A multi linear regression analysis [67] is performed using Minitab 15 [68] to compute the correlation coeffi- cients C, C1, C2, C 3 and C4 by plotting experimental PA along the abscissa and the values of the quantum me- chanical descriptors along the ordinate. The computed correlation coefficients C, C1, C2, C3 and C4, for the Set 1, Set 2, Set-3 and Set-4 are tabulated in Table 1. Thereafter, we have computed the P.A’s of four sets of molecules invoking the suggested ansatz, Equation (6), and putting the quantum mechanical descriptors and the respective correlation coefficients of each set of mole- cules under our study. The comparative study of theo- retically evaluated and experimentally determined PA’s of the Set 1 - Set 4 is performed in the Tables 2-5 re- spectively. Table 1. Correlation coefficients and R2 value for the Set 1, Set 2, Set 3 and Set 4. Sets C C1 C 2 C 3 C 4 R 2 1 450 18.0 24100 –40539 8019 0.992 2 –113 –4.66–1810 3561 –705 0.818 3 17.1 0.666–0.1 –0.1 –0.150.995 4 –129 +7.94147 167 –11.10.916 Table 2. Experimental P.A (eV), calculated P.A (eV) and R2 for the Set 1. Molecule Experimental P.A Calculated P.A R2 Methane 5.63294 6.01418 0.99 Ethane 6.17932 6.56332 Propane 6.48286 6.83883 Butane* 6.83237 7.07331 Isobutane 7.02491 7.34303 Pentane* 6.86533 7.13276 Hexane* 7.01407 7.37095 *P.A calculated by Wróblewski et al.45 Table 3. Experimental P.A (eV), calculated P.A (eV) and R2 for the Set 2. Molecule Experimental P.A Caculated P.A R2 Formaldehyde 7.38916 7.90889 0.817 Formic acid 7.68837 8.13938 Methanol 7.81846 8.57263 Ketene 8.55564 8.96532 Acetaldehyde 7.9659 8.34277 Ethanol 8.04829 8.57857 Acetic acid 8.12201 8.55023 Acetone 8.41254 8.90774 Propanol 8.15236 8.47112 Propionic acid8.26077 8.50291 Methyl acetate8.28679 8.61904 Butanol 8.17838 8.46674 Copyright © 2011 SciRes. JQIS  91 S. K. RAJAK ET AL. Table 4. Experimental P.A (eV), calculated P.A (eV) and R2 for the Set 3. Molecule Experimental P.A Caculated P.AR2 NH3 8.846181 8.860042 0.995 CH3NH2 9.284153 9.341572 CH3CH2NH2 9.409908 9.399806 (CH3)2CHNH2 9.47929 9.499455 (CH3)2NH 9.566017 9.583402 (CH3)3CNH2 9.57469 9.596479 (CH3)3N 9.761153 9.794852 Table 5. Experimental P.A (eV), calculated P.A (eV) and R2 for the Set 4. Molecules Experimental P.A (eV)) Calculated P.A (eV) R2 3-H3C6H4N(C2H5)2 9.925935 9.722904 0.91 4-H3C6H4N(C2H5)2 9.912926 9.706435 C6H5N(C3H7)2 9.912926 9.673925 C6H5N(CH3)(C2H5) 9.84788 9.522402 C6H5NH(C2H5) 9.618053 9.592654 C6H5NHCH3 9.457608 9.44481 C6H5CH2NH2 9.401235 8.976198 2-(OH)C6H4NH2 9.28849 9.197386 3-(OH)C6H4NH2 9.28849 9.197251 4-CH3C6H4NH2 9.266808 9.06326 3-CH3C6H4NH2 9.253799 9.04584 3-CH3C6H4N(CH3)2 9.253799 9.044886 1,2-C6H4(NH2)2 9.22778 9.031081 4-ClC6H4NH2 9.045653 8.720894 3-BrC6H4NH2 9.023971 8.683775 4-FC6H4NH2 9.023971 8.763088 3-CF3C6H4NH2 8.854853 8.674228 For better visualization of the comparative study, the results of the theoretically computed and experimentally determined proton affinities of the Set 1 - Set 4 are de- picted in the Figures 1-4 respectively. 4. Results and Discussion A deeper look on the Table 2 and Figure 1 (for Set 1), and the Table 4 and Figure 3 (for Set 3) reveals that there are excellent correlation between the theoretically computed proton affinities of the seven hydrocarbons (Set 1) and seven aliphatic amines (Set 2) respectively. The R2 value for the correlation of Set1 and Set 3 are 0.99 and 0.995 respectively. A close look at the Figure 1 and Figure 3 reveals that the two sets of PA’s—experi- mental and theoretical of the two groups of molecules are so close to each other that one curve just superimposes upon the other. A look at the Table 3 and Figure 2 (for Set 2), and Table 5 and Figure 4 (for Set 4) reveals that there is fairly a good correlation between the theoretically com- puted and experimentally determined proton affinities of as many as twelve compounds containing alcohols, car- bonyls, carboxylic acids and esters(Set 2), and seventeen aromatic amines (Set 4) respectively. The R2 value for correlation of Set 2 and Set 4 are 0.817 and 0.91 respec- tively. Figure 1. Plot of calculated P.A Vs Experimental P.A and P.A calculated by Wróblewski et al. for Set 1. Figure 2. Plot of calculated P.A Vs experimental P.A for Set 2. Copyright © 2011 SciRes. JQIS 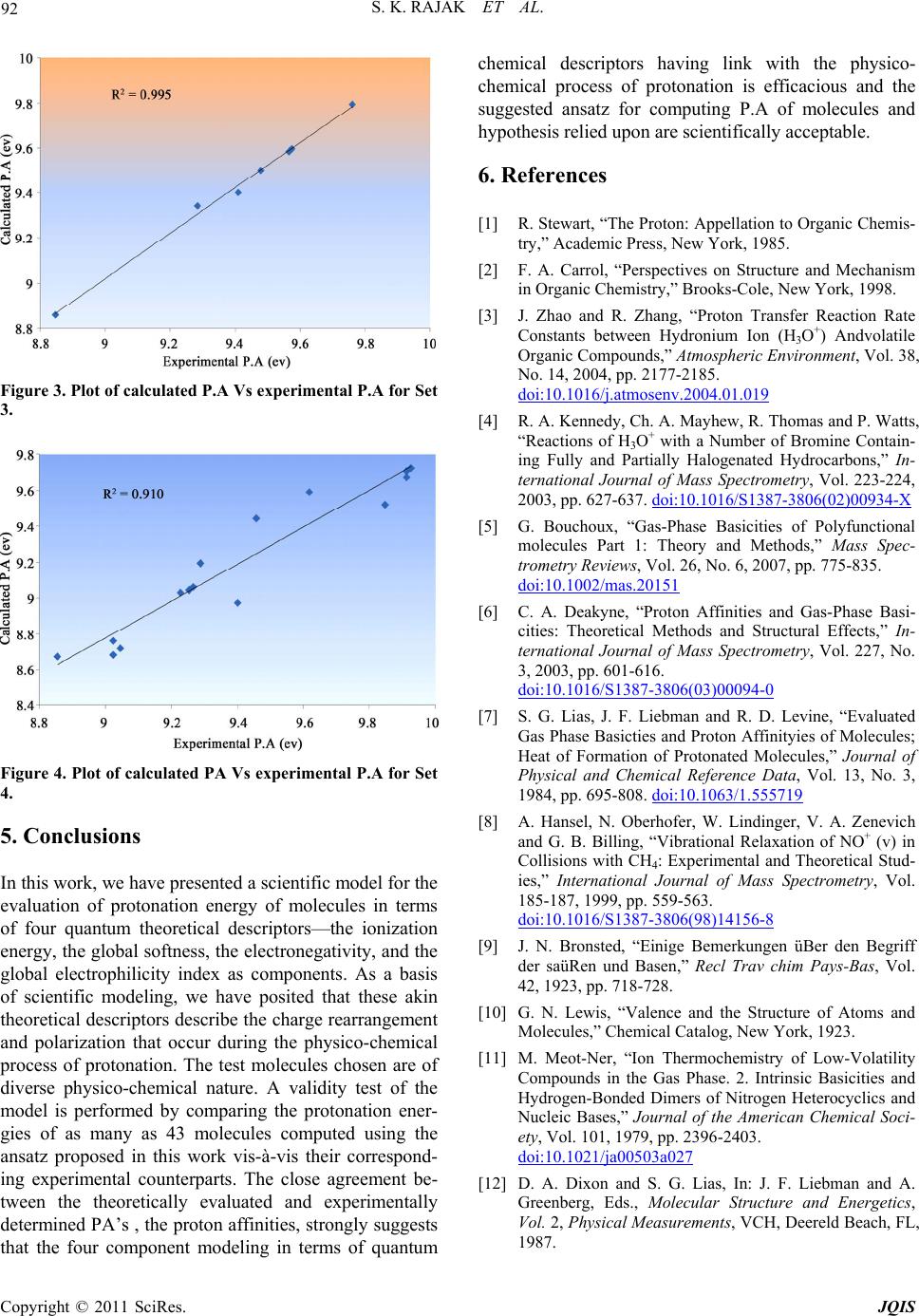 S. K. RAJAK ET AL. 92 Figure 3. Plot of calculated P.A Vs experimental P.A for Set 3. Figure 4. Plot of calculated PA Vs experimental P.A for Set 4. 5. Conclusions In this work, we have presented a scientific model for the evaluation of protonation energy of molecules in terms of four quantum theoretical descriptors—the ionization energy, the global softness, the electronegativity, and the global electrophilicity index as components. As a basis of scientific modeling, we have posited that these akin theoretical descriptors describe the charge rearrangement and polarization that occur during the physico-chemical process of protonation. The test molecules chosen are of diverse physico-chemical nature. A validity test of the model is performed by comparing the protonation ener- gies of as many as 43 molecules computed using the ansatz proposed in this work vis-à-vis their correspond- ing experimental counterparts. The close agreement be- tween the theoretically evaluated and experimentally determined PA’s , the proton affinities, strongly suggests that the four component modeling in terms of quantum chemical descriptors having link with the physico- chemical process of protonation is efficacious and the suggested ansatz for computing P.A of molecules and hypothesis relied upon are scientifically acceptable. 6. References [1] R. Stewart, “The Proton: Appellation to Organic Chemis- try,” Academic Press, New York, 1985. [2] F. A. Carrol, “Perspectives on Structure and Mechanism in Organic Chemistry,” Brooks-Cole, New York, 1998. [3] J. Zhao and R. Zhang, “Proton Transfer Reaction Rate Constants between Hydronium Ion (H3O+) Andvolatile Organic Compounds,” Atmospheric Environment, Vol. 38, No. 14, 2004, pp. 2177-2185. doi:10.1016/j.atmosenv.2004.01.019 [4] R. A. Kennedy, Ch. A. Mayhew, R. Thomas and P. Watts, “Reactions of H3O+ with a Number of Bromine Contain- ing Fully and Partially Halogenated Hydrocarbons,” In- ternational Journal of Mass Spectrometry, Vol. 223-224, 2003, pp. 627-637. doi:10.1016/S1387-3806(02)00934-X [5] G. Bouchoux, “Gas-Phase Basicities of Polyfunctional molecules Part 1: Theory and Methods,” Mass Spec- trometry Reviews, Vol. 26, No. 6, 2007, pp. 775-835. doi:10.1002/mas.20151 [6] C. A. Deakyne, “Proton Affinities and Gas-Phase Basi- cities: Theoretical Methods and Structural Effects,” In- ternational Journal of Mass Spectrometry, Vol. 227, No. 3, 2003, pp. 601-616. doi:10.1016/S1387-3806(03)00094-0 [7] S. G. Lias, J. F. Liebman and R. D. Levine, “Evaluated Gas Phase Basicties and Proton Affinityies of Molecules; Heat of Formation of Protonated Molecules,” Journal of Physical and Chemical Reference Data, Vol. 13, No. 3, 1984, pp. 695-808. doi:10.1063/1.555719 [8] A. Hansel, N. Oberhofer, W. Lindinger, V. A. Zenevich and G. B. Billing, “Vibrational Relaxation of NO+ (v) in Collisions with CH4: Experimental and Theoretical Stud- ies,” International Journal of Mass Spectrometry, Vol. 185-187, 1999, pp. 559-563. doi:10.1016/S1387-3806(98)14156-8 [9] J. N. Bronsted, “Einige Bemerkungen üBer den Begriff der saüRen und Basen,” Recl Trav chim Pays-Bas, Vol. 42, 1923, pp. 718-728. [10] G. N. Lewis, “Valence and the Structure of Atoms and Molecules,” Chemical Catalog, New York, 1923. [11] M. Meot-Ner, “Ion Thermochemistry of Low-Volatility Compounds in the Gas Phase. 2. Intrinsic Basicities and Hydrogen-Bonded Dimers of Nitrogen Heterocyclics and Nucleic Bases,” Journal of the American Chemical Soci- ety, Vol. 101, 1979, pp. 2396-2403. doi:10.1021/ja00503a027 [12] D. A. Dixon and S. G. Lias, In: J. F. Liebman and A. Greenberg, Eds., Molecular Structure and Energetics, Vol. 2, Physical Measurements, VCH, Deereld Beach, FL, 1987. Copyright © 2011 SciRes. JQIS  93 S. K. RAJAK ET AL. [13] L. A. Curtiss, K. Raghavachari and P. A. Pople, “Gaus- sian-2 Theory Using Reduced Moller-Plesset Orders,” Journal of Chemical Physics, Vol. 98, No. 2, 1993, pp. 1293-1298. doi:10.1063/1.464297 [14] J. E. Del Bene, “Molecular Orbital Study of the Protona- tion of DNA Bases,” The Journal of Physical Chemistry, Vol. 87, 1983, pp. 367-371. doi:10.1021/j100225a040 [15] B. J. Smith and L. Radom, “Calculation of Proton Affini- ties Using the G2(MP2,SVP) Procedure,” The Journal of Physical Chemistry, Vol. 99, No. 17, 1995,pp. 6468-6471. doi:10.1021/j100017a028 [16] B. S. Jursic, “Density Functional Theory and Complete Basis Set Ab Initio Evaluation of Proton Affinity for Some Selected Chemical Systems,” Journal of Molecular Structure: THEOCHEM, Vol. 487, No. 1, 1999, pp. 193- 203. doi:10.1016/S0166-1280(99)00154-2 [17] S. Hammerum, “Heats of Formation and Proton Affinities by the G3 Method,” Chemical Physics Letters, Vol. 300, No. 3-4, 1999, pp. 529-532. doi:10.1016/S0009-2614(98)01439-0 [18] W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, “Ab Initio Molecular Orbital Theory,” John Willey and Sons, New York, 1986. [19] J. K. Labanowskiy, R. A. Hill, D. J. Heisterbergy, D. D. Miller, C. F. Bender and J. W. Andzelm, “Proton Affini- ties Calculated by Traditional Ab Initio Approaches and by Density Functional Methods,” http://www.ccl.net/cca/documents/proton-affinity/affiniti es.pdf [20] J. L. Ozment and A. M. Schmiedekamp, “Proton Affini- ties of Molecules Containing Nitrogen and Oxygen: Comparing Ab Initio and Semiempirical Results to Ex- periments,” International Journal of Quantum Chemistry, Vol. 43, No. 6, 1992, pp. 783-800. doi:10.1002/qua.560430606 [21] M. Eckert-Maksic, M. Klessinger and Z. B. Maksic, “Theoretical Calculations of Proton Affinities in Phenol,” Chemical Physics Letters, Vol. 232, No. 5, 1995, pp. 472- 478. doi:10.1016/0009-2614(94)01383-7 [22] N. Russo, M. Toscano, A. Grand and T. Mineva, “Proton Affinity and Protonation Sites of Aniline. Energetic Be- havior and Density Functional Reactivity Indices,” The Journal of Physical Chemistry A, Vol. 104, No. 17, 2000, pp. 4017-4021. doi:10.1021/jp991949e [23] R. Margabandu and K. Subramani, “Comparative Study of Various Quantum Mechanical Descriptors for Predic- tion of Ionization Constant (pKa) of Substituted Ani- lines,” International Journal of ChemTech Research, Vol. 2, No. 3, 2010, pp. 1507-1513. [24] R. W. Taft, M. Taagepera, J. L. M. Abboud, J. F. Wolf, D. J. DeFrees, W. J. Hehre, J. E. Bartmess and Jr. R. T. McIver, “The Separation of Polarizability and Inductive Effects in Gas- and Solution-Phase Proton-Transfer Equi- libriums,” Journal of the American Chemical Society, Vol. 100, No. 24, 1978, pp. 7765-7767. doi:10.1021/ja00492a075 [25] Z. B. Maksic and R. Vianello, “Quest for the Origin of basicity: Initial vs Final State Effect in Natural Nitrogen Bases,” The Journal of Physical Chemistry A, Vol. 106, No. 2, 2002, pp. 419-430. doi:10.1021/jp013296j [26] P. Perez, A. Toro-Labbe and R. Contreras, “A Semiquan- titative Description of Electrostatics and Polarization Substituent Effects: Gas-Phase Acid-Base Equilibria as Test Cases,” The Journal of Physical Chemistry A, Vol. 104, No. 51, 2000, pp. 11993-11998. doi:10.1021/jp0025734 [27] P. Geerlings, F. De Proft and W. Langenaeker, “Concep- tual Density Functional Theory,” Chemical Reviews, Vol. 103, 2003, pp. 1793-1873. doi:10.1021/cr990029p [28] M. Berkowitz, S. K. Ghosh and R. G. Parr, “On the Con- cept of Local Hardness in Chemistry,” Journal of the American Chemical Society, Vol. 107, 1985, pp. 6811- 6814. doi:10.1021/ja00310a011 [29] S. K. Ghosh, M. Berkowitz and R. G. Parr, “Transcrip- tion of Ground-State Density-Functional Theory into a Local Thermodynamic,” Proceedings of the National Academy of Sciences, Vol. 81, No. 24, 1984, pp. 8028- 8031. doi:10.1073/pnas.81.24.8028 [30] C. Hansch and A. Leo, “Substituent Constants for Corre- lation Analysis in Chemistry and Biology,” John Wiley & Sons, New York, 1979. [31] M. V Putz, N. Russo and E. Sicilia, “Atomic Radii Scale and Related Size Properties from Density Functional Electronegativity Formulation,” The Journal of Physical Chemistry A, Vol. 107, 2003, pp. 5461-5465. doi:10.1021/jp027492h [32] M. V. Putz, N. Russo and E. Sicilia, “About the Mulliken Electronegativity in DFT,” Theoret Chim Acta, Vol. 114, 2005, pp. 38-45. doi:10.1007/s00214-005-0641-4 [33] M. V. Putz, “Systematic Formulations for Electronegativ- ity and Hardness and Their Atomic Scales within Density Functional Softness Theory,” International Journal of Quantum Chemistry, Vol. 106, No. 2, 2006, pp. 361-389. doi:10.1002/qua.20787 [34] M. V. Putz, “Semi Classical Electronegativity and Che- mical Hardness,” Journal of Theoretical and Computa- tional Chemistry, Vol. 6, No. 1, 2007, pp. 33-47. [35] M. V. Putz, “Density Functionals of Chemical Bonding,” International Journal of Molecular Sciences, Vol. 9, No. 6, 2008, pp. 1050-1095. [36] M. V. Putz, “Electronegativity: Quantum Observable,” International Journal of Quantum Chemistry, Vol. 109, No. 4, 2009, pp. 733-738. doi:10.1002/qua.21957 [37] L. Tarko and M. V. Putz, “On Electronegativity and Chemical Hardness Relationships with Aromaticity,” Journal of Mathematical Chemistry, Vol. 47, No. 1, 2010, pp. 487-495. doi:10.1007/s10910-009-9585-6 [38] M. V. Putz, “Chemical Action and Chemical Bonding,” Journal of Molecular Structure: THEOCHEM, Vol. 900, No. 1-3, 2009, pp. 64-70. doi:10.1016/j.theochem.2008.12.026 [39] R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, “Electronegativity: The Density Functional Viewpoint,” Copyright © 2011 SciRes. JQIS  S. K. RAJAK ET AL. 94 Journal of Chemical Physics, Vol. 68, No. 8, 1978, pp. 3801-3807. doi:10.1063/1.436185 [40] R. G. Parr and R. G. Pearson, “Absolute Hardness: Com- panion Parameter to Absolute Electronegativity,” Journal of the American Chemical Society, Vol. 105, No. 26, 1983, pp. 7512-7516. doi:10.1021/ja00364a005 [41] R.G. Parr and W. Yang, “Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity,” Journal of the American Chemical Society, Vol. 106, No. 14, 1984, pp. 4049-4050. doi:10.1021/ja00326a036 [42] W. Yang and R. G. Parr, “Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis,” Proceedings of the National Academy of Sci- ences, Vol. 82, No. 20, 1985, pp. 6723-6726. doi:10.1073/pnas.82.20.6723 [43] R. G. Parr and W. Yang, “Density Functional Theory of Atoms and Molecules,” Oxford University Press, New York, 1989. [44] R. G. Parr, L. V. Szentpaly and S. Liu, “Electrophilicity Index,” Journal of the American Chemical Society, Vol. 121, No. 9, 1999, pp. 1922-1924. doi:10.1021/ja983494x [45] D. C. Ghosh, “A Theorrtical Study of Some Selected Molecules and Their Protonation by the Application of CNDO Method,” Premchand Roychand Research Stu- dentship Award, University of Calcutta, 1976. [46] T. Chakraborty and D. C. Ghosh, “Computation of the Atomic Radii through the Conjoint Action of the Effec- tive Nuclear Charge and the Ionization Energy,” Molecu- lar Physics, Vol. 108, No. 16, 2010, pp. 2081-2092. [47] B. E. Mills, R. L. Martin and D. A. Shirley, “Further Studies of the Core Binding Energy-Proton Affinity Cor- relation in Molecules,” Journal of the American Chemi- cal Society, Vol. 98, No. 9, 1976, pp. 2380-2385. doi:10.1021/ja00425a002 [48] L. L. Lohr, “Protonic Counterpart of Electronegativity as an Organizing Principle for Acidity and Basicity,” The Journal of Physical Chemistry, Vol. 88, 1984, pp. 3607- 3611. doi:10.1021/j150660a046 [49] R. G. Pearson, “Absolute Electronegativity and Hardness Correlated with Molecular Orbital Theory,” Proceedings of the National Academy of Sciences, Vol. 83, No. 22, 1986, pp. 8440-8441. doi:10.1073/pnas.83.22.8440 [50] P. Chaquin, “Absolute Electronegativity and Hardness: An Analogy with Classical Electrostatics Suggests an In- terpretation of the Parr ‘Electrophilicity Index’ as a ‘Global Energy Index’ Leading to the ‘Minimum Elec- trophilicity Principle,” Chemical Physics Letters, Vol. 458, 2008, pp. 1439-1444. [51] S. Noorizadeh, “Is There a Minimum Electrophilicity Principle in Chemical Reactions?” Chinese Journal of Chemistry, Vol. 27, 2007, pp. 1439-1444. doi:10.1002/cjoc.200790266 [52] N. Islam and D. C. Ghosh, “A New Algorithm for the Evaluation of Equilibrium Inter nuclear Bond Distance of Heteronuclear diatomic Molecule Based on the Hardness Equalization Principle,” The European Physical Journal D—Atomic, Molecular, Optical and Plasma Physics, Vol. 61, No. 2, 2010, pp. 341-348. [53] N. Islam and D. C. Ghosh, “A New Radial Dependent Electrostatic Algorithm for the Evaluation of the Elec- trophilicity Indices of the Atoms,” International Journal of Quantum Chemistry, Vol. 111, No. 14, 2010, pp. 3556- 3564. doi:10.1002/qua.22861 [54] N. Islam and D. C. Ghosh, “Determination of Some De- scriptors of the Real World Working on the Fundamental Identity of the Basic Concept and the Origin of the Elec- tronegativity and the Global Hardness of Atoms. Part 2: Computation of the Dipole Moments of Some Heteronu- clear Diatomics,” International Journal of Quantum Chemistry, Vol. 111, No. 12, 2010, pp. 2802-2810. doi:10.1002/qua.22651 [55] N. Islam and D. C. Ghosh, “Evaluation of Global Hard- ness of Atoms Based on the Commonality in the Basic Philosophy of the Origin and the Operational Signifi- cance of the Electronegativity and the Hardness. Part I. The Gordy’s Scale of Electronegativity and the Global Hardness,” European Journal of Chemistry, Vol. 1, No. 2, 2010, pp. 83-89. [56] N. Islam and D. C. Ghosh, “Charge transfer associated with the physical process of hardness equalization and the chemical event of the molecule formation and the dipole moments,” International Journal of Quantum Chemistry, 111, No. 12, 2010, pp. 2811-2819. [57] N. Islam and D. C. Ghosh, “Determination of Some De- scriptors of the Real World Working on the Fundamental Identity of the Basic Concept and the Origin of the Elec- tronegativity and the Global Hardness of Atoms, Part 1: Evaluation of Internuclear Bond Distance of Some Het- eronuclear Diatomics,” International Journal of Quantum Chemistry, 2010, in Press. doi:10.1002/qua.22500 [58] N. Islam and D. C. Ghosh, “A Quest for the Algorithm for Evaluating the Molecular Hardness,” International Journal of Quantum Chemistry, Vol. 111, No. 9, 2010, pp. 1931-1941. doi:10.1002/qua [59] N. Islam and D. C. Ghosh, “Whether There Is a Hardness Equalization Principle Analogous to the Electronegativity qualization Principle—A Quest,” International Journal of Quantum Chemistry, Vol. 111, No. 9, 2010, pp. 1961- 1969. doi:10.1002/qua.22508 [60] N. Islam and D. C. Ghosh, “Whether Electronegativity and Hardness Are Manifest Two Different Descriptors of the One and the Same Fundamental Property of Atoms— A Quest,” International Journal of Quantum Chemistry, Vol. 111, No. 1, 2009, pp. 40-51. doi:10.1002/qua.22415 [61] N. Islam and D. C. Ghosh, “A New Algorithm for the Evaluation of the Global Hardness of Poly Atomic Molecules,” Molecular Physics, 2011, Accepted. [62] E. P. L. Hunter and S. G. Lias, “Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An up- date,” Journal of Physical and Chemical Reference Data, Vol. 27, No. 3, 1998, pp. 413-656. doi:10.1063/1.556018 [63] National Institute of Standards and Technology. http://webbook.nist.gov/chemistry/pa-ser.html. [64] T. Wróblewski, L. Ziemczonek, A. M. Alhasan and G. P. Copyright © 2011 SciRes. JQIS  S. K. RAJAK ET AL. Copyright © 2011 SciRes. JQIS 95 Karwasz. http://www.fizyka.umk.pl/~karwasz/.../2007_Ab_initio_an d _density_functional.pdf. [65] S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin and W. G. Mallard, “Gas-Phase Ion and Neutral Thermochemistry,” Journal of Physical and Chemical Reference Data, Vol. 17, Suppl. 1, 1988, pp. 1-861. doi:10.1063/1.555819 [66] PQSMol 1.2-20-win, Parallel Quantum Solutions, LLC. http://www.pqs-chem.com. [67] C. Nantasenamat, C. Isarankura-Na-Ayudhya, T. Naenna and V.Prachayasittikul, “A Practical Overview of Quan- titative Structure—Activity Relationship,” EXCLI Jour- nal, Vol. 8, 2009, pp. 74-88. [68] MINITAB, a Statistical Software of STATSOFT Inc U.S.A.
|