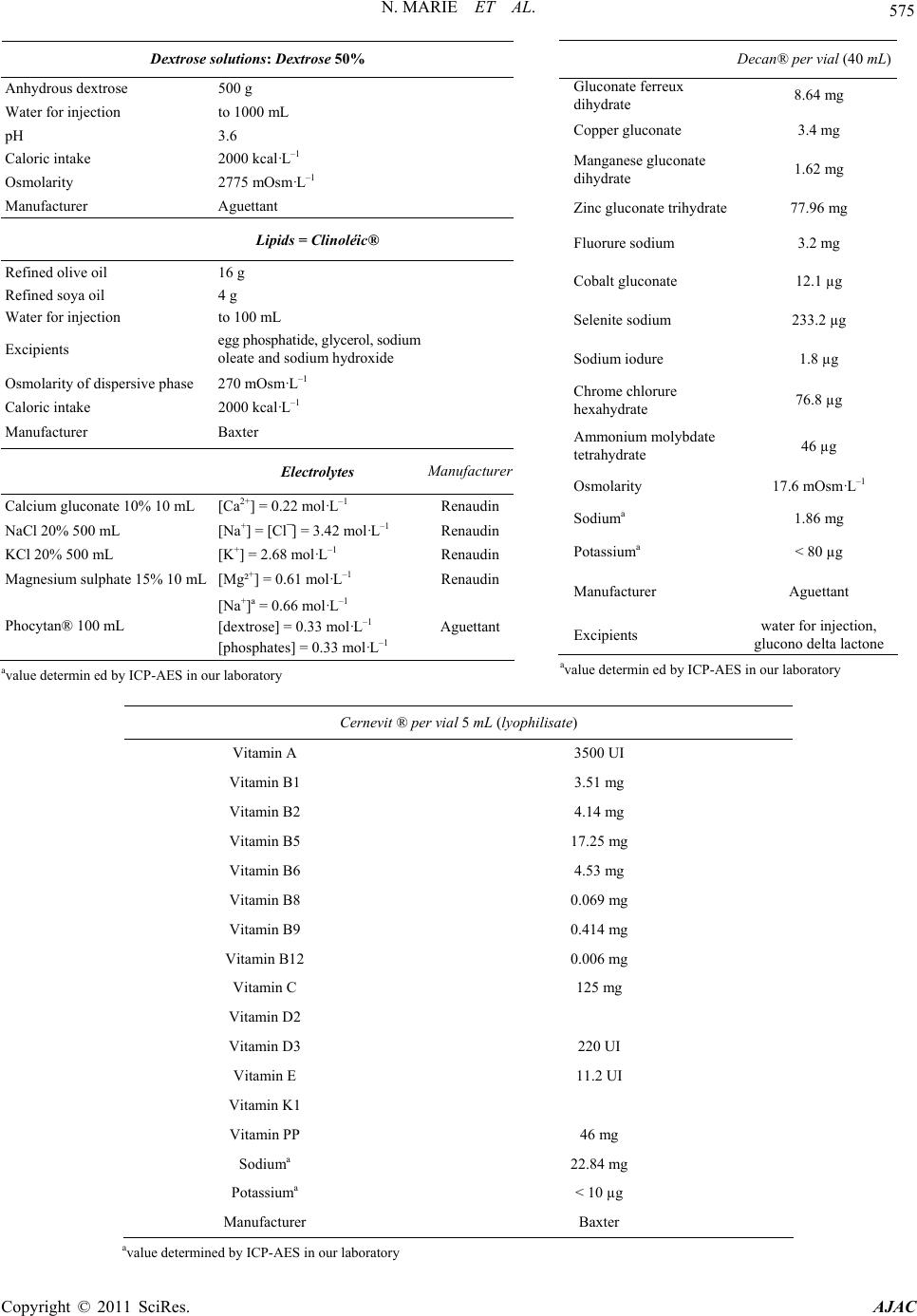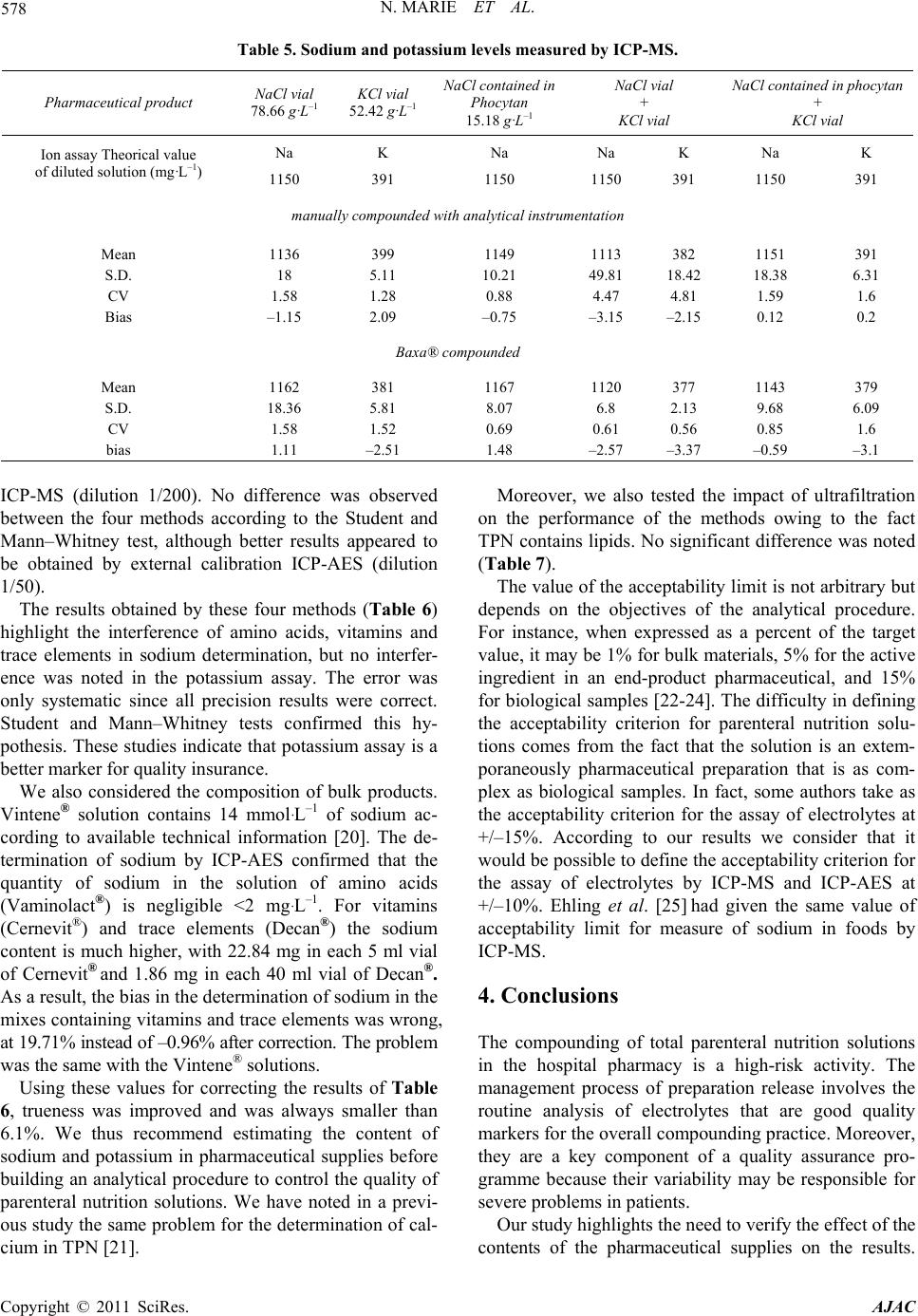
580 N. MARIE ET AL.
Table 7. Results obtained on parenteral nutrition mixes after ultrafiltration.
Methods ICP AES External
Calibration 1/100 dilution ICP MS
1/100 dilution
Compounds Theoretical value
(mg·L-1)
Na
1150
K
391
Na
1150
K
391
Assay without ultrafiltration
(mg·L-1) 1305 409 1317 466
Ions + D + AA + La Assay after ultrafiltration
(mg·L-1) 1314 410 1328 469
Assay without ultrafiltration
(mg·L-1) 1499 396 1506 454
Ions + D+ AA + L + vit + TEa Assay after ultrafiltration
(mg·L-1) 1496 399 1562 469
aD = dextrose, AA = amino acids, L = lipids, vit = vitamins, TE = trace elements
In our case, we recommend using the potassium assay as
a quality marker because no supplies contain this elec-
trolyte.
To reduce the risk and to improve the quality of com-
pounding, we recommend using an automated com-
pounding device instead of gravity-fill TPN system but,
even in this case, the acceptance criterion for sodium and
potassium determination was not <10%.
5. Acknowledgements
Françoise Lacroix and Severine Durand from EHESP/
LERES are gratefully acknowledged for their technical
support throughou t this study.
6. References
[1] P. Bonnabry, I. Cingria, F. Sadeghipour, H. Ing, C. Fonzo
Christe and R. E. Pfizer, “Use of a Systematic Risk
Analysis Method to Improve Safety in the Production of
Paediatric Parenteral Nutrition Solutions,” Quality Safety
Health Care, Vol. 14, No. 2, 2005, pp. 93-98.
doi:org/10.1136/qshc.2003.007914
[2] “Safe Practices for Parenteral Nutrition Formulations,”
National Advisory Group on Standards and Practice
Guidelines for Parenteral Nutrition, Journal Parenteral
Enteral Nutrition, Vol. 22, No 3, 1998, pp. 49-66.
[3] H. Price, “New Technology Assists in Meeting ASPEN
and ASHP Safety Guidelines to Minimize Total Par-
enteral Nutrition Compounding Errors,” Journal of Par-
enteral Enteral Nutrit i o n, V ol . 2 5, No 3 , 2001, pp. 100-102.
[4] C. M. Crill, E. B. Hak and R. A. Helms, “Accuracy of
Parenteral Nutrition Solutions Compounded with Auto-
mated Systems and by Hand,” American Journal of Health-
System Pharmacy, Vol. 62, No. 15, 2005, pp. 2345-2348.
doi:org/10.2146/ajhp050322
[5] S. Nussbaumer, S. Fleury-Souverain, L. Bouchoud, S.
Rudaz, P. Bonnabry and J.-L.Veuthey, “Determination of
Potassium, Sodium, Calcium and Magnesium in Total
Parenteral Nutrition Formulations by Capillary Electro-
phoresis with Contactless Conduc tivity Detection,” Journal
of Pharmaceutical and Biomedical Analysis, Vol. 53,
2010, pp. 130-136.
[6] C. St Laurent, L. Roulet and A. Dupuis, “Aspects
Pharmacotechniques de la Nutrition Parentérale,”Actualités
Pharmaceutiques Hospitalières, Vol. 14, 2008, pp. 44-50.
[7] H. Zegbeh, F. Pirot, T. Quessada,T. Durand, F .Vételé, A.
Rose, V. Bréant and G.Aulagner, “Exactitude, Precision
et Rapidité de Fabrication des Poches de Mélanges
Nutritifs Parentéraux :C omp a rai so n de Te ch ni que s Aut o -
matisée et Manuelle,” Ann Pharm Fr Journal, Vol. 69,
2011, pp. 38-44.
[8] P. Rucart, D. Balayssac, V. Sautou-Miranda, A. Boyer
and J. Chopineau, “Enquête Nationale Sur Les Contrôles
Des Mélanges Nutritifs Parentéraux Fabriqués Dans Les
Pharmacies à Usage Intérieur des Centres Hospitaliers
Universitaires,” Nutrition Clinique et Métabolisme, Vol.
21, No. 2, 2007, pp. 47-48.
[9] D. J. Douglas, “Some Current Perspectives on ICP-MS,”
Canadian Journal of Spectroscopy, Vol. 34, No. 2, 1989,
pp. 38-49. doi:org/10.1007/s00216-009-2658-3
[10] H. Xie, K. Huang, J. Liu, X. Nie and L. Fu “Determina-
tion of Trace Elements in Re sidual Oil by H i g h- R e so l u ti o n
Inductively Coupled Plasma Mass Spectrometry,” Ana-
lytical Bioanalytical Chemistry, Vol. 393, No. 4, 2009, pp.
2075-2080. doi:org/10.1007/s00216-009-2658-3
[11] J. M. Mermet and E. Poussel, “Couplage plasma induit
par haute fréquence-spectrométrie de masse,” Techniques
de l’Ingénieur, Vol. TA4, 09, 1999, p. 2720.
[12] J. P. Cristol, B. Balint, B. Canaud and M. F. Daurés,
“Méthodes de Dosage du Sodium Dans les Liquides
Biologiques,” Néphrologie et Thérapeutique, V ol . 3, 2007,
pp. S104-S111.
[13] G. B. Levy, “Determination of Sodium with Ion-Selective
Electrodes,” Clinical Chemistry, Vol. 27, No. 8, 1981, pp.
1435-1438.
[14] H. T. Delves, “Atomic Absorption Spectroscopy in Clinical
Analysis,” Annales of Clincal Biochemistry, Vol. 24, No.
Copyright © 2011 SciRes. AJAC