B. S. SUNDAR ET AL.
537
The best-fit linear equation obtained was Y = 36422x
+ 338053. At all concentration leve ls, standard deviation
of peak area was significantly low and RSD was below
1.0%. Analysis of residuals indicated that residuals were
scattered within ± 2.0% with respect to 100% concentra-
tion response.
3.3.3. Accu racy
The percentage recovery of amlexanox in bulk drug
samples ranged from 98.86% - 101.05% [Table 2].
3.3.4. Robustness
Close observation of analysis results for deliberately
changed chromatographic conditions (flow rate, pH and
column temperature) revealed that the resolution be-
tween closely eluting degradant in basic condition and
amlexanox was always greater than 2.5, illustrating the
robustness of the method [Table 3].
3.3.5. Solution Stability and Mobile Phase Stability
The %RSD of assay of amlexanox during solution stabil-
ity and mobile phase stability experiments was within
1.0% RSD. No significant changes were observed in the
content of amlexanox during solution stability and mo-
bile phase stability experiments. The solution stability
and mobile phase stability experiments data confirms
that sample solutions and mobile phase used during as-
say and related substance determination were stable up to
the study.
3.3.6. Ass ay Analysis
Analysis was performed for different batches of amlex-
anox in bulk drug samples (n = 3) ranged from 99.5% -
100.2%.
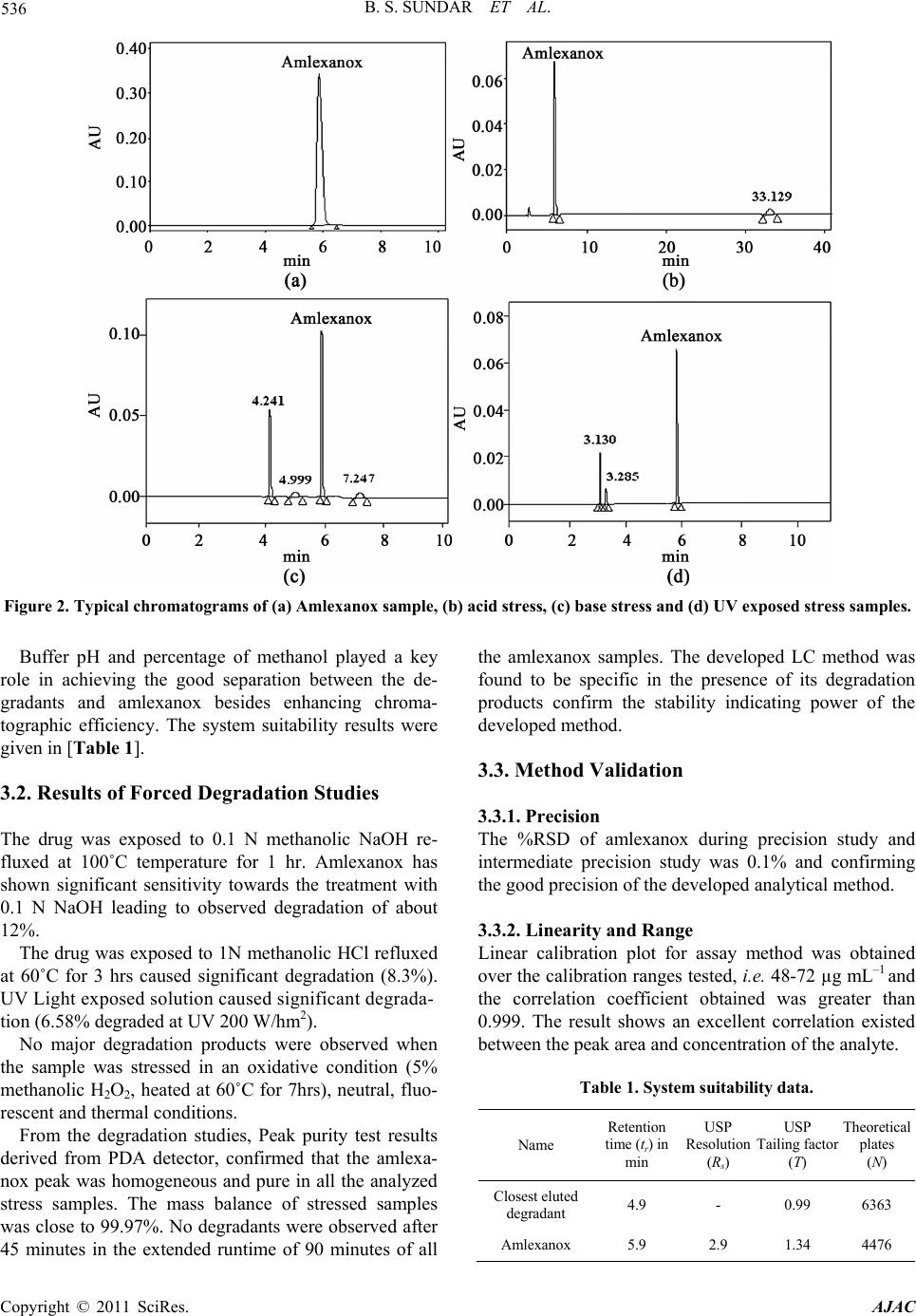
Table 2. Results of Accuracy study for Bulk drugs.
Added (g)
(n = 3) %Recovery for
Bulk drugs %RSD for
Bulk drugs
48 101.05 0.23
60 100.27 0.39
72 98.86 0.04
n =3, Number of determinations
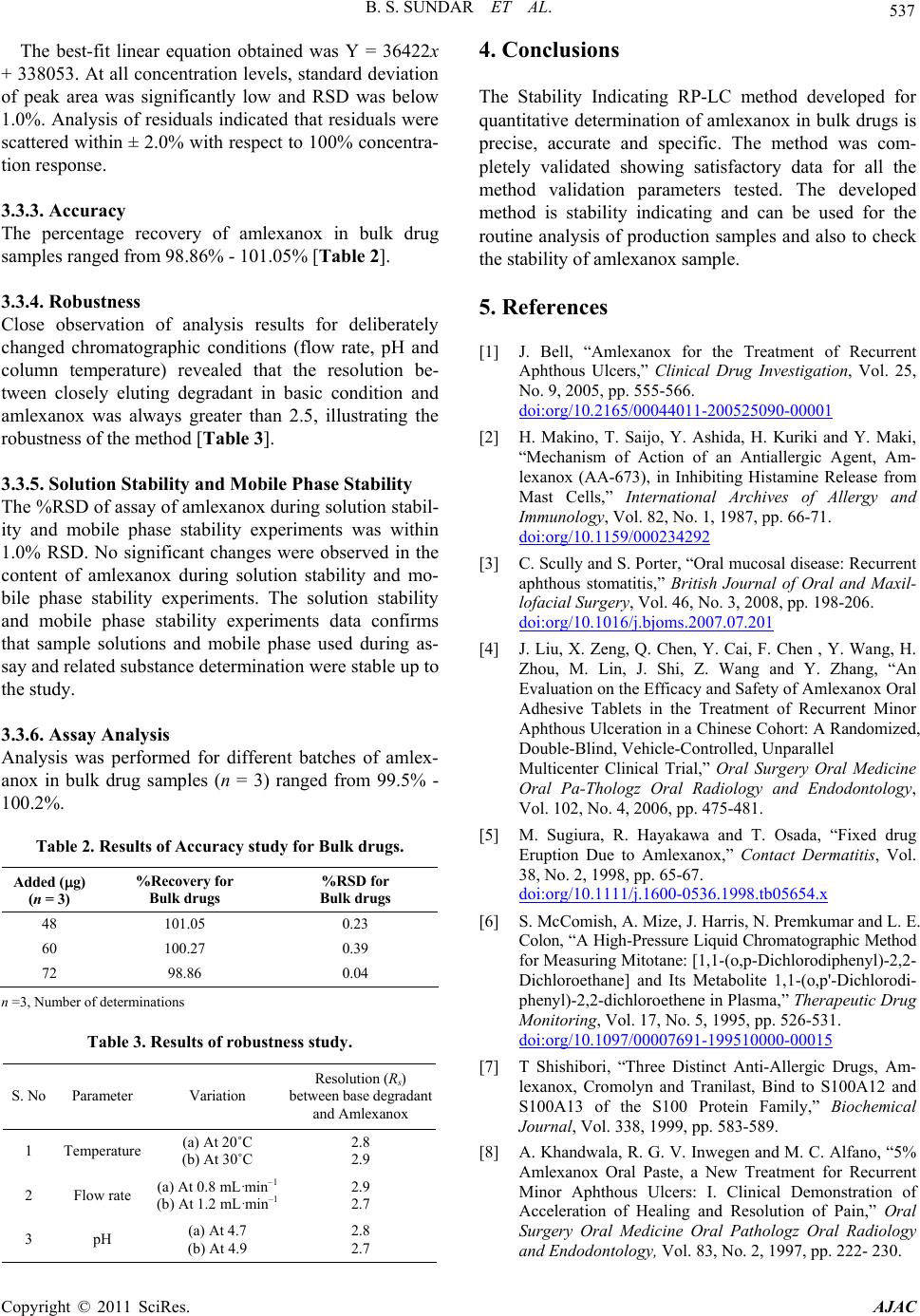
Table 3. Results of robustness study.
S. No Parameter Variation Resolution (Rs)
between base degradant
and Amlexanox
1 Temperature (a) At 20˚C
(b) At 30˚C 2.8
2.9
2 Flow rate
(a) At 0.8 mL·min–1
(b) At 1.2 mL·min–1 2.9
2.7
3 pH (a) At 4.7
(b) At 4.9 2.8
2.7
4. Conclusions
The Stability Indicating RP-LC method developed for
quantitative determinatio n of amlexanox in bu lk drugs is
precise, accurate and specific. The method was com-
pletely validated showing satisfactory data for all the
method validation parameters tested. The developed
method is stability indicating and can be used for the
routine analysis of production samples and also to check
the stability of amlexanox sample.
5. References
[1] J. Bell, “Amlexanox for the Treatment of Recurrent
Aphthous Ulcers,” Clinical Drug Investigation, Vol. 25,
No. 9, 2005, pp. 555-566.
doi:org/10.2165/00044011-200525090-00001
[2] H. Makino, T. Saijo, Y. Ashida, H. Kuriki and Y. Maki,
“Mechanism of Action of an Antiallergic Agent, Am-
lexanox (AA-673), in Inhibiting Histamine Release from
Mast Cells,” International Archives of Allergy and
Immunology, Vol. 82, No. 1, 1987, pp. 66-71.
doi:org/10.1159/000234292
[3] C. Scully and S. Porter, “Oral mucosal disease: Recurrent
aphthous stomatitis,” British Journal of Oral and Maxil-
lofacial Surgery, Vol. 46, No. 3, 2008, pp. 198-206.
doi:org/10.1016/j.bjoms.2007.07.201
[4] J. Liu, X. Zeng, Q. Chen, Y. Cai, F. Chen , Y. Wang, H.
Zhou, M. Lin, J. Shi, Z. Wang and Y. Zhang, “An
Evaluation on the Efficacy and Safety of Amlexanox Oral
Adhesive Tablets in the Treatment of Recurrent Minor
Aphthous Ulceration in a Chinese Cohort: A Randomized,
Double-Blind, Vehicle-Controlled, Unparallel
Multicenter Clinical Trial,” Oral Surgery Oral Medicine
Oral Pa-Thologz Oral Radiology and Endodontology,
Vol. 102, No. 4, 2006, pp. 475-481.
[5] M. Sugiura, R. Hayakawa and T. Osada, “Fixed drug
Eruption Due to Amlexanox,” Contact Dermatitis, Vol.
38, No. 2, 1998, pp. 65-67.
doi:org/10.1111/j.1600-0536.1998.tb05654.x
[6] S. McComish, A. Mize, J. Harris, N. Pre mkumar and L. E.
Colon, “A High-Pressure Liquid Chromat ographic Method
for Measuring Mitotane: [1,1-(o,p-Dichlorodiphenyl)-2,2-
Dichloroethane] and Its Metabolite 1,1-(o,p'-Dichlorodi-
phenyl)-2,2-dichloroethene in Plasma,” Therapeutic Drug
Monitoring, Vol. 17, No. 5, 1995, pp. 526-531.
doi:org/10.1097/00007691-199510000-00015
[7] T Shishibori, “Three Distinct Anti-Allergic Drugs, Am-
lexanox, Cromolyn and Tranilast, Bind to S100A12 and
S100A13 of the S100 Protein Family,” Biochemical
Journal, Vol. 338, 1999, pp. 583-589.
[8] A. Khandwala, R. G. V. Inwegen and M. C. Alfano, “5%
Amlexanox Oral Paste, a New Treatment for Recurrent
Minor Aphthous Ulcers: I. Clinical Demonstration of
Acceleration of Healing and Resolution of Pain,” Oral
Surgery Oral Medicine Oral Pathologz Oral Radiology
and Endodontology, Vol. 83, No. 2, 1997, pp. 222- 230.
Copyright © 2011 SciRes. AJAC