 American Journal of Analytical Chemistry, 2011, 2, 511-521 doi:10.4236/ajac.2011.25061 Published Online September 2011 (http://www.SciRP.org/journal/ajac) Copyright © 2011 SciRes. AJAC Liquid Chromatography Mass Spectrometer (LC-MS/MS) Study of Distribution Patterns of Base Peak Ions and Reaction Mechanism with Quantification of Pesticides in Drinking Water Using a Lyophilization Technique Sukesh Narayan Sinha* National Institute of Nutrition (ICMR, New Delhi), Jamai-Osmania, Hyderabad, India E-mail: *sukeshnr_sinha@yahoo.com Received May 30, 2011; revised July 1, 2011; accepted July 15, 2011 Abstract In the process of the development of agriculture, pesticides have become an important tool as an insecticide to kill the insect from plant for boosting food production. Therefore the insecticides/pesticides and herbicides have been used in India for agriculture setting. In this connection a sensitive method for the quantification of 5 pesticides in drinking water samples to the µg·L–1 level has been developed. The paper also describes the effect of dissociation energy on ion formation and sensitivity of pesticides in water samples. The structure, ion formations, distribution of base peak and fragmentation schemes were correlated with the different dis- sociation energies. The new ion was obtained at different mass to charge ratio, which was the characteristic ion peak of targeted pesticide. Additionally, a simple solvent lyophilization followed by selective analysis using a liquid chromatography-mass spectrometry method was used. This method was accurate (≥98%) as it possesses limits of detection in the 6 - 38 ng·L–1 range, and the percentage relative standard deviations are less than 8.62% at the low µg·L–1 end of the method‟s linear range. The percentage recovery of all the pesti- cides at the 0.1 µg ·L–1 levels of detection ranges from 92% - 104%. This method was used for the quantifica- tion of pesticides in water samples collected from different parts from urban city of Hyderabad, India. In this study, 13 water samples were analyzed in which all samples showed detectable level of the malathion and alachlor. The concentration of pesticides ranged from 0.004 µg·L–1 to 0.691 µg·L–1 exceeded to the maxi- mum residual limit of Indian standard. Keywords: Water, LC-MS/MS, Lyphilization, Pesticides, Dissociation Energy 1. Introduction Agricultural development continues to remain the most important objective of Indian planning and policy. In the process of development of agriculture, pesticides have become an important tool as a plant protection agent for boosting food production. Currently, India is the largest producer of pesticides in Asia and ranks twelfth in the world for the use of pesticides [1]. Humans are exposed to pesticides through soil, water, air and food by different routes of exposure such as inhalation, ingestion and der- mal contact [2]. For instance, dietary intake represents the major source of pesticide exposure to children, and this exposure may increase pesticide-related health risks in children in comparison to adults [3]. Increasing inci- dences of cancer, chronic kidney diseases, suppression of the immune system, sterility among males and females, endocrine disorders and neurological and behavioral dis- orders, especially among children, have been attributed to chronic pesticide poisoning [1]. The presence of pesticide residues in various compo- nents of the environment and food commodities is a matter of concern all over the world [4-6]. In India sev- eral methods have also been used for pesticide residual analysis in different food commodities (e.g., vegetables, fruits and other products of food) using a GC method [7-9]. We also analyzed pesticide levels using different method in food and biological samples [10-14]. Fur- thermore, a method was reported [15] for the analysis of pesticide residues using a quick, cheap, effective, rugged,  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC and safe (QuEChERS) multi-residue method in combina- tion with gas and liquid chromatography (LC-MS/MS) and tandem mass spectrometric detection. A mixture of 38 pesticides was quantitatively recovered from spiked lemon, raisins, wheat and flour using GC-MS/MS, while 42 pesticides were recovered from oranges, red wine, red grapes, raisins and wheat flour using LC-MS/MS for determination [15]. A multi-analyte method for the quan- tification of contemporary pesticides in human serum and plasma using high-resolution mass spectrometry was reported [16]. I have used a very accurate, simple and reproducible LC-MS/MS method for the quantification of pesticide residue at low levels in drinking water samples collected from different part of urban areas. 2. Experimental Sections 2.1. Materials All pesticide standards were purchased from Sigma-Al- drich, Inc. (USA). Methanol acetonitrile (LC-MS grade), water (LC-MS grade) were obtained from Sigma Aldrich GmbH. Formic acid was purchased from Sigma Aldrich (USA). All reagents were made freshly in LC-MS grade water or solvent before use. 2.2. Stock Solutions Individual stock solutions at 1 mgL–1 of pesticides (alach- lor, malathion, dimethoate, chlorpyrifos and metribuzin) were prepared in acetonitrile. The stock solutions were divided into aliquots, sealed in ampoules and stored at –40˚C. 2.3. Calibration Standard From the stock solutions, eleven working standard sets for alachlor, malathion, dimethoate, chloripyrifos and metribuzin (0.1, 0.2, 1, 2, 5, 10, 30, 50, 100, 150 and 250 ngml–1) were prepared to encompass the entire linear range of the method by using serial dilution technique. These standards were then used for the validation of me- thod (determination of limit of detection (LOD), limit of quantification (LOQ), recovery experiment and linearity experiment). The standard sets were divided into aliquots, sealed in ampoules and stored at –40˚C until use. 2.4. Laboratory Reagent Blanks Before extraction of water samples, the purchased water samples were tested by LC-MS/MS using a similar ex- traction method that was used for the recovery experi- ment, and the water was found to be free from pesticide residues. 2.5. Recovery Experiment by GC-MS/MS The water sample was spiked with the standard of each compound, alachlor, malathion, dimethoate, chlorpyrifos and metribuzin at the different level (0.1, 0.2, 1, 2, 5, 10 and 30 µg·mL–1). 2.6. Sampling Thirteen water samples were included in the sampling of water for the purpose of pesticide residue analysis. One liter water samples were collected from different part of the urban city. Five ml water has been taken for lyophi- lization. 3. Sample Preparations Unknown water and reagent blanks were prepared iden- tically. Five mL of pure water was pipetted into 20 mL test tubes. The water was spiked with the mixtures of different pesticides at different concentrations (0.1, 0.2, 1, 2, 5, 10 and 30 ng·mL–1 of alachlor, malathion, dime- thoate, chlorpyrifos and metribuzin). Then water was mixed and allowed to equilibrate for approximately 30 minutes. The tubes were then placed in a methanol bath and held at –100˚C for at least 15 min. Once the samples were frozen, they were placed in a lyophilizer at –109˚C. The vacuum status was checked and the samples were left for 6 hours to ensure complete dryness. The samples were then removed from the lyophilizer for extraction. Four milliliters of acetonitrile was added at neutral pH (7) to each tube, mixed for 3 min on a vertex shaker and supernatant was transferred into 20 ml centrifuge tubes. In the second step, samples were extracted with 4 milli- liters acetonitrile for 3 min. and supernatant was then transferred to the first extract. There after all of the ex- tracted tubes were centrifuged for 10 min at 3000 rpm. Next, the supernatant solution was transferred into a new set of 20 ml tubes for drying and placed in a TurboVap at room temperature under 5-psi nitrogen and completely dried. The dry residues were reconstituted in 1 mL ace- tonitrile for analysis. 4. Instrumental Analysis 4.1. Chromatographic Condition Ten micro-liter of the concentrated extract was analyzed using, 4000-QTRAP triple-quadrupole hybrid mass spec- trometer in MRM mode. The analysis of all pesticides was performed using a liquid chromatograph (LC, Shi- 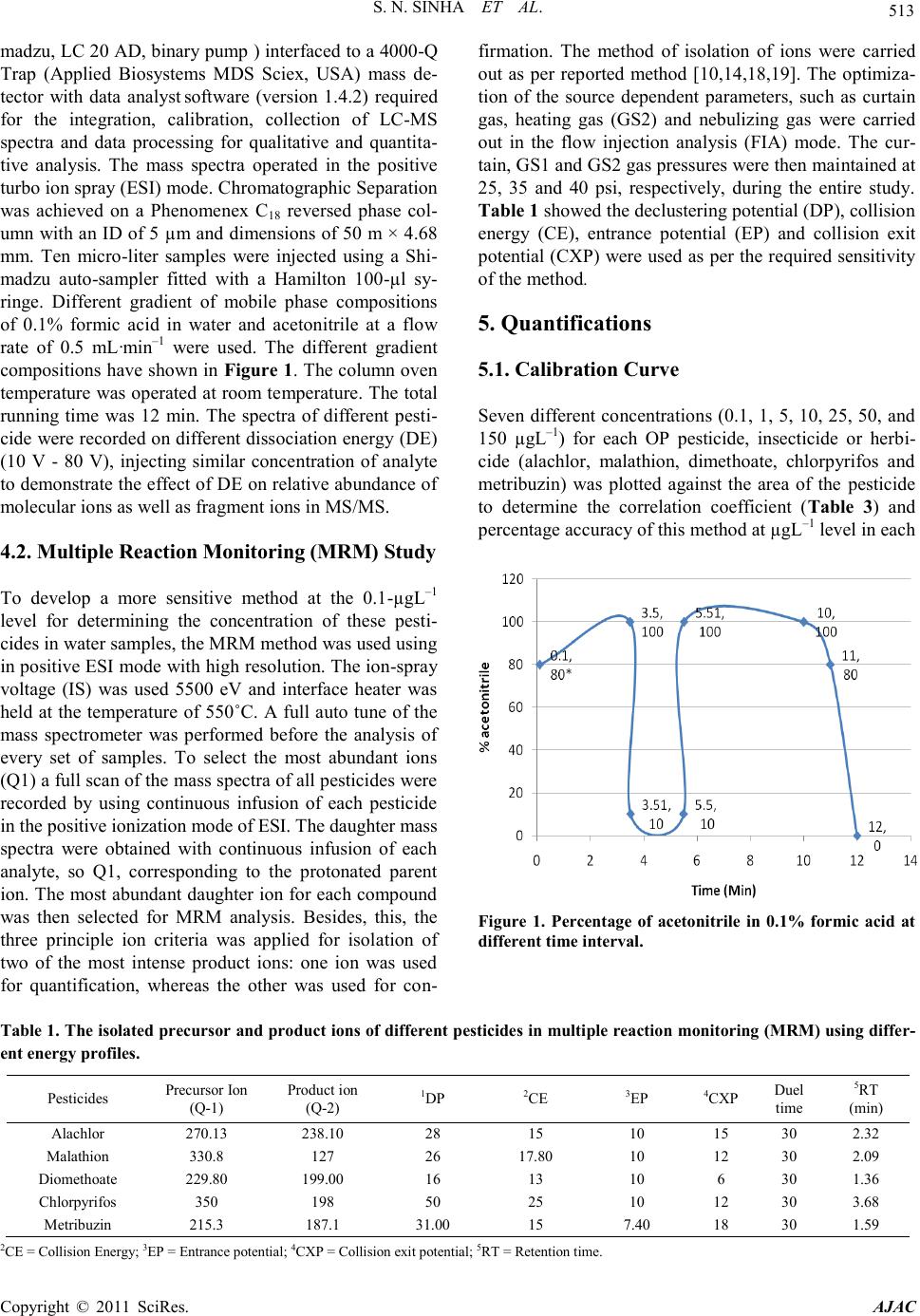 S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC madzu, LC 20 AD, binary pump ) interfaced to a 4000-Q Trap (Applied Biosystems MDS Sciex, USA) mass de- tector with data analyst software (version 1.4.2) required for the integration, calibration, collection of LC-MS spectra and data processing for qualitative and quantita- tive analysis. The mass spectra operated in the positive turbo ion spray (ESI) mode. Chromatographic Separation was achieved on a Phenomenex C18 reversed phase col- umn with an ID of 5 µm and dimensions of 50 m × 4.68 mm. Ten micro-liter samples were injected using a Shi- madzu auto-sampler fitted with a Hamilton 100-µl sy- ringe. Different gradient of mobile phase compositions of 0.1% formic acid in water and acetonitrile at a flow rate of 0.5 mL·min–1 were used. The different gradient compositions have shown in Figure 1. The column oven temperature was operated at room temperature. The total running time was 12 min. The spectra of different pesti- cide were recorded on different dissociation energy (DE) (10 V - 80 V), injecting similar concentration of analyte to demonstrate the effect of DE on relative abundance of molecular ions as well as fragment ions in MS/MS. 4.2. Multiple Reaction Monitoring (MRM) Study To develop a more sensitive method at the 0.1-µgL–1 level for determining the concentration of these pesti- cides in water samples, the MRM method was used using in positive ESI mode with high resolution. The ion-spray voltage (IS) was used 5500 eV and interface heater was held at the temperature of 550˚C. A full auto tune of the mass spectrometer was performed before the analysis of every set of samples. To select the most abundant ions (Q1) a full scan of the mass spectra of all pesticides were recorded by using continuous infusion of each pesticide in the positive ionization mode of ESI. The daughter mass spectra were obtained with continuous infusion of each analyte, so Q1, corresponding to the protonated parent ion. The most abundant daughter ion for each compound was then selected for MRM analysis. Besides, this, the three principle ion criteria was applied for isolation of two of the most intense product ions: one ion was used for quantification, whereas the other was used for con- firmation. The method of isolation of ions were carried out as per reported method [10,14,18,19]. The optimiza- tion of the source dependent parameters, such as curtain gas, heating gas (GS2) and nebulizing gas were carried out in the flow injection analysis (FIA) mode. The cur- tain, GS1 and GS2 gas pressures were then maintained at 25, 35 and 40 psi, respectively, during the entire study. Table 1 showed the declustering potential (DP), collision energy (CE), entrance potential (EP) and collision exit potential (CXP) were used as per the required sensitivity of the method. 5. Quantifications 5.1. Calibration Curve Seven different concentrations (0.1, 1, 5, 10, 25, 50, and 150 µgL–1) for each OP pesticide, insecticide or herbi- cide (alachlor, malathion, dimethoate, chlorpyrifos and metribuzin) was plotted against the area of the pesticide to determine the correlation coefficient (Table 3) and percentage accuracy of this method at µgL–1 level in each Figure 1. Percentage of acetonitrile in 0.1% formic acid at different time interval. Table 1. The isolated precursor and product ions of different pesticides in multiple reaction monitoring (MRM) using differ- ent energy profiles. 2CE = Collision Energy; 3EP = Entrance potential; 4CXP = Collision exit potential; 5RT = Retention time.  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC analytical run. Linear regression analyses were performed on plots of the calculated concentrations versus expected concentrations. With this analysis, a slope of 0.999 would be indicative of 99% accuracy (Table 3) 5.2. Recovery Experiment The recoveries of the method were determined by spik- ing water samples free of pesticides with different known concentrations of reference standards. The recovery of each pesticide was calculated at each of the known con- centration levels by comparing the measured concentra- tions with the spiked concentrations, as per the reported method [17,18,]. A ratio of 1.00 indicated 100% recov- ery. LC mixtures of 0.10, 0.20, 1, 2, 5, 10, 30 ppb for alachlor, malathion, dimethoate, chlorpyrifos and me- tribuzine) in acetonitrile were prepared using the pesti- cide reference standards previously described. The per- centage recovery of each pesticide was calculated by comparing the peak area ratio of the spiked standards with those of the pure standards. Water samples were fortified with the mixture of the five pesticides at differ- ent concentration (0.10, 0.20, 1, 2, 5, 10, 30 µg·L–1) and allowed to standing for 30 min so that all of the pesti- cides were absorbed thoroughly by the samples before making the extraction. Seven un-spiked water samples and 7 reagent blanks served as the negative control for quality assurance purposes. All the samples were ex- tracted as previously described. 5.3. Limit of Detection (LOD) The point at which the measured value was considered reliable was when it was larger than the uncertainty as- sociated with it, also called the LOD. In this method, the analytical LOD was calculated as per the earlier reported method [17,18]. 5.4. Lower Limits of Method Validation (LLMV) The LLMV by LC-MS/MS for alachlor, malathion, di- methoate, chlorpyrifos and metribuzin were 0.1 µg ·L–1. 6. Results and Discussions This method was developed to confirm and accurately quantify pesticides in water samples. The lyophilization followed by extraction process was simple, accurate and easy. In the case of water samples, several variations of the extraction procedure were attempted. In many cases these extractions were not optimal, and good recovery of the analytes was not achieved due to the polar nature of OPs pesticides. Therefore, I used a simple lyophilization process for the complete dryness of the samples to mi- nimize the matrix effect. The extraction of the analytes from dry samples was easy and overall good recovery was achieved. In this method, 5 mL of water samples was lyophilized and was extracted at neutral pH using 5 mL of a acetonitrile twice with two minutes of shaking each time. The percentage recoveries and percentage RSD obtained were well within previously prescribed analytical method [16]. The acetonitrile resulted in 82% - 104% extraction efficiency for alachlor, malathion, di- methoate, chlorpyrifos and metribuzin in the water sam- ples. The different solvent gradient was fixed to accom- modate the physical and chemical properties of the pesti- cides (Figure 1). The specificity of 4000 Q-trap mass spectrometry allows for the elimination of interfering components in the water sample extracts, which in turn provided the low detection limits of the method. These specificity requirements precluded the use of single qua- drupole mass spectrometry. Thus, this method was applied in MRM mode to increase the sensitivity for quantifica- tion at the µg ·L–1 level. The extracted ion chromatograms of alachlor, malathion, dimethoate, chlorpyrifos and me- tribuzin (10 µg ·L–1 spikes) are shown in Figure 2. The isolation of ions of OP pesticides was carried out in a similar fashion as per the reported method [10,14,17, 18]. In first series of experiment the full scan spectra were recorded, using manual tuning in FIA mode after that the characteristic stable ions were isolated for MRM transition for confirmation and quantification of five pes- ticides in water samples. The detail isolated ion for quan- tification, different energy parameters (DE, EP, FP and CE) and retention times (RT) for MRM transitions are shown in Table 1. The confirmation ions were isolated at m/z 162, 99, 171, 125 and 131 for alachlor, malathion, dimethoate, chlorpyrifos and metribuzin, respectively, by using different energy set up. The percentage recovery and RSD has been shown in Table 2. The selected molecular ion, and selected product ion scan were performed and different collision dissociation energies were applied in MS/MS mode to obtain different fragmentation patterns. The ion formation of study sam- ple of dimethoate is shown in Figure 3 The m/z 198 was obtained due to the elimination of ethylene (–CH2=CH2–) molecule from parent ion molecule m/z 229.9, because the oxy-gen atom donates the lone pair to hydrogen atom by remote charge mechanism. Similarly, -N=CH2 mole- cule was removed from m/z 198 leading to the formation of structure at m/z 170. The dimethoate possesses a suf- ficient long chain to permit transfer of hydrogen namely loss due to hydrogen rearrangement mechanism. Similar pattern noted previously with triazofos, chlorpyrifos and phenolate ion [10,14]. The structure was formed at m/z 124 due to the removal of –C3H6SNO group from m/z 229.9. This new structure has been isolated due to struc-  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC ture reactivity and ion reaction mechanism of dimethoate. The fragmentation scheme of chlorpyrifos has been shown in Figure 4. The m/z 321.1 was obtained due to the eli- mination of ethylene (–CH2=CH2–) molecule from par- ent ion molecule m/z 350, because the oxy-gen atom donates the lone pair to hydrogen atom by remote charge mechanism. Additionally, the phosphorous atom is sta- bile through dл-pл bonding, therefore ethylene molecule removed from the m/z 321.1 leading to the formation of new structure at m/z 293.4. Similarly, –C4H10O2PS mo- lecule was removed from parent ion m/z 350 leading to the formation of structure at m/z 197.8. The new stable structure was formed at m/z 152.8 due to the removal of –CCl group from m/z 197.8. This new structure has been isolated due to rearrangement and ion reaction me- chan- ism of chlorpyrifos. The ion formation and reaction activity of alachlor has been shown in Figure 5. The m/z 238 was obtained due to the elimination of methyl alcohol (–CH3OH) molecule from parent ion molecule m/z 270, because the oxy-gen atom donates the lone pair to hydrogen atom by remote charge mechanism. Additionally, the nitrogen atoms ob- served steric hindrance, which deactivate the whole mole- cule and therefore C2HOCl molecule removed from the m/z ion 238 leading to the formation of new stable mo- lecule at m/z 162. Similarly, a new structure was formed at m/z 110 due to expulsion of acetylene molecule from m/z 162. Additionally, a new structure at m/z 137 was formed due to removal of C2H molecule from m/z 162. Figure 2. Extracted ion chromatogram (EIC) of spiked water samples at 10 PPB each (1) alachlor, (2) malathion, (3) dime- thoate (4) chlorpyrifos (5) metribuzin.  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC Table 2. Percentage recovery (mean) and % RSD of pesticides at different spiked concentrations. Spiked concentration (ngmL–1) RSD = Relative Standard Deviation; SD = Standard Deviation; N= Number of replicate The fragmentation schemes and ion reaction mechan- ism were observed in case of metribuzin, which has been shown in Figure 6. The m/z 186 was obtained due to the elimination of ethylene (–CH2=CH2–) molecule from parent ion molecule m/z 215, because the oxy-gen atom donates the lone pair to hydrogen atom by remote charge mechanism. Additionally, the three nitrogen atoms are present in aromatic ring, which deactivate the whole molecule and therefore ethylene molecule removed from the parent ion molecule [14]. Similarly, C5H9N2O mole- cule was removed from m/z 215 leading to the formation of structure at m/z 72.1. The new structure was formed at m/z 117 due to the removal of –C5H12O group from m/z 215. This new structure has been isolated due to structure reactivity and ion reaction mechanism of metribuzin. The removal of ions and formation of new structure was observed in this study due to remote charge me- chanism, rearrangement, nucleophilic and electrophilic reaction. Similar pattern noted previously with triazofos, chlorpyrifos and phenolate ion [10,14]. The pesticide-free water samples were spiked with dif- ferent concentrations of standard (i.e., alachlor, malathion, dimethoate, chloripyrifos and metribuzin,). The inter-day percentage recoveries, the relative standard deviation (RSD), limit of quantification (LOQ) and the limit of detection (LOD) were determined as per the reported methodology [17,18], and the results are shown in Table 4. The obtained percent recoveries for all these pesticides were found to be in the range of 96% - 103% of the standard value (Table 2) [19,20]. The obtained RSD was below 8% for all compounds, which further reinforced the importance, sensitivity, precision and selectivity of this method. The different behaviours of base peak pattern recorded on different dissociation energy (DE) are illustrated in Figure 7. These results reveal the m/z at 350 was ob- tained at DE 10 V, while m/z at 197, 125 and 97 were obtained at DE 20, 30, and 40, respectively for chlorpy- rifos. The result clearly indicates that the m/z 125 and m/z 97 were used for confirmation of chlorpyrifos in water samples. Additionally, the m/z at 270,110, 83 and 70 were obtained at DE 10, 30, 60 and 70 respectively 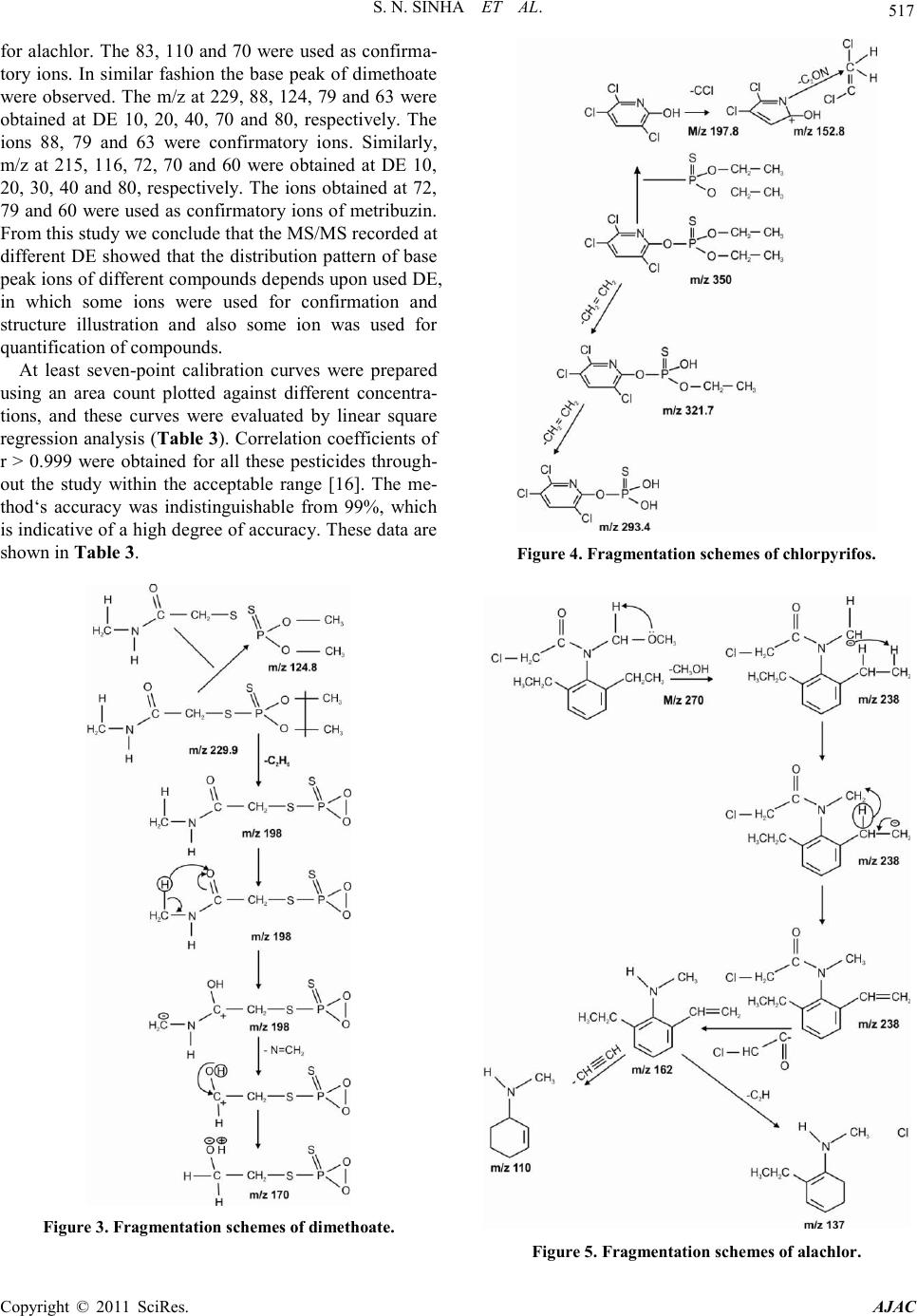 S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC for alachlor. The 83, 110 and 70 were used as confirma- tory ions. In similar fashion the base peak of dimethoate were observed. The m/z at 229, 88, 124, 79 and 63 were obtained at DE 10, 20, 40, 70 and 80, respectively. The ions 88, 79 and 63 were confirmatory ions. Similarly, m/z at 215, 116, 72, 70 and 60 were obtained at DE 10, 20, 30, 40 and 80, respectively. The ions obtained at 72, 79 and 60 were used as confirmatory ions of metribuzin. From this study we conclude that the MS/MS recorded at different DE showed that the distribution pattern of base peak ions of different compounds depends upon used DE, in which some ions were used for confirmation and structure illustration and also some ion was used for quantification of compounds. At least seven-point calibration curves were prepared using an area count plotted against different concentra- tions, and these curves were evaluated by linear square regression analysis (Table 3). Correlation coefficients of r > 0.999 were obtained for all these pesticides through- out the study within the acceptable range [16]. The me- thod„s accuracy was indistinguishable from 99%, which is indicative of a high degree of accuracy. These data are shown in Table 3. Figure 3. Fragmentation schemes of dimethoate. Figure 4. Fragmentation schemes of chlorpyrifos. Figure 5. Fragmentation schemes of alachlor.  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC Figure 6. Fragmentation schemes of metribuzin. Table 3. Accuracy determination using the correlation coefficient of spiked samples at different concentrations with uncer- tainties parameter (slope, intercept and standard error in slope). 0.1, 1, 5, 10, 25, 50, 150 0.1, 1, 5, 10, 25, 50, 150 0.1, 1, 5, 10, 25, 50, 150 0.1, 1, 5, 10, 25, 50, 150 ar = correlation coefficient; br2 = Determinations of coefficient; RSD = Relative Standard Deviation; N = Number of replicate; cSES = Standard error in slope Table 4. LOD, LOQ, % accuracy, and coefficient of deter- mination for eight pesticides. LOD = Limit of Determination; LOQ = Limit of Quantification The mean concentration of chloripyrifos Malathion, Alachlor, dimethoate and metribuzin in bore water were 0.283 (ranged from 0.029 to 0.691 µg·L–1), 0.246 (ranged from 0.032 to 0.566), 0.157 (ranged from 0.038 to 0.231 µg ·L–1), 0.102 (ranged from 0.041 to 0.233 µg ·L–1), 0.227 (ranged from 0.051 to 0.51 µg ·L–1) µg ·L–1, respectively. The averaged concentration of chlorpyrifos malathion, alachlor, dimethoate and metribuzin in MC water is 0.095 (ranged from 0.054 to 0.19 µg ·L–1), 0.100 (ranged from 0.057 to 0.21 µg ·L–1), 0.0986 (ranged from 0.05 to 0.162 µg ·L–1), 0.092 (ranged from 0.083 to 0.105 µg ·L–1), 0.027 (ranged from 0.004 to 0.051 µg·L–1) µg ·L–1 , respectively. The percentage of pesticide showed in Figure 8. 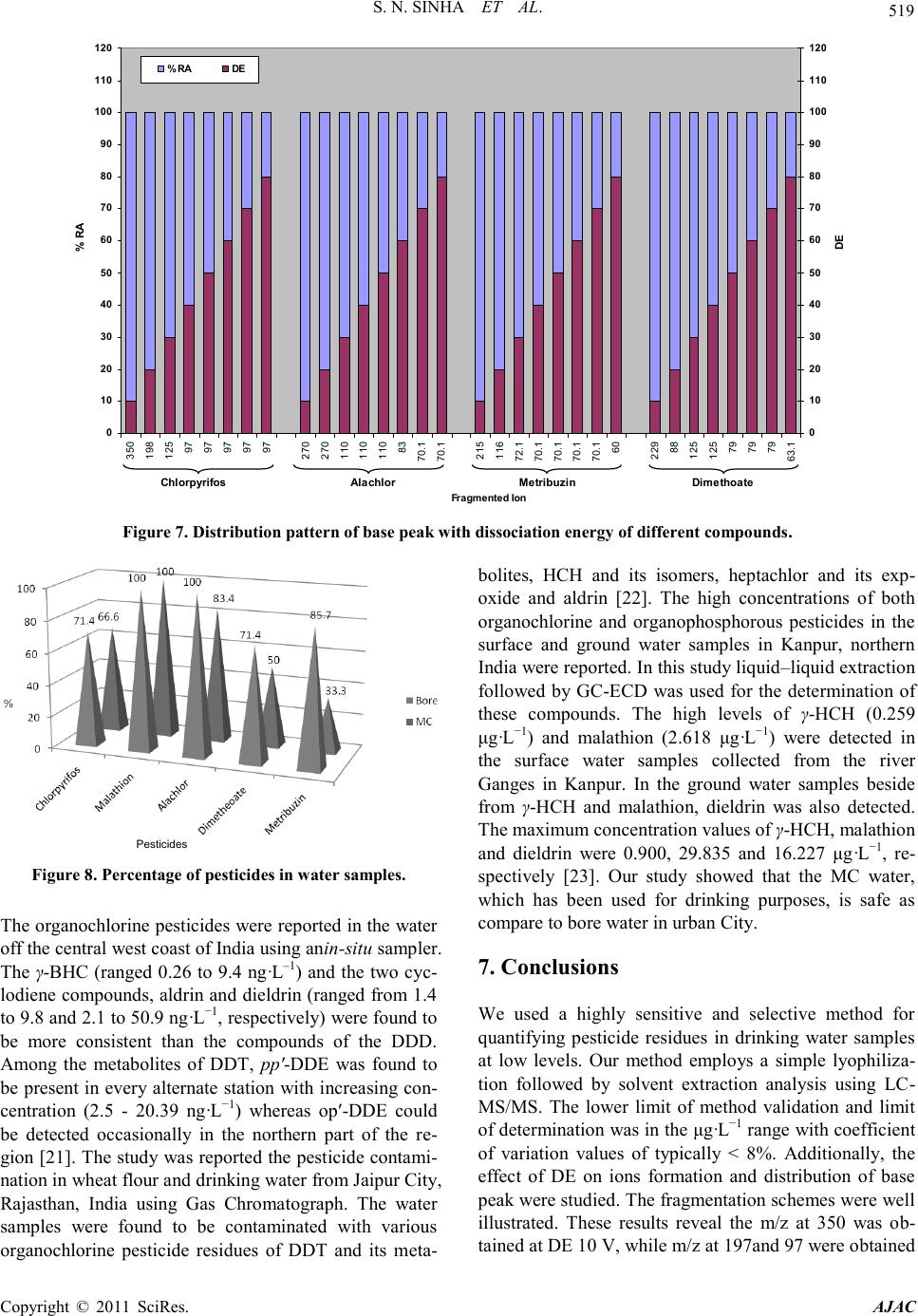 S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC 0 10 20 30 40 50 60 70 80 90 100 110 120 350 198 125 97 97 97 97 97 270 270 110 110 110 83 70.1 70.1 215 116 72.1 70.1 70.1 70.1 70.1 60 229 88 125 125 79 79 79 63.1 0 10 20 30 40 50 60 70 80 90 100 110 120 Figure 7. Distribution pattern of base peak with dissociation energy of different compounds. Figure 8. Percentage of pesticides in water samples. The organochlorine pesticides were reported in the water off the central west coast of India using anin-situ sampler. The γ-BHC (ranged 0.26 to 9.4 ng·L–1) and the two cyc- lodiene compounds, aldrin and dieldrin (ranged from 1.4 to 9.8 and 2.1 to 50.9 ng·L−1, respectively) were found to be more consistent than the compounds of the DDD. Among the metabolites of DDT, pp′-DDE was found to be present in every alternate station with increasing con- centration (2.5 - 20.39 ng·L−1) whereas op′-DDE could be detected occasionally in the northern part of the re- gion [21]. The study was reported the pesticide contami- nation in wheat flour and drinking water from Jaipur City, Rajasthan, India using Gas Chromatograph. The water samples were found to be contaminated with various organochlorine pesticide residues of DDT and its meta- bolites, HCH and its isomers, heptachlor and its exp- oxide and aldrin [22]. The high concentrations of both organochlorine and organophosphorous pesticides in the surface and ground water samples in Kanpur, northern India were reported. In this study liquid–liquid extraction followed by GC-ECD was used for the determination of these compounds. The high levels of γ-HCH (0.259 μg·L−1) and malathion (2.618 μg·L−1) were detected in the surface water samples collected from the river Ganges in Kanpur. In the ground water samples beside from γ-HCH and malathion, dieldrin was also detected. The maximum concentration values of γ-HCH, malathion and dieldrin were 0.900, 29.835 and 16.227 μg·L−1, re- spectively [23]. Our study showed that the MC water, which has been used for drinking purposes, is safe as compare to bore water in urban City. 7. Conclusions We used a highly sensitive and selective method for quantifying pesticide residues in drinking water samples at low levels. Our method employs a simple lyophiliza- tion followed by solvent extraction analysis using LC- MS/MS. The lower limit of method validation and limit of determination was in the μg·L−1 range with coefficient of variation values of typically < 8%. Additionally, the effect of DE on ions formation and distribution of base peak were studied. The fragmentation schemes were well illustrated. These results reveal the m/z at 350 was ob- tained at DE 10 V, while m/z at 197and 97 were obtained  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC at DE 20, and 40, respectively for chlorpyrifos. The re- sult clearly indicates that the m/z 125 and m/z 97 were used for confirmation of chlorpyrifos in water samples. Additionally, the m/z at 270,110, 83 and 70 were ob- tained at DE 10, 30, 60 and 70 respectively for alachlor. The 83, 110 and 70 were used as confirmatory ions. In similar fashion the base peak of dimethoate were ob- served. The m/z at 229, 88, 124, 79 and 63 were obtained at DE 10, 20, 40, 70 and 80, respectively. The ions 88, 79 and 63 were confirmatory ions. Similarly, m/z at 215, 116, 72, 70 and 60 were obtained at DE 10, 20, 30, 40 and 80, respectively. The ions obtained at 72, 79 and 60 were used as confirmatory ions of metribuzin. Thirteen water samples were collected from the different parts of the urban city, and they were each analyzed showing residual pesticide at detectable concentrations. These data indicate that drinking water (MC) is less contami- nated with pesticide residues than that of bore water at lower levels. We plan to further explore pesticide residue analysis in marketed water samples. Additionally, we will apply this method for measuring pesticides in water samples collected from different places in India. 8. Acknowledgements The authors are thankful to the Indian Council of Medi- cal Research for financial assistance. They would also like to take this great opportunity to express their heart- felt gratitude to the Director General of the Indian Coun- cil of Medical Research for granting an opportunity to work on this project. They are extremely thankful to the Director of the National Institute of Nutrition (Hydera- bad) for giving the necessary facilities and kind support to carry out this work at the National Institute of Nutri- tion, Hyderabad. The authors are thankful to all the tech- nical staff especially Mr Vasudev, scientific staff and the statistician for the technical and statistical support during this work. 9. References [1] P. C. Abhilash and N. Singh, “Pesticide Use and Applica- tion: An Indian Scenario,” Journal of Hazardous Mate- rials, Vol. 165, No. 1-3, 2009, pp. 1-12. HHUdoi:10.1016/j.jhazmat.2008.10.061U [2] V. K. Bhatnagar, “Pesticides Pollution: Trends and Pers- pectives,” ICMR Bulltin, Vol. 31, 2001, pp. 87-88. [3] D. Atkinson, F. Burnett, G. N. Foster, A. Litterick, M. Mullay and C. A. Watson, “The Minimization of Pesti- cide Residues in Food: A Review of the Published Lite- rature,” Food Standards Agency, London, 2003. [4] B. Kumari, R. Gulati, T. S. Kathpal, “Monitoring of Pes- ticidal Contamination in Honey,” The Korean Journal of Apiculture, Vol. 18, No. 2, 2003, pp. 155-160. [5] B. Kumari, V. K. Madan, J. Singh, S. Singh and T. S. Kathpal, “Monitoring of Pesticidal Contamination of Farmgate Vegetables from Hisar Environmental Moni- toring and Assessment,” Earth and Environmental Science, Vol. 90, No. 1-3, 2004, pp. 65-77. HHUdoi:10.1023/b:emas.0000003566.63111U [6] B. Kumari, J. Singh, S. Singh and T. S. Kathpal, “Moni- toring of Butter and Ghee (Clarified Butter Fat) for Pesti- cidal Contamination from Cotton Belt of Haryana, India,” Environmental Monitoring and Assessment, Vol. 105. No. 1-3, 2005, pp. 111-120. HHUdoi:10.1007/s10661-005-3159-2U [7] B. Kumari, T. S. Kathpal and E. M. Assess. “Monitoring of Pesticide Residues in Vegetarian Diet,” Environmental Monitoring Assessment, Vol. 151, 2009, p. 1926. [8] B. Kumari, V. K. Madan and T. S. Kathpal, “Monitoring of Pesticide Residues in Fruits,” Environmental Monitor- ing and Assessment, Vol. 123, No. 1-3, 2006, pp. 407-412. HHUdoi:10.1007/s10661-006-1493-7U [9] B. Kumari, V. K. Madan, R. Kumar and T. S. Kathpal, “Monitoring of Seasonal Vegetables for Pesticide Resi- dues,” Environmental Monitoring and Assessment, Vol. 74, No. 3, 2002, pp. 263-270. HHUdoi:10.1023/A:1014248827898U [10] S. N. Sinha, R. Pal, A. Dewan, M. M. Mansuri and H. N. Saiyed, “Effect of Dissociation Energy on Ion Formation and Sensitivity of An Analytical Method for Determina- tion of Chlorpyrifos in Human Blood, Using Gas Chro- matography-Mass Spectrometer (GC-MS in MS/MS),” International Journal of Mass Spectrometry, Vol. 253, No. 1-2, 2006, pp. 48-57. HHUdoi:10.1016/j.ijms.2006.02.020U [11] V. K. Dua, G. Rosy, S. N. Sinha and A. P. Das, “Alleth- rin in the Air during the Use of a Heated Mosquito Re- pellent Mat,” Bulletin of Environmental Contamination and Toxicology, Vol. 75, No. 4, 2005, pp. 747-750. HHUdoi:10.1007/s00128-005-0814-9U [12] A. Dewan, V. K. Bhatnagar, M. L. Mathur, T. Chakma, R. Kashyap and G. H. Sadhu, et al., “Repeated Episodes of Endosulfan Poisoning,” Journal of Toxicology-Clinical Toxicology, Vol. 42, No. 4, 2004, pp. 1-7. HHUdoi:10.1081/CLT-120039542UHH [13] S. N. Sinha, T. S. Patel, N. M. Desai, M. M. Mansuri, A. Dewan and H. N. Saiyed, “GC-MS Study of Endosulfan in Biological Samples,” Asian Journal of Chemistry, Vol. 16, No. 3-4, 2004, pp. 1685-1690. [14] S. N. Sinha, “Effect of Dissociation Energy: Signal to Noise Ratio on Ion Formation and Sensitivity of Analyti- cal Method for Quantification and Confirmation of Tria- zofos in Blood Samples Using Gas Chromatography-Mass Spectrometer (GC-MS/MS),” International Journal of Mass Spectrometry, Vol. 296, No. 1-3, 2010, pp. 47-52. HHUdoi:10.1016/j.ijms.2010.08.014U [15] P. Paya, M. Anastassiades, D. Mack, I. Sigalova, B. Tas- delen and J. Oliva, et al., “Analysis of Pesticide Residues Using the Quick Easy Cheap Effective Rugged and Safe (QuEChERS) Pesticide Multiresidue Method in Combi- nation with Gas and Liquid Chromatography and Tandem Mass Spectrometric Detection,” Analytical Bioanalytical  S. N. SINHA ET AL. Copyright © 2011 SciRes. AJAC Chemistry, Vol. 389, No. 6, 2007, pp. 1697-1714. HHUdoi:10.1007/s00216-007-1610-7U [16] D. B. Barr, J.R. Barr, V. L. Maggio, Jr. R. D. Whitehead, M. A. Sadowski and R. Whyatt, et al. “A Multi-Analyte Method for the Quantification of Contemporary Pesti- cides in Human Serum and Plasma Using High-Resolution Mass Spectrometry,” Journal of Chromatography B, Vol. 778, 2002, pp. 9-11. [17] S. N. Sinha, K. Vasudev, M. V. Rao and M. Odetokun, “Quantification of Organophosphate Insecticides in Drin- king Water in Urban Areas Using Lyophilization and High-Performance Liquid Chromatography-Electrospray Ionization-Mass Spectrometry Techniques,” International Journal of Mass Spectrometry, Vol. 300, 2011, pp. 12-20. HHUdoi:10.1016/j.ijms.2010.11.006U [18] S. N. Sinha, V. K. Bhatnagar, P. Doctor, G. S. Toteja, N. P. Agnihotri and R. L. Kalra, “A Novel Method for Pesti- cide Analysis in Refined Sugar Samples Using a Gas Chromatography-Mass Spectrometer (GC-MS/MS) and Simple Solvent Extraction Method,” Food Chemistry, Vol. 126, No. 1, 2011b, pp. 379-386. HHUdoi:10.1016/j.foodchem.2010.10.110U [19] K. D. Miller and P. Milne, “Determination of Low-Level Pesticides Residues in Soft Drinks and Sport Drinks by Liquid Chromatography with Tandem Mass Spectrometry: Collaborative Study,” Journal of AOAC International, Vol. 91 No. 1, 2008, pp. 181-201. [20] K. D. Miller and P. Milne, “Determination of (0.5 µg/L) in Soft Drinks and Sport Drinks by Gas Chro Low-Level Pesticides Residues Monograph with Mass Spectrometry: Collaborative Study,” Journal of AOAC International, Vol. 91, No. 1, 2008b, pp. 202-236. [21] A. Sarkar and R. Gupta Sen, “Determination of Orga- nochlorine Pesticides in Indian Coastal Water Using a Mooredin-Situ Sampler,” Water Research, Vol. 23, No. 8, 1989, pp. 975-978. HHUdoi:10.1016/0043-1354(89)90170-XU [22] N. Bakore, P. J. John and P. Bhatnagar, “Organochlorine Pesticide Residues in Wheat and Drinking Water Samples from Jaipur, Rajasthan, India,” Environmental Monitor- ing and Assessment, Vol. 98, No. 1-3, 2002, pp. 381-389. HHUdoi:10.1023/B:EMAS.0000038197.76047.83U [23] N. Sankararamakrishnan, A. K. Sharma and R. Sanghi, “Organochlorine and Organophosphorous Pesticide Re- sidues in Ground Water and Surface Waters of Kanpur, Uttar Pradesh, India,” Environment International, Vol. 31, No. 1, 2005, pp. 113-120. HHUdoi:10.1016/j.envint.2004.08.001U
|