Journal of Computer and Communications
Vol.04 No.15(2016), Article ID:72316,9 pages
10.4236/jcc.2016.415005
A Novel Prediction Method of Protein Structural Classes Based on Protein Super-Secondary Structure
Longlong Liu, Jing Cui*, Jie Zhou
Department of Mathematical Sciences, Ocean University of China, Qingdao, China




Received: September 26, 2016; Accepted: November 25, 2016; Published: November 28, 2016
ABSTRACT
At present, the feature extraction of protein sequences is the most basic issue to predict protein structural classes and is also the key problem to decide the quality of prediction. In order to predict protein structural classes accurately, we construct a 14-dimensional feature vector based on protein secondary and super-secondary structure information to reflect the content and spatial ordering of the given protein sequences. Among the vector, seven features about  -helix bundle, hairpin
-helix bundle, hairpin 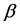 motifs, Rossman folds,
motifs, Rossman folds,  -plaits and other super-secondary structure information are first proposed in our paper. Experiments show that our method improves overall accuracy of lower similarity datasets 1189 and 640 by 0.9% - 3.8% and 0.5% - 4.2% respectively compared with other methods and has a competitive advantage for predicting proteins in
-plaits and other super-secondary structure information are first proposed in our paper. Experiments show that our method improves overall accuracy of lower similarity datasets 1189 and 640 by 0.9% - 3.8% and 0.5% - 4.2% respectively compared with other methods and has a competitive advantage for predicting proteins in 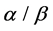 and
and 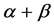 classes.
classes.
Keywords:
Proteins, Super-Secondary Structure, Low Similarity, New Features

1. Introduction
Modern molecular biological studies indicate that the function of a protein is determined by its spatial structure. Therefore, it’s very important to predict the structural classes of the newly discovered protein accurately [1]. The types and order of 20 kinds of amino acids are the basic information in a protein sequence, then a large number of initial prediction methods use features based on amino acid composition (AAC) and the position information, which are the easiest and most intuitive methods [2] [3] [4] [5]. However, these methods have advantage to predict protein sequences with high degree of similarity and have disadvantage to predict protein sequences with similarity less than 40%. Due to the low-similarity protein sequences always have high-similarity secondary structural contents and spatial arrangements, so a large number of researchers tried to extract features from the secondary structure of proteins predicted by PSI-PRED[6].Under the guidance of this idea, SPRED model [2] and MODAS model [7] were constructed. Currently novel computational prediction methods [8] [9] build feature vectors by using the protein secondary structure information and protein sequences are predicted into four classes (All-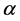 , All-
, All-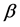 ,
, 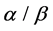 ,
, ) using SVM (support vector machine) classifier. Overall prediction accuracy on several datasets of these methods reach to 80% - 90%, but the prediction of
) using SVM (support vector machine) classifier. Overall prediction accuracy on several datasets of these methods reach to 80% - 90%, but the prediction of 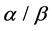 and
and  classes is not ideal, especially for
classes is not ideal, especially for 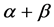 class with accuracy just about 70%.
class with accuracy just about 70%.
In order to improve the prediction accuracy of  and
and 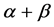 classes, we will extract seven different features reflecting general contents and spatial arrangements of the secondary structural elements from super-secondary structure of a given protein sequence. We use SVM to predict protein structural classes after features are extracted. Finally, our paper evaluated our model objectively.
classes, we will extract seven different features reflecting general contents and spatial arrangements of the secondary structural elements from super-secondary structure of a given protein sequence. We use SVM to predict protein structural classes after features are extracted. Finally, our paper evaluated our model objectively.
2. Materials and Methods
2.1. Materials
Currently proposed methods widely use low-similarity benchmark datasets named 1189, FC699, 640 and 25PDB.The sequence similarity of 25PDB [10], 1189 [11], FC699 [1] and 640[12] are lower than 25%, 40%, 40% and 25% respectively. In order to compare with other methods, we choose 25PDB dataset as training set for SVM classifier, while the other datasets 1189, FC699 and 640 are test sets.
2.2. Feature Vector
Nowadays, a lot of methods are used to predict amino acids sequences into secondary structural sequences (SSS) constructed by three secondary structural elements,  -helix (H),
-helix (H), 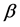 -strand (E), and random coil(C). Firstly, we obtain corresponding secondary structural sequences by PSI-PRED (version 2.6) [6]. It’s difficult to distinguish
-strand (E), and random coil(C). Firstly, we obtain corresponding secondary structural sequences by PSI-PRED (version 2.6) [6]. It’s difficult to distinguish 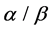 and
and 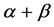 classes for both of them contain
classes for both of them contain  -helices and
-helices and  -strands.
-strands.  -helices and
-helices and  -strands are usually separated in
-strands are usually separated in  class, while they are usually interspersed in
class, while they are usually interspersed in  class. In order to better represent the distribution of
class. In order to better represent the distribution of  -helices and
-helices and  -strands, every segment H, E and
-strands, every segment H, E and  in secondary structure sequence(SSS) is replaced by
in secondary structure sequence(SSS) is replaced by ,
,  and
and  respectively, the new sequence is secondary structure segment sequence(SS), all element
respectively, the new sequence is secondary structure segment sequence(SS), all element  are removed from SS to form a new sequence represented by SSW[13].
are removed from SS to form a new sequence represented by SSW[13].
Our novel method mapped each protein sequence into a 14-dimentional vector that can be defined as
 (1)
(1)
where T is a transpose symbol,  (
( ) is one of the features in feature vector P. The elements
) is one of the features in feature vector P. The elements  of Equation (1) are based on super- secondary structure information and are first proposed in this paper. What’s more,
of Equation (1) are based on super- secondary structure information and are first proposed in this paper. What’s more,  are proposed specially to make a distinction between
are proposed specially to make a distinction between  and
and  classes. A number of secondary structure elements interact with each other and form a regular combination of secondary structure, which acts as a structural member of tertiary structure in much protein and is known as super-secondary structure (motif). Some super-secondary structures are related to specific functions and there are three basic forms of combination:
classes. A number of secondary structure elements interact with each other and form a regular combination of secondary structure, which acts as a structural member of tertiary structure in much protein and is known as super-secondary structure (motif). Some super-secondary structures are related to specific functions and there are three basic forms of combination: ,
,  and
and . In order to tap the structural characteristics of each class, we extract several typical features of folds and combinations.
. In order to tap the structural characteristics of each class, we extract several typical features of folds and combinations.
1) The easiest  structure is hairpin
structure is hairpin  motif which is connected by a short loop. Multiple hairpin
motif which is connected by a short loop. Multiple hairpin  motifs together will form a stable and widespread
motifs together will form a stable and widespread  -turns, so extracting the number of hairpin
-turns, so extracting the number of hairpin  motifs (defined as
motifs (defined as ) are very meaningful. Super-secondary structure
) are very meaningful. Super-secondary structure  is a
is a  helix bundle and is often formed by two intertwined spiral parallel or ant parallel
helix bundle and is often formed by two intertwined spiral parallel or ant parallel ?helices. Structure
?helices. Structure  named Rossman folds is one of the most special structures of
named Rossman folds is one of the most special structures of  class. So we extract the number of super-secondary structure
class. So we extract the number of super-secondary structure  (
( ) and
) and  (
( ) as features. Then the corresponding features can be defined as
) as features. Then the corresponding features can be defined as ,
,  ,
, .
.
2) In proteins of  class, -strands and
class, -strands and  -helices are parallel,
-helices are parallel,  -helix and
-helix and  - sheets appear alternatively, such as
- sheets appear alternatively, such as . Proteins of
. Proteins of  class are two types. Such as “
class are two types. Such as “ -plaits” in which
-plaits” in which  -helices and
-helices and  -strands appear alternatively, this characteristic is same to proteins in
-strands appear alternatively, this characteristic is same to proteins in  class. In
class. In  -plaits, the alternative form of
-plaits, the alternative form of  -helices and
-helices and  -strands is
-strands is  or
or  . In another type
. In another type  -helices and
-helices and  -strands are separated. Therefore the number of occurrences of
-strands are separated. Therefore the number of occurrences of  segments (
segments ( ) and
) and  (
( ) segments is an obvious characteristic to distinguish proteins in
) segments is an obvious characteristic to distinguish proteins in  and
and  classes.
classes.
Features mentioned above can be represented as:
 ,
, 
where  and
and  are the maximal lengths of
are the maximal lengths of  segments and
segments and  segments in SSW.
segments in SSW.
3) In proteins of the  class,
class,  -helices and
-helices and  -strands alternate more frequently than in proteins of the
-strands alternate more frequently than in proteins of the  class. Based on this characteristic, we can design a feature
class. Based on this characteristic, we can design a feature , where
, where  is the alternating frequency of
is the alternating frequency of  -helices and
-helices and  -strands in SSS.
-strands in SSS.
4) Because the length of the secondary structural segments will affect the assignment of the structural class, we define new features
 and
and 
where  and
and  are the maximal lengths of
are the maximal lengths of  -helix and
-helix and  -strand segments in SSS.
-strand segments in SSS.
5) Position information of SS is also the deciding factor. Herein, the position of a segment is defined as a starting position of the segment. The corresponding features can be defined as
 ,
, 
where  (
( ) is the starting order of the
) is the starting order of the  -helix (
-helix ( -strand) segment in SSS,
-strand) segment in SSS,  and
and  are the occurrences of
are the occurrences of  -helix and
-helix and  -strand segments in SSS.
-strand segments in SSS.
6) To reflect the position information of protein secondary structure, two features  can be defined as
can be defined as

where  and
and  are the j-th order of H and E in SSS,
are the j-th order of H and E in SSS,  and
and  are the number of H and E in protein secondary structure sequence (SSS).
are the number of H and E in protein secondary structure sequence (SSS).
7) The probability of content C can be ignored due to the sum of the three probabilities of H, E and C is 1 [1]. Hence, the two features are expressed as

where ,
, .
.
2.3. Classification Algorithm Construction
Protein secondary structure prediction is a multiclass classification problem. With high prediction accuracy, support vector machine (SVM) has been widely used for protein secondary structure classification [4] [5]. Here we use of the "one to one" multiclass classification method that construct a multiclass classifier by combining six binary classifiers. We choose Gaussian radial basis function (RBF) as the kernel function for SVM [14]. Using a grid search on the training set (25PDB) by tenfold cross-validation, we can find out the penalty parameter  and kernel parameter
and kernel parameter , the final parameters are
, the final parameters are ,
, .
.
2.4. Performance Measures
In this paper, we use an independent testing dataset cross-validation. There are many indicators to evaluate model’s performance, sensitivity (Sens), specificity (Spec), Matthew’s correlation coefficient (MCC) and overall accuracy (OA) are widely used in protein structure prediction [15]. The total number of proteins, classes and proteins in k-th
class are denoted by N, k and  respectively, so
respectively, so . Usually, four parameters are used by studies for examining a predictor’s effectiveness:
. Usually, four parameters are used by studies for examining a predictor’s effectiveness:
The number of proteins which is correctly predicted as kth class and non-kth class are denoted by  and
and . The number of proteins which is incorrectly predicted as kth class and non-kth class are denoted by
. The number of proteins which is incorrectly predicted as kth class and non-kth class are denoted by  and
and . Where
. Where ,
, . Using these parameters, we can obtain Equation (2):
. Using these parameters, we can obtain Equation (2):

(2)
3. Results and Discussion
3.1. Structural Class Prediction Accuracies
In our experiment, we use 25PDB dataset as a training set and other three datasets as testing sets. We not only report the values of Sens, Spec, MCC and overall accuracy (OA) of every structural class of testing set, but also report the average of Sens, Spec, MCC and overall accuracy (OA). The detail results can be seen in Table 1. The overall accuracy is more than 84% for each test set and it reaches 90% for FC699 dataset. What’s more, the average overall accuracy of 3 test sets is up to 86.6%. The Sens and

Table 1. The prediction quality of our method on test datasets.
MCC values of  class are the lowest. This is not strange because
class are the lowest. This is not strange because  class is complicated and it not only contains
class is complicated and it not only contains  and
and  classes but also contains
classes but also contains  -plaits structure, so the prediction accuracy of
-plaits structure, so the prediction accuracy of  class is lower.
class is lower.
3.2. Feature Vector Analysis
To better verify the effect of the new proposed seven features, we do the following experiment with FC699, 1189 and 640 datasets. The comparison of obtained accuracies between our method including 14 features and our method including 7 features can be seen in Table 2. After added new features, the average overall accuracy increases by 2.6% up to 86.6%. For FC699 dataset, the overall accuracy and accuracies of All- , All-
, All- and
and  classes are improved by 2.7%, 1.5%, 4.4% and 2.7% respectively. For 1189 and 640 datasets, the overall accuracy increases by 2.6% and 2.3%, respectively. However, the results of
classes are improved by 2.7%, 1.5%, 4.4% and 2.7% respectively. For 1189 and 640 datasets, the overall accuracy increases by 2.6% and 2.3%, respectively. However, the results of  and
and  classes are not obvious because of the interference of other classes [13].
classes are not obvious because of the interference of other classes [13].
To further validate effect of super-secondary structure features, we do experiment just on proteins in  and
and  classes with a 14-dimensional feature vectors. The 25PDBS, FC699S, 1189S and 640S sets are the subsets formed by removing all the proteins in the All-
classes with a 14-dimensional feature vectors. The 25PDBS, FC699S, 1189S and 640S sets are the subsets formed by removing all the proteins in the All- and All-
and All- classes from 25PDB, FC699, 1189 and 640 datasets respectively. Hence, we use the 25PDBS to train SVM classifier and other subsets to test. The parameters
classes from 25PDB, FC699, 1189 and 640 datasets respectively. Hence, we use the 25PDBS to train SVM classifier and other subsets to test. The parameters  and
and  (
( ,
, ) are selected by tenfold cross-validation on 25PDBS with a grid search method. The corresponding experimental results are shown in Table 3. In Table 3, the overall accuracies of all datasets predicted by our method are higher than 80%. The overall accuracies and accuracies of
) are selected by tenfold cross-validation on 25PDBS with a grid search method. The corresponding experimental results are shown in Table 3. In Table 3, the overall accuracies of all datasets predicted by our method are higher than 80%. The overall accuracies and accuracies of  class predicted by our method are the highest compared with other competitive methods. The prediction accuracies of all structural classes are higher than 90% on FC699S subset. The accuracies of
class predicted by our method are the highest compared with other competitive methods. The prediction accuracies of all structural classes are higher than 90% on FC699S subset. The accuracies of  class and the overall accuracy are increased by 9.2% - 27.4% and 2.4% - 7.3% on 1189S respectively. For 640S, the accuracies of
class and the overall accuracy are increased by 9.2% - 27.4% and 2.4% - 7.3% on 1189S respectively. For 640S, the accuracies of  class and the overall accuracy are improved by 2.9% - 11.7% and 0.6% - 5.2%
class and the overall accuracy are improved by 2.9% - 11.7% and 0.6% - 5.2%
Table 2. Comparison of the accuracies between the method including 14 features and one including only 7 features.
Table 3. The accuracy of differentiating between the  and
and  class.
class.
respectively, however, the accuracy of  class is consistent with the result produced by PKS-PPSC model. Experimental results show that our method for predicting proteins in
class is consistent with the result produced by PKS-PPSC model. Experimental results show that our method for predicting proteins in  and
and  classes is very effective.
classes is very effective.
3.3. Comparison with Other Prediction Method
It’s known to all, SCPRED [2] and MODAS [7] are famous in predicting protein secondary structure and are often used as baseline for comparison. From Table 4 we can see, our method improves the overall accuracies by 0.9% - 3.8% and 0.5% - 4.2% on 1189 and 640 datasets compared with other competing prediction methods including SCPRED and MODAS. And only for FC699 dataset, the overall accuracy is lower than Kong et al. and is not the highest, but it is increased by 0.8% - 2.9% compared with the rest methods. Compared with model SCPRED and the method of Liu and Jia, the overall accuracy predicted by our method is improved by 2.9% and 0.8% on FC699 dataset, respectively, besides the accuracy of  class is increased by 4.8% and 19.5%. Our method obtains the highest accuracies for the All-
class is increased by 4.8% and 19.5%. Our method obtains the highest accuracies for the All- and
and  classes which reach to 87.8% and 79.3% on 1189 dataset and the overall accuracy is the highest than other exiting methods. For 640 dataset, the overall accuracy and the accuracy of All-
classes which reach to 87.8% and 79.3% on 1189 dataset and the overall accuracy is the highest than other exiting methods. For 640 dataset, the overall accuracy and the accuracy of All- class are the highest.
class are the highest.
Therefore our method extracting features based on super-secondary structure has the ability to reflect the realistic characteristics of proteins more accurately. Specially, our method improved the accuracies of  and
and  classes greatly. According Table 4, we find our method is not always the best. The reason is that some methods not only extract features based on protein secondary structure but also combine other information. In contrast, our method is aimed to predict secondary structural classes by extracting features more effectively just on the basis of secondary structure.
classes greatly. According Table 4, we find our method is not always the best. The reason is that some methods not only extract features based on protein secondary structure but also combine other information. In contrast, our method is aimed to predict secondary structural classes by extracting features more effectively just on the basis of secondary structure.
Table 4. Performance comparison of difference methods on 3 test datasets.
4. Conclusion
In this paper, a novel method is proposed based on protein super-secondary structure information. Seven new features related to  -helix bundle, hairpin
-helix bundle, hairpin  motifs, Rossman folds,
motifs, Rossman folds,  -plaits and other information are very useful to predict protein secondary structural classes. We adopt advanced SVM classifier which use little computational time and space, is accurate and is very suitable for large-scale protein sequence databases. Finally, experimental results show that this new prediction method not only improve the overall prediction accuracy but also improve the accuracies of all structural classes, especially, the accuracies of
-plaits and other information are very useful to predict protein secondary structural classes. We adopt advanced SVM classifier which use little computational time and space, is accurate and is very suitable for large-scale protein sequence databases. Finally, experimental results show that this new prediction method not only improve the overall prediction accuracy but also improve the accuracies of all structural classes, especially, the accuracies of  and
and  classes are improved greatly. Hence, the new extracted features can reflect the characteristics of different structural classes more accurately and our method is more effective than previous methods.
classes are improved greatly. Hence, the new extracted features can reflect the characteristics of different structural classes more accurately and our method is more effective than previous methods.
Acknowledgements
The authors would like to thank all of the researchers who made publicly available data used in this study and thank the National Natural Science Foundation of China (No: 61303145) for the support to this work.
Cite this paper
Liu, L.L., Cui, J. and Zhou, J. (2016) A Novel Prediction Method of Protein Structural Classes Based on Protein Super-Secondary Structure. Journal of Computer and Communications, 4, 54-62. http://dx.doi.org/10.4236/jcc.2016.415005
References
- 1. Kurgan, L.A., Zhang, T., Zhang, H., Shen, S. and Ruan, J. (2008) Secondary Structure-Based Assignment of the Protein Structural Classes. Amino Acids, 35, 551-564. http://dx.doi.org/10.1007/s00726-008-0080-3
- 2. Kurgan, L., Cios, K. and Chen, K. (2008) Scpred: Accurate Prediction of Protein Structural Class for Sequences of Twilight-Zone Similarity with Predicting Sequences. BMC Bioinformatics, 9, 815-818. http://dx.doi.org/10.1186/1471-2105-9-226
- 3. Olyaee, M.H., Yaghoubi, A. and Yaghoobi, M. (2016) Predicting Protein Structural Classes Based on Complex Networks and Recurrence Analysis. Journal of Theoretical Biology, 404, 375-382. http://dx.doi.org/10.1016/j.jtbi.2016.06.018
- 4. Zhang, S. (2015) Accurate Prediction of Protein Structural Classes by Incorporating PSSS and PSSM into Chou’s General PseAAC. Chemometrics & Intelligent Laboratory Systems, 142, 28-35. http://dx.doi.org/10.1016/j.chemolab.2015.01.004
- 5. Ding, S., Yan, L., Shi, Z. and Yan, S. (2014) A Protein Structural Classes Prediction Method Based on Predicted Secondary Structure and Psi-Blast Profile. Biochimie, 97, 60-65. http://dx.doi.org/10.1016/j.biochi.2013.09.013
- 6. Jones, D.T. (1999) Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. Journal of Molecular Biology, 292, 195-202. http://dx.doi.org/10.1006/jmbi.1999.3091
- 7. Mizianty, M.J. and Lukasz, K. (2009) Modular Prediction of Protein Structural Classes from Sequences of Twilight-Zone Identity with Predicting Sequences. BMC Bioinformatics, 10, 1-24. http://dx.doi.org/10.1186/1471-2105-10-414
- 8. Liu, T. and Jia, C. (2010) A High-Accuracy Protein Structural Class Prediction Algorithm Using Predicted Secondary Structural Information. Journal of Theoretical Biology, 267, 272-275. http://dx.doi.org/10.1016/j.jtbi.2010.09.007
- 9. Zhang, S., Ding, S. and Wang, T. (2011) High-Accuracy Prediction of Protein Structural Class for Low-Similarity Sequences Based on Predicted Secondary Structure. Biochimie, 93, 710-714. http://dx.doi.org/10.1016/j.biochi.2011.01.001
- 10. Kurgan, L.A. and Homaeian, L. (2006) Prediction of Structural Classes for Protein Se-quences and Domains—Impact of Prediction Algorithms, Sequence Representation and Homology, and Test Procedures on Accuracy. Pattern Recognition, 39, 2323-2343. http://dx.doi.org/10.1016/j.patcog.2006.02.014
- 11. Wang, Z. and Zheng, Y. (2000) How Good Is Prediction of Protein Structural Class by the Component-Coupled Method? Proteins Structure Function & Bioinformatics, 38, 165-175. http://dx.doi.org/10.1002/(SICI)1097-0134(20000201)38:2<165::AID-PROT5>3.0.CO;2-V
- 12. Yang, J.Y., Peng, Z.L. and Xin, C. (2010) Prediction of Protein Structural Classes for Low- Homology Sequences Based on Predicted Secondary Structure. BMC Bioinformatics, 11, 1-10. http://dx.doi.org/10.1186/1471-2105-11-s1-s9
- 13. Liang, K., Zhang, L. and Lv, J. (2014) Accurate Prediction of Protein Structural Classes by Incorporating Predicted Secondary Structure Information into the General Form of Chou's Pseudo Amino Acid Composition. Journal of Theoretical Biology, 344, 12-18. http://dx.doi.org/10.1016/j.jtbi.2013.11.021
- 14. Zheng, Y., Bailey, T.L. and Teasdale, R.D. (2005) Prediction of Protein b-Factor Profiles. Proteins Structure Function & Bioinformatics, 58, 905-912. http://dx.doi.org/10.1002/prot.20375
- 15. Baldi, P., Brunak, S., Chauvin, Y., Andersen, C.A. and Nielsen, H. (2000) Assessing the Accuracy of Prediction Algorithms for Classification: An Overview. Bioinformatics, 16, 412- 424. http://dx.doi.org/10.1093/bioinformatics/16.5.412