Open Access Library Journal
Vol.03 No.11(2016), Article ID:72047,7 pages
10.4236/oalib.1103091
An Observation of One Case of Adult Partial Atrio-Ventricular Defect
Georges Kinda1,2, Koudougou Jonas Kologo1,3, Aimé Bama2, Georges Rosario Christian Millogo1,3, Salimata Traoré2, Lassina Dao1,2, Solange Ouédraogo Yougbaré1,2, Aïssata Kaboré1,2, Sonia Kaboret1,2, Kisito Nagalo1,2, Laure Toguyéni Tamini1,2, Relwendé Aristide Yaméogo4, Fla Kouéta1,2, Diarra Yé1,2, Léonie Claudine Lougué Sorgho1,5, Patrice Zabsonré1,3
1University Department of Medical Science (UFR/SDS), University Ouaga 1 Professor Joseph KI-ZERBO, Ouagadougou, Burkina Faso
2Medical Pediatric Department of University Health Center Charles De Gaulle, Ouagadougou, Burkina Faso
3Cardiology Department of University Health Center Yalgado Ouédraogo, Burkina Faso
4Medical Department of Regional Health Center, Koudougou, Burkina Faso
5Laboratory of Medical Imagery of University Health Center Charles De Gaulle, Ouagadougou, Burkina Faso

Copyright © 2016 by authors and Open Access Library Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: September 30, 2016; Accepted: November 13, 2016; Published: November 16, 2016
ABSTRACT
The newborn with partial atrio-ventricular defect is born quickly normal without functional signs, and then cardiac insufficiency appears in the first weeks of life respiratory unrests. The spontaneous evolution makes itself toward a complication of the shunts left-right with cardiac insufficiency, Osler illness, the death or toward the pulmonary obstructive illness about 6 months old. In order to fear the evolutionary possibilities and debate difficulties of hold in charge in countries with limited resources such as Burkina Faso, we return a clinic observation of a case of partial atrio- ventricular defect in a sixty years old man.
Subject Areas:
Cardiology
Keywords:
Atrioventricular Canal, Congenital Heart Diseases, Echocardiography, Pulmonary Arterial Hypertension, Ouagadougou

1. Introduction
We call abusively atrio-ventricular canal, cardiac diseases due to faulty development of the atrio-ventricular canal; in the major form (complete atrio-ventricular canal: 20% of the atrio-ventricular canals) all the elementary lesions are present; with the incomplete forms, the evolution of the embryo is longer with a late completion of the development of the embryo: ventricular septal defect, auricle ostium primum defect + cardiac mitral valve cleft lip gives a partial atrio-ventricular canal (80% of the atrio-ventricular canals), isolated ostium primum atrial septal defect, isolated abnormalities of the tricuspid valve; no primum septum (single auricle ) major form of atrial septal defect). We report a case of adult partial atrio-ventricular defect where consent was obtained from the patient.
2. Observation
It is a case of a sixty years old man who was examined for dyspnea on exertion phase III of NYHA and a cough with white expectorations evolving through relapsing-remitting for more than ten (10) years but which worsened a year ago. His brother noticed that he has always been sick during his childwood and was often exempted from family chores. At the clinical examination, we could notice a retarded development of height and weight with a weight of 45 kg, a height of 148 cm representing a body mass index of 20.54 kg/m2, a high-pressure of 110/65 mmHg, a saturation in oxygen with natural air of 95%.
The thorax is normal, the heart beats are regular and slow with a frequency of 50 cycles per minutes associated with a systolic murmur at the fifth (5th) left inter-rating space having an intensity of 3/6, with apexo-axilliary irradiation, a xiphoidian systolic murmur 3/6 and a systolic murmur at the third (3rd) left inter-rating space having an intensity of 3/6 with a pulmonary first sound bright. We could also notice a spontaneous jugular turgescence and a farm and regular hepatomegaly (hepatic arrow = 13 cm) ands wollen legs. There is no sign of finger cubbing, of cyanosis and no sign in favor of a trysomy of chromosome 21.
At the biological level, the complete blood count, the serum electrolytes, the hepatic transaminases and the blood creatinine were normal.
The front chest X-ray shows a cardiomegaly with a cardiothoracic indice which was sharp, sub-diapragmatic and which equals 0.72, a double bump aspect of the right inferior arc, and a left-overhanging middle arc, a pulmonary hypervascularisation with vascular redistribution towards the tops (Figure 1).
The eletrocardiography shows a regular atrial fibrillation with a frequency of 49 cycles/mn, an undetermined axis and a complete right bundle branch block (Figure 2).
The echography of the heart shows a non-dilated left ventricle (TDLVD = 28.4 mm, TSLVD = 20.7 mm) with a paradoxal ventricular septum. The left ventricular lining are normal, the Ejection Fraction is estimated to 54% at Simpson. We could notice the presence of a 14 mm diameter inlet ventricular septal defect with a maximum gradient left ventricle-right ventricle of 4.8 mmHg, a maximum speed of 1.10 m/s and a double atrioventricular valve having a thick and soft mitral valve which causes a grade II mitral insufficiency (maximum gradient left ventricle-left auricle = 74 mmHg, maximum speed = 4.29 m/s). The mitral surface is estimated to 2.3 cm2 with planimetrics. The

Figure 1. A front chest X-ray showing a cardiomegaly with a cardiothoracic indice of 0.72, a right lower contour with a double bump aspect.
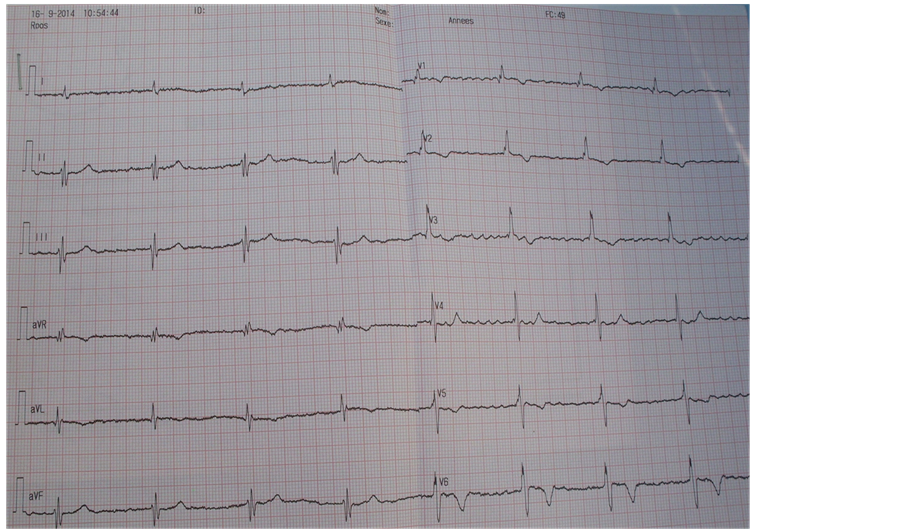
Figure 2. The surface eletrocardiography shows a regular auricle fibrillation with a frequency of 49 cycles/mn, an undetermined countour and a complete right bundle branch block.
aortic sigmoid are thick and soft with a grade I aortic failure. The aortic surface is estimated to 1.83 cm2 with planimetrics. The right ventricle is dilated (TDRVD = 48.6 mm), the right ventricle linings are normal. We could notice a dysfunctioning of right ventricle-left auricle and left ventricle-right auricle of weak gradient (Figure 3 and Figure 4).

Figure 3. Ventricular septal defect in trans thoracic echography.

Figure 4. Trans thoracic echography with color flow shows mitral and tricuspid insuficiency.
The left auricle is dilated (diameter of 52 mm, surface of 23 cm2), the left auricle/Ao ratio is 2.08; The right auricle is dilated too (diameter of 54 mm, 24 cm2 of surface). We could notice a large Ostium Primum atrial septal defect of a diameter of 25.9 cm without a perceptible shunt between the two auricles. The pulmonary sigmoids are calcified without stenosis but with a grade II pulmonary insufficiency of maximum gradient right ventricle-pulmonary artery equalin 11.4 mmHg and a maximum speed of 1.69 m/s. The tricuspid is thick without stenosis and a grade II tricuspid dysfonctioning (max gradient right ventricle-right auricle = 34 mmHg, max speed 2.91 m/s). The vena cava inferior is dilated (22 mm), the systolic pulmonary artery pressure is estimated to 49 mmHg from the tricuspid insufficiency. Moreover, we could notice a pericardial effusion which was of mean abundance (17.9 mm in relation with the infero- lateral lining of the left ventricle) (Figure 5).
3. Discussion
Atrioventricular channel represents around 7.4% of all congenital heart malformations [1] . From a pathologic point of view, people who suffer from it can be classified in three groups [2] : Those associated with Trisomy 21 represent 45% of all cases, those associated with other genetic syndromes represent 30% and those who have only an isolated non-syndromic atrioventricular canal represent 25% (group to which belongs our patient). Indeed our patient has no obvious clinical sign of Trisomy, the caryotype was not achieved. It is well-known that eusomic patients’ atrioventricular canal has a prevalence of left obstructive abnormities [3] such as abnormities of the mitral valve, a

Figure 5. Trans-thoracic echography, shows an ostium primum atrial septal defect, 2 atrio- ventricular valves, a bi-auricular dilatation and right ventricular dilatation.
left ventricle hypoplaxy, a sub-aortic stenosis, a aorta coartication due an inexistence of ventricular deficiency. No left obstructive abnormitie was found with our patient. The surgical treatment (intervention of Rastelli) [4] is imperative because of the precocious risk of pulmonary arterial hypertension. The survival of trisomic persons with un repaired intra-cardiac shuntleading to Eisenmenger syndrome is the same as with patients having a normal “caryotype” and is from 96% to 20 years [5] . Our patient shows evidence of a big longevity at the cost of iterative cardiac insufficiency. The Atrioventricular channel can be diagnosed in intra-uterine and as for a big number of cardiac malformations tracked down before the birth, the prenatal advice and the prognostic information must not be generalized. A precise analysis of the anatomy of the congenital heart malformation must be achieved. For that to make foetal cardiac scans must be repeated possibly to follow the anatomical and functional evolution of the cardiac anomaly during pregnancy. 3D cardiac scan should permit to refine the anatomical description of the cardiac malformation and therefore to specify information given to parents before the birth [6] [7] .
4. Conclusion
The atrioventricular channel must be repaired in the childhood, because of good results in adult age of the patients and the gravity of the Eisenmenger syndrome, auricular dysrythmias and complete auriculo-ventricular block in case of abstention as it is the case of our patient. He is steady enough in stage of cardiac insufficiency stage III under Furosemide, Hydrochlorothiazide, Captopril and the Molsidomin. Our patient shows evidence of a big longevity and comes to remind us the difficulty to define long-term prognosis of the complex congenital heart diseases and congenital heart diseases which cannot be precociously repaired.
Cite this paper
Kinda, G., Kologo, K.J., Bama, A., Millogo, G.R.C., Traoré, S., Dao, L., Yougbaré, S.O., Kaboré, A., Kaboret, S., Nagalo, K., Tamini, L.T., Yaméogo, R.A., Kouéta, F., Yé, D., Sorgho, L.C.L. and Zab- sonré, P. (2016) An Observation of One Case of Adult Partial Atrio-Ventricular Defect. Open Access Library Journal, 3: e3091. http://dx.doi.org/10.4236/oalib.1103091
References
- 1. Carmi, R., Boughman, J.A. and Ferencz, C. (1992) Endocardial Cushion Defect: Further Studies of “Isolated” versus “Syndromic” Occurrence. American Journal of Medical Genetics, 43, 569-575.
http://dx.doi.org/10.1002/ajmg.1320430313 - 2. Digilio, M.C., Marino, B., Toscano, A., Giannotti, A. and Dallapiccola, B. (1999) Atrioventricular Canal Defect without Down Syndrome: A Heterogeneous Malformation. American Journal of Medical Genetics, 85, 140-146.
http://dx.doi.org/10.1002/(SICI)1096-8628(19990716)85:2<140::AID-AJMG8>3.0.CO;2-A - 3. De Biase, L., Di Ciommo, V., Ballerini, L., Bevilacqua, M., Marcelletti, C. and Marino, B. (1986) Prevalence of Left-Sided Obstructive Lesions in Patients with Atrioventricular Canal without Down’s Syndrome. Journal of Thoracic and Cardiovascular Surgery, 91, 467-469.
- 4. Rastelli, G.C., Ongley, P.A., Kirklin, J.W. and McGoon, D.C. (1968) Surgical Repair of the Complete Form of Persistent Common Atrioventricular Canal. Journal of Thoracic and Cardiovascular Surgery, 55, 299-308.
- 5. Bini, R. (2003) Partial Atrioventricular Canal. Orphanet Encyclopedia.
http://www.orpha.net/data/patho/GB/uk-PAVC.pdf - 6. Acar, P., Saliba, Z., Aggoun, Y., Butera, G., Bonnet, D. and Iserin, L. (1999) Nouvelle approche anatomique des valvulopathies congénitales par l’échographie tridimentionnelle. Archives des Maladies du Coeur et Vaisseaux, 92, 583-590.
- 7. Acar, P., Dulac, Y., Taktak, A. and Villaceque, M. (2004) Echographie 3D temps réel des cardiopathies congénitales. Archives des Maladies du Coeur et Vaisseaux, 97, 472-478.