Open Access Library Journal
Vol.03 No.10(2016), Article ID:71513,24 pages
10.4236/oalib.1103114
The Probable Structure of “Form IV” (Alkali-Denatured Circular DNA)
Ken Biegeleisen
Physician in Private Practice, 19 E. 80 St., Suite 1E, New York, USA

Copyright © 2016 by author and Open Access Library Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 2, 2016; Accepted: October 22, 2016; Published: October 26, 2016
ABSTRACT
A detailed molecular model for alkali-denatured duplex circular DNA (“Form IV”) is proposed. The illustrative biological example used is the replicative form of fx174, a 5 kb duplex circular chromosome. The model explains all of Form IV’s known and peculiar features. In a sedimentation coefficient vs. pH titration, Form IV begins to appear at pH 12.3, at which point it can be persuasively argued that no further supertwists can be added to the already-highly-supertwisted chromosome. Therefore a new structure must appear. The sedimentation coefficient s then undergoes a massive, but initially reversible increase as the pH is raised further, culminating at pH 12.8 with a 250% increase. This degree of compactness can only be explained by a 4-stranded tetraplex structure, consisting of a pair of duplexes whose base pairs are mutually intercalated. Above pH 12.8, the structural changes become irreversible, suggesting a further conformational change. It is proposed that this involves an axial rotation of the component duplex strands, so that the bases now stack on the outside, and the phosphate groups lie in the core, where they bond ionically by means of salt bridges. When the irreversibly denatured compact structure is neutralized at moderate-to-high salt concentrations, a third novel structure appears, which has a sedimentation coefficient midway between the native 21 s and the denatured 50 s. It is proposed that this is a hybrid structure; part tetraplex, part duplex. To return to a fully-duplex form, it is necessary to both neutralize the solution, and also to greatly reduce the ionic strength, i.e., to the range 0.001 - 0.01 M. Since the DNA, under those conditions, cannot possibly have normal complementary base-pairing, the duplex structure must either be tautomerically base-paired, or else stabilized solely by base-stacking, with no base-pairing at all.
Subject Areas:
Molecular Biology
Keywords:
DNA, Circular, Alkali, Denatured, Denaturation, Renatured, Renaturation, Sedimentation Coefficient

1. Introduction
In 1963, Dulbecco and Vogt discovered that the chromosome of the mammalian oncogenic virus polyoma was circular [1] , thus extending the finding of circularity in John Cairns’ famous autoradiographs of the chromosome of the bacterium E. coli, published earlier that year [2] . Shortly thereafter, the discovery of circularity was further extended to the “RF” (replicative form) of the chromosome of the bacterial virus fx174, by Kleinschmidt et al. [3] .
Again in the same year, 1963, Weil and Vinograd [4] reported that the also-circular chromosome of the mammalian oncogenic virus polyoma, when subjected to alkali denaturation, underwent an extraordinary increase in compactness, to a new form they referred to as a “cyclic coil”. A subsequent detailed examination disclosed, for polyoma, four distinct forms of the viral chromosome, which those authors referred to by Roman numeral names. These are the names and sedimentation coefficients of the four forms [5] :
Form I: The native duplex supertwisted circular form; 20 s.
Form II: The native duplex, after introduction of a single-stranded nick; 16 s.
Form III: The native duplex, after linearization by full-duplex cleavage; 14 s.
Form IV: The intact (i.e., un-cleaved) native duplex after alkali denaturation; 53 s.
Although the Roman numeral terminologies “Form I” to “Form III” were first proposed by Vinograd et al. [5] , I should emphasize that those authors did not themselves employ the terminology “Form IV”, but rather the terminology noted above, namely “double-stranded cyclic coil”. Subsequent authors invented their own terminologies for this form, such as denDNA, Form I’ and Form Id [6] . In order to bring coherence and consistency to these form names, Strider [6] [7] introduced the terminology “Form IV” for the ultra-compact 53 s form, which species name we shall adhere to.
At that time, in order to more precisely characterize all these different DNA forms, Rush & Warner [8] undertook a detailed sedimentation coefficient vs. pH titration for circular DNA, employing the RF chromosome of fx174. Figure 1(a) is an adaptation of their results.
Their findings have proven applicable to a number of other small circular DNA species [5] [8] , and therefore appear to be general. Starting with the lower denaturation curve (o-o-o), note that from pH 7 to about pH 11.6 the sedimentation coefficient of Form I (i.e., the native form, as extracted from the virus capsid), is constant, at about 21 s. Then, between pH 11.6 and 11.8, there is a dramatic drop in the sedimentation coefficient, which reaches a minimum of about 16 s. With further increases of pH, the sedimentation coefficient begins to increase again, until, at a pH of about 12.2, it has resumed its native value of 21 s.

Figure 1. (a) Velocity sedimentation coefficient vs. pH titration of the replicative form (“RF”) of the chromosome of the bacterial virus fx174. As explained in the text, although the primary variable here is pH, it may be that some of the conformational changes shown are the result of changes in the salt concentration. The bulk solvent was 0.2 M NaCl, 0.025 M EDTA, 0.05 M NaHCO3, i.e., over 0.2 M salt. At pH 13, the additional salt added, in the form of NaOH, caused the [Na+] concentration to rise to about 0.4 M. The drawings above the lower curve indicate the state of superhelical twisting as a function of pH. Note that for graphic clarity, no secondary helical twisting is shown; the drawings are only meant to illustrate tertiary (superhelical) twisting. At pHs below 11.8, the supertwisting is right-handed (“negative”); above that pH the winding sense reverses, and becomes left-handed (“positive”). At pH 11.8 there is no superhelical winding; the chromosome is an “open circle”, whose larger surface area causes s to drop to a minimum. The lower curve (o-o-o) shows the stages in denaturation, culminating at pH 13 with an approximately 250% increase in s. This is “Form IV”. The upper curve (・-・-・) is a neutralization curve for the Form IV. Note that even at pH 7, the sedimentation coefficient has not returned to the native 21 s, but remains quite elevated, at about 36 s, midway between the native form and the maximally-compact form seen at pH 13. (b) Electron micrograph of Form IV, revealing a surprisingly non-compact, open-circular appearance. What happened to the 250% increase in sedimentation coefficient depicted in Panel A?! This unexpected result may be due to the extremely low ionic strength employed in preparation of DNA samples for electron microscopy, namely 0.001 - 0.01 M salt.
The explanation for these data, which has been accepted without argument by all authors on the subject, is succinctly summarized by the little pictures above the curve (which, for graphic simplicity, show only supertwisting, not secondary helical winding). These pictures illustrate, in a highly-oversimplified and apocryphal fashion, that the native Form I is supertwisted in the right-handed sense (usually referred to by workers in this field as “negative” supertwisting), but that with increasing pH the supertwists are progressively unwound, culminating, at pH 11.8, in an open-circular form which, due to its larger surface area, sediments more slowly than the native chromosome in velocity sedimentation experiments such as this. Then, as the pH is further increased, the chromosome begins to supertwist in the opposite, i.e., left-handed (or “positive”) sense. This continues until the chromosome has returned to the native supertwist count, only in the opposite winding sense, at pH 12.2.
This explanation for the events in the pH range 7 - 12.2 is so well-known that it is now found in college textbooks. Nevertheless, there are many subtleties here. These, however, are beyond the scope of this manuscript. Some of these subtleties have been reviewed in detail elsewhere [9] .
As we move up the pH scale (Figure 1(a)), to pH 12.3, we see that there is a shoulder in the curve, marked chi (χ). This shoulder is found in comparable curves for several other DNA species that have been studied [5] [8] , and its role in the alkali denaturation process is therefore firmly established. No one has ever attempted to explain this shoulder.
Above pH 12.3, the s value enters into a phase of dramatic increase, up to pH 13, at which point it finally levels off. This dramatic increase is smooth and continuous, i.e., it has no additional shoulders, suggesting that a single process is taking place within this pH range. Insofar as the data shows, the increase in s, at least when plotted against pH, is approximately linear, and does not seem to suggest a cooperative change such as is seen in the heat denaturation of linear DNA.
Rush and Warner [8] showed that in the pH range 12.3 - 12.8, the conformation change―whatever that change may be―is reversible, meaning that if the pH is reduced back to neutrality, the native Form I structure is restored. But what exactly is happening to the structure between pH 12.3 - 12.8? Neither they, nor any other authors have ever offered any explanation.
Above pH 12.8, the conformational change suddenly becomes irreversible. It is this new conformation that is known as “Form IV”, which is the subject of the present manuscript.
Something of the nature of Form IV is depicted by the upper curve, consisting of the black circles (・-・-・), which is a neutralization curve. These upper-curve data show that merely neutralizing the alkali-denatured Form IV does not restore the native Form I conformation. Instead, at the pH 7 end of the upper curve, we see a persistently- compact form, whose compactness (about 36 s) is considerably below that of the ultra- compact form at pH 13 (about 50 s), but still almost twice as great as that of the native chromosome (21 s). What is this form? Is it still Form IV, only with some sort of further conformational change? Or has it converted to something else entirely? Insofar as the Form IV literature reveals, these questions have never been either asked or answered.
To deepen the mystery, several authors have performed electron microscopic examinations of Form IV (Figure 1(b)) [10] . One might naively expect to see some sort of extensively-compacted or greatly-twisted structure in these electron micrographs, but, as Figure 1(b) shows, we are quite astounded to see an open-circular form, no different in appearance than that of the native Form I after being nicked! The nicked native form, as noted above, is called “Form II”. The introduction of even a single nick into the native Form I causes an unwinding of all superhelical twists, resulting in an open-circular form, similar in appearance to that which is seen in Figure 1(b). What on earth is going on here? How can something which is ultra-compact have an electron microscopic appearance which suggests that it is, in fact, ultra-extended?!
The data on these phenomena were all published between 1963 and 1981. All those authors agreed on the events in the pH range 7 - 12.3. But in the 35 years that have passed since then, no explanation for anything from the pH 12.3 “χ” shoulder and beyond, has ever been offered by any author. It is the purpose of the present manuscript to attempt to begin to fill these gaps in our knowledge, by proposing plausible structures for all these fx174 chromosomal forms.
2. Methods
The molecular models described herein were created employing Maestro (Release 2015-1) (Version 10.1.013), a software package that is part of the Schrödinger Suite. The Maestro component has recently been made available to academic users as freeware.
The graphic images of the models were exported from Discovery Studio DS Visualizer (v4.0.100.13345), which is provided as freeware by Accelrys, Inc.
The method whereby the Form IV model was constructed by rotations and translations of the strands of the Wu tetraplex DNA model are explained in Figure 8.
3. Results
3.1. Events at the pH 12.3 “χ” Shoulder; The Supertwist Limit
Although I have never seen any discussion of it, it is very clear that circular DNA, under typical laboratory conditions, has a supertwist limit. That limit is apparently a supertwist count equal to about 0.5% of the number of base pairs in the chromosome. Thus, for the fx174 RF, which has about 5000 base pairs, the supertwist limit is about 25, which has been verified experimentally for a number of 5kb species of DNA, as well as observed directly by electron microscopy [11] [12] [13] .
The existence of this supertwist limit is implied, although not forcefully, in the Rush-Warner data of Figure 1(a). Let us return to the lower denaturation curve (o-o-o) in that figure. Note that the sedimentation coefficient of the right-handed superhelical native form at pH 7, about 21 s, is the same as that of the left-handed superhelical form at pH 12.2. This implies that the number of superhelical twists is the same at both pHs, and that they differ only in the direction of helical winding. Although the changes in the shape of the denaturation curve at pHs above 12.3 do not rigorously prove that no further supertwisting is possible, they argue against it. But there are more compelling evidences of a supertwist limit than this.
Consider next a typical topoisomerase experiment. When a circular chromosome of about 5 kb in size is allowed to come to equilibrium in the presence of that enzyme, and the products are examined by electrophoresis, a family of approximately 25 topoisomers is observed (Figure 2).
At equilibrium, each topoisomer differs from its neighbor by ±1 supertwist. Now, ordinarily, no topoisomer ever has an electrophoretic mobility greater than that of the

Figure 2. Typical topoisomerase experiment for a 5 kb circular chromosome [14] . Topoisomerase nicks and re-seals DNA, giving rise to a set of so-called “topoisomers” that differ by ±1 in their superhelical winding. The arrow indicates the direction of electrophoretic migration. The native chromosome co-migrates with the fastest-moving topoisomers. The nicked chromosome, having no supertwists, co-migrates with the slowest-moving topoisomers. The gel on the left has been treated to maximize separation of the slow-moving topoisomers. The gel on the right has been treated to maximize separation of the rapidly-moving topoisomers. Between the two gels the complete set of topoisomers can be seen. The drawings on the top of the figure are accurate, indicating that the topoisomers with the lowest supertwist counts will clump together at the top of the gel. The drawings in the middle and bottom are apocryphal, since the highest supertwist count would actually be approximately 25, of which we’ve only drawn about half, for graphic clarity. Topoisomerase-generated bands always contain a mixture of right-handed and left- handed topoisomers. These will co-migrate as long as the absolute value of the supertwist count is the same, regardless of the direction of superhelical twisting. The point of this figure is to emphasize that the supertwist count in the native chromosome is always the same as that of the most highly-twisted topoisomer, regardless of the direction of superhelical winding of the topoisomer. That supertwist count is therefore the limit of superhelical winding that is possible (unless an external force is applied to artificially overwind or underwind the chromosome).
native chromosome. This means that the supertwist count in the native chromosome is the greatest number of supertwists possible for that DNA, i.e., that the only way to increase that supertwist count would be through the application of some sort of external force, to drive the supertwisting beyond the natural limit. We may therefore say that, in the absence of such an external force, the native chromosome may be well-described as being maximally supertwisted in the right-handed (“negative”) sense. But each band in a topoisomerase experiment is a 50 - 50 mixture of the right- and left-handed forms having the same absolute supertwist count (i.e., ±1, ±2, ±3, etc.). It follows logically that the absolute number of supertwists in the native chromosome is, in fact, the supertwist limit for that DNA, whether in the native state where the supertwisting is right-handed, or in a band in a topoisomerase experiment where the supertwisting may be either to the right or the left.
Another line of evidence comes from the ethidium bromide vs. s titration of the chromosome of the oncogenic virus SV-40 (also about 5 kb), performed by Bauer and Vinograd in 1968 [15] (Figure 3).
Ethidium bromide (EtBr) is a fluorescent intercalating dye, whose planar molecules insert themselves between DNA base pairs. This stretches the DNA out, causing circular DNA right-handed (“negative”) supertwists to unwind. But, as the figure shows, after the right-handed supertwists are all unwound, further EtBr causes the supertwists to rewind, only in the opposite sense, i.e., left-handed (“positive”). This is illustrated in the small drawings I’ve inserted above the data, which are the very same drawings I used in Figure 1(a). Insofar as s reflects the state of superhelicity, we see once again that there is a supertwist limit: the maximum number of left-handed supertwists, at high dye concentrations, is the same as the number of right-handed supertwists found in the native chromosome (only opposite in superhelical winding sense), which, for a chromosome of this size, would be about 25.
This supertwist limit cannot be accounted for by presuming EtBr binding saturation. Quite to the contrary, note from the abscissa that only about 10% of the inter-base positions are occupied by the intercalating agent at the supertwist limit. There’s lots of

Figure 3. Effect of ethidium bromide on the sedimentation coefficient of Forms I [o-o-o] and II [―] of SV-40 DNA. The effect of the dye on Form II is minimal, but the effect on Form I is similar in every respect to the effect of pH on the s value of Form I of fx174 shown in Figure 1(a). As the amount of bound dye per nucleotide residue increases, the native right-handed (“negative”) supertwists unwind, until the dye/nucleotide ratio reaches approximately 0.05, at which point the chromosome has become an open circle. Further addition of dye then causes the winding in of left-handed (“positive”) supertwists, up to what appears to be a supertwist limit, which, based upon the sedimentation coefficient (21 s), is equal in absolute value to the supertwist count of the native chromosome, only in the opposite winding sense. Note that at the supertwist limit, the fraction of nucleotides with an intercalated dye molecule is only 0.11 - 0.12, barely more than one dye molecule per 10 nucleotide residues! Since approximately 90% of the nucleotides have room for additional ethidium bromide molecules, it must indeed be the case that there is a supertwist limit, and that the further binding of dye could only be achieved by exceeding that limit, which will not happen. Adapted from Bauer & Vinograd [15] .

Figure 4. Effect of increasing ethidium bromide (EtBr) concentration on the electrophoretic mobility of various DNA forms. (We shall be ignoring the lowest row of bands, “Form V”, which is not pertinent to the present discussion). The large blue arrow shows the direction of migration. The apparatus shown holds 28 lanes of agarose, each one with a different concentration of EtBr. The concentration of EtBr increases toward the right. Form II (the uppermost row of bands) is essentially unaffected by increasing EtBr concentration. Form I, on the other hand (highlighted by the blue guideline), displays the same unwinding and re-winding of superhelical turns that were seen in Figure 3, except that here the EtBr concentration was raised considerably beyond that which pertains to the supertwist limit. The supertwist limit is reached in the 14th gel from the left (yellow arrow). All the gels from 14 - 27 reveal approximately the same Form I electrophoretic mobility, showing that unnaturally-high degrees of supertwisting cannot be forced by merely increasing the EtBr bromide concentration.
room in the chromosome to bind more dye, but that would cause further supertwisting, which is evidently impossible.
One might object that in this experiment, the dye concentration was not “pushed” high enough, and that perhaps more dye might drive more supertwists. That experiment, however, was done by Stettler et al. in 1979 [16] (Figure 4).
In that experiment, several DNA forms were examined by gel electrophoresis, in the apparatus shown, containing 28 separate agarose gels, each having a different amount of EtBr. The EtBr concentration increases to the right. As we have previously seen, the effect of increasing ethidium bromide concentration on Form II is nil. The Form I data, however (which I’ve highlighted with a blue guideline), again show the unwinding of right-handed supertwists, and rewinding into left-handed supertwists; exactly as we’ve seen twice before. The difference here is that the EtBr concentration was pushed far past the supertwist limit, which is marked by the yellow arrow. Note, however, that in spite of the “pushing” of the EtBr concentration, no further supertwisting took place. Once again, we see that when a left-handed superhelically-wound chromosome reaches the absolute value of the supertwist count seen in the native form, no further supertwisting is possible. And for fx174, that number is about ±25.
Returning now to Figure 1(a), we may say with much more confidence that the χ shoulder is a place where a conformational change is taking place; a conformation which is intermediate between a maximally-supertwisted starting form at pH 12.3, and a mysterious new form which appears at higher pHs.
3.2. Events in the pH Range 12.3 - 12.8, The Region of Reversible Denaturation
As the pH climbs into the range 12.3 - 12.8, the sedimentation coefficient dramatically increases. Why? There are two things we can say about this immediately:
1) This is not merely further supertwisting, since we have already passed the super- twist limit.
2) Whatever this new structure is, it is at least partially, if not totally base-paired.
How do we know that the new structure is base-paired? We know this because up to pH 12.8, the denaturation is reversible, and the native structure can be restored by simple neutralization [8] . But at pH 13, where the bases are all nearly 100% deprotonated, and where essentially all base-pairing is therefore abolished, renaturation becomes almost impossible [6] [7] .
This inability to readily renature, at high pH, cannot be the result of a sugar- phosphate backbone change, because the pK of DNA phosphate is less than 2. But the bases all have pKs in the 12 - 13 range. The resistance to renaturation at pH 13 must therefore be due to the loss of base-pairing. Pending some other unfathomable alternative explanation, we therefore must conclude that the ability of fx174 DNA to renature in the pH range 12.3 - 12.8 is due to persistence of base pairing.
If the structure we propose for DNA in this pH range is to be selected from among structures which have been previously described, then the choice narrows down to exactly one: the tetraplex structure, consisting of a pair of DNA duplexes merged into a single 4-stranded structure by mutual intercalation of their base pairs. This is the structure iconoclastically proposed for all DNA by Tai Te Wu [17] [18] (Figure 5).
Although there is, at the present time, no direct physical evidence that the Wu structure exists in nature, there is good indirect evidence, in that the structure of the complex between DNA and protamine, in sperm cells, can only be solved by putting the DNA into the Wu conformation [19] [20] [21] . The negative charges of DNA phosphate, when the DNA is in the classic Watson-Crick conformation, simply cannot anastomose with the positive charges in protamine, the latter of which are found on the many arginine residues which constitute over 50% of the protamine amino acid sequence. But with the DNA in the Wu conformation, and the protamine modeled as a small β-sheet (from the most-favorable part of the Ramachandran Plot for β-sheets), the negative and positive charges align with all the precision of soldiers in drill formation.
Moreover, Wu, in presenting his tetraplex model, at the same time also presented multiple evidences that the strands of certain plasmid chromosomes can be non-destructively separated, which would be impossible if they had the “classic”, plectonemic double-helix structure [17] .
There is actual physical evidence for the existence of the tetraplex type of DNA structure, consisting of two duplexes with mutually-intercalated base pairs, in the form of the Gehring tetramer, whose structure has been solved by high-resolution X-ray crystallography [22] [23] (Figure 6).
But the Gehring tetramer only forms at very low pH, and, being stabilized by what are known as C-C+ base pairs, depends upon a base sequence which is almost entirely poly-C. Moreover, the Gehring tetramer consists of a pair of antiparallel duplexes, the individual strands of which, however, are parallel; a strand disposition not known to occur in nature. The Wu tetramer is therefore the model of choice for fx174 in the pH range 12.3 - 12.8, if we are to explain the phenomena in this pH range in terms of a known DNA structure.
If we invoke the Wu structure, then the events between pH 12.3-12.8 (Figure 1(a))― if we may speak anthropomorphically―can be interpreted to mean that, as the chromosome “tries” to supertwist beyond the 25 supertwist limit for this species, the back-
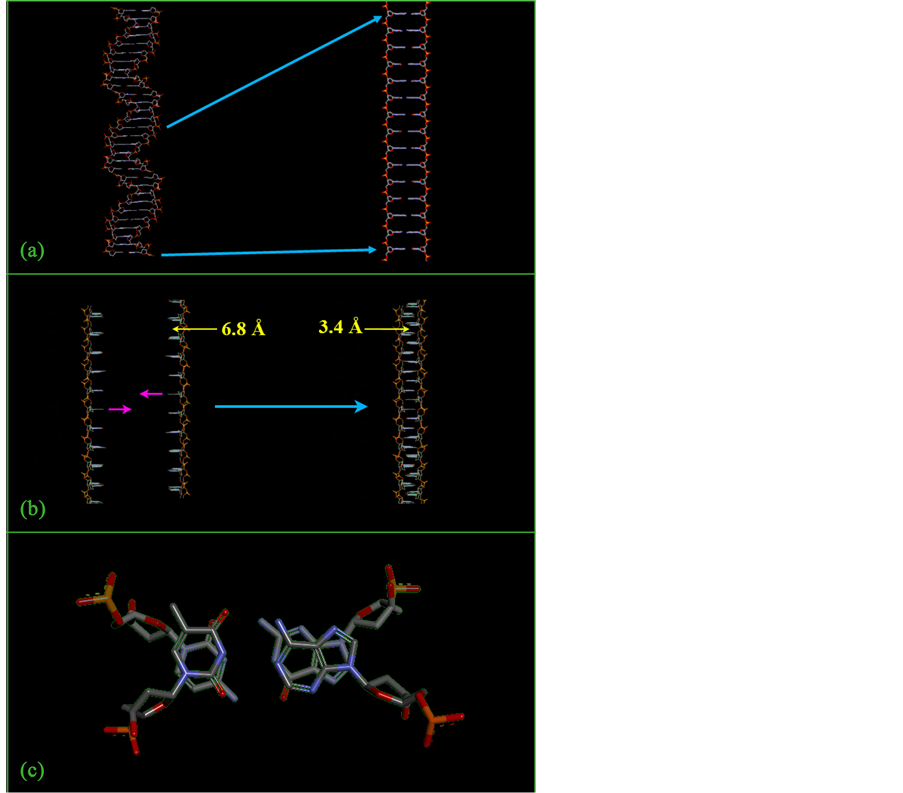
Figure 5. Intercalated tetraplex structure of Tai Te Wu. (a) The sugar-phosphate backbone of a helical DNA duplex is hypothetically stretched to its full length, yielding a “straight ladder” duplex with an impossible 6.8 Å base pair spacing. (b) A pair of such “straight ladder” duplexes is merged into a single tetraplex structure, which restores the normal 3.4 Å base pair spacing. (c) Axial view of the tetraplex structure. In contrast to a hypothetically-overly-supertwisted circular DNA duplex, which could not exist in the real world because of phosphate-phosphate charge repulsions, here the phosphate groups are on the periphery, where they do not clash.
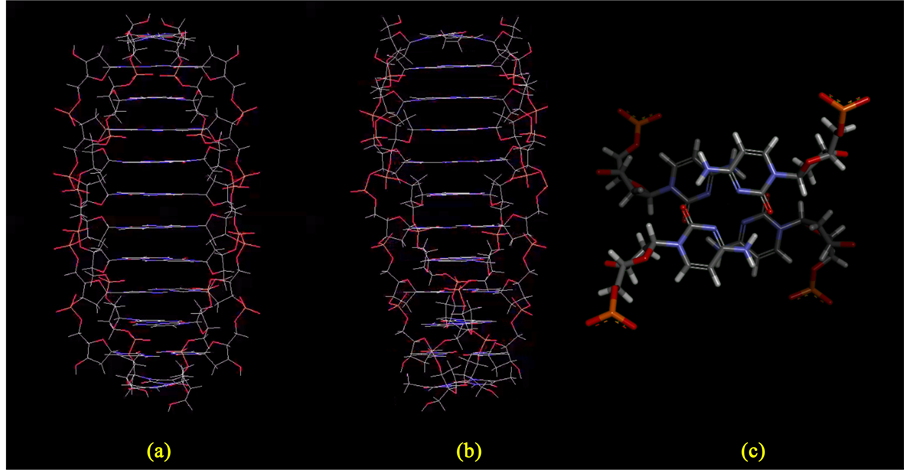
Figure 6. Gehring Tetramer, three views. This is a 4-stranded DNA structure with a right-handed helical twist. The structure is difficult to portray in a flat drawing (a rotating virtual model may be seen in reference [23] ). (a) Viewed from this rotational perspective, the structure looks similar to Watson-Crick duplex DNA. (b) In this rotation, the fact that there are 4 sugar-phosphate backbones is fairly evident. (c) In axial view, the extreme rotational offset between adjacent base pairs (90˚) is evident. The C-C+ base pairs can also be seen.
bone phosphate groups, which are negatively charged, are brought into an excessively- close juxtaposition, causing the chromosome to “seek” another structure. The Wu tetraplex solves the problem in two ways: (1) it creates the sort of compact structure that can potentially account for the great increase in s, and (2) it removes the rapidly-developing steric hindrances due to phosphate-phosphate charge repulsions and Van der Waals violations, by placing the phosphate groups on the outside of the structure, where they do not adversely interact, and where they can instead favorably interact with the surrounding water molecules (Figure 5(c)).
But can the Wu structure fully-account for the extraordinary increase in the sedimentation coefficient between the structure seen at pH 12.8 (50 s), and that which was seen at pH 12.3 (21 s); a ratio of [50 s/21s] = 2.4? An analysis of this problem, based upon the Svedberg equation for calculating s values, has been presented elsewhere [24] . This analysis revealed that the predicted ratio of s values was 2.2, very close to the observed ratio of 2.4.
The proposed transition from the duplex left-handed superhelical form at pH 12.3, to the Wu intercalated tetraplex form at pH 12.8, is clearly a stepwise, gradual transition, as opposed to a cooperative transition (such as is seen, for example, in a DNA melting curve). If we may again speak anthropomorphically, the DNA quite evidently does not particularly “like” the tetraplex structure, but only “accepts” it under duress, and therefore only converts to it a little at a time; converting, at each point in the pH scale, only the minimum length necessary to avoid the torsional strain which would otherwise exist at that pH due to superhelical overwinding.
In other words, in the pH 12.3 - 12.8 region, the DNA must have a hybrid structure, starting out (at pH 12.3) all duplex, ending up (at pH 12.8) mostly or all tetraplex, and having a mixed, or hybrid structure in between.
At the end of this pH region of the denaturation curve, i.e., at pH 12.8, the structure has maximized its compactness, but is still capable of renaturation. But when the pH is further increased to 13, the renaturation becomes irreversible. The s value at pH 13, however, does not significantly increase. Clearly there must be yet another conformational change at that elevated pH. But what is it?
3.3. Appearance of the Irreversible Form IV Structure at pH 13
In order to begin to understand what the irreversible Form IV structure might be, we need first to consider what forces remain, at pHs in excess of 12.8, to stabilize that structure. Among those forces known to stabilize nucleoprotein structures, there are essentially only two available in this setting: base stacking, and salt bridging.
Although there is no significant hydrogen-bonding between bases at pH 13, the phenomenon of base stacking is not only “alive and well”, but perhaps even enhanced, since base stacking is a hydrophobic phenomenon, and the surrounding environment at pH 13 has increased in hydrophilicity, which might increase the tendency of the bases to sequester themselves from the solvent by stacking.
Since base stacking is currently regarded as being the most important stabilizing force in DNA under physiological conditions [25] , substantially exceeding the free energy contribution of base-pairing, one could speculate that at pH 13, fx174 DNA might simply remain in the Wu conformation, i.e., the 4-stranded, mutually-intercalated conformation, only without base pairing. This unlikely possibility is illustrated in Figure 7, which suggests that no change has taken place in the Wu structure other than that the bases have moved apart a bit, due to charge repulsions of the now-deprotonated moieties that were formerly the recipients of the hydrogen bonds that stabilize DNA in nature.
But if this sort of thing actually happened, why then wouldn’t the structure instantly renature when the pH was restored to neutrality?
One might explain the failure to renature, at neutral pH, as being the result of a circular base “drift”, i.e., by presuming that while at pH 13, the two single-stranded circular DNA strands underwent mutually-contrary rotation, causing the correct base pairs to fall out of alignment. But if that was the case, then one might further expect that at neutral pH, that same circular “drift” would continue, resulting in rapid, or at least fairly-rapid renaturation, and such has not been observed [6] [7] . (I am assuming here that the anomalous base-pairing of randomly-juxtaposed bases will not prevent this circular “drift”, which presumption may not be entirely true). The un-pairing of bases at pH 13 is instantaneous, whereas the reappearance of base pairs, when the Form IV structure is neutralized, does not occur at any noticeable rate.
This suggests that the Form IV structure is probably not, after all, an inherently unstable one in which the strands are continuously and aimlessly undergoing a circular

Figure 7. Unlikely conception of Wu structure before and after denaturation. (a) Axial view of the Wu tetraplex, at pHs up to 12.8. The yellow lines symbolize hydrogen bonding, which, in part at least, persists up to that pH. (b) The same structure at pH 13, the point of irreversible denaturation. This hypothetical candidate for Form IV structure consists merely of the Wu tetraplex from panel A, only without hydrogen bonds. The negative charge repulsions, due to deprotonation, drive the base pairs apart, and the structure simply remains in that conformation, stabilized solely by base stacking.
“drift”, but more likely a single definite structure which is stable as long as the prevailing conditions of pH, temperature and ionic strength continue. But what structure might that be?
Other than base stacking, the only other well-known stabilizing force remaining at pH 13 is the salt bridge. Every DNA residue has a negatively-charged phosphate group, which is “ready and willing” to electrostatically bind to any positively-charged group which is available. In the Rush-Warner pH vs. s titration shown in Figure 1(a), the bulk solvent had a sodium concentration of about 0.2 M at pH 7, rising to about 0.4 M at pH 13. To put this in perspective, this is nearly 3x the physiological salt concentration of the human body, the latter being about 0.15 M (=0.09% sodium, w/v), wherefore we might regard the conditions at pH 13 as being well-described as “high salt” conditions.
I would therefore propose that, at pH 13, the DNA strands undergo a full-length, approximately 90˚ axial rotation, so that the bases stack on the outside of the tetraplex structure, while the phosphate groups move to the inside, bound by salt bridges with the ambient sodium ions (Figure 8).
I first proposed a naïve version of this sort of structure in 2006 [26] [27] [28] . Because the 2006 structure has now proven to be incorrect, and because it cannot now be rescinded, it is appropriate to acknowledge it, for the purpose of explaining its shortcomings. Because of the unaffordable cost of virtual modeling software at that time

Figure 8. Steps in the conceptual development of the fx174 Form IV model. (a) The starting structure is the Wu tetraplex, which can exist up to pH 12.8. The yellow arrows indicate the directions of the ~90˚ rotations (with the centers of rotation coinciding approximately with the phosphorus atoms) which will convert this to Form IV when the pH rises to 13 (at which pH all base pairing is abolished). (b) A conceptual intermediate, with the bases now “sandwich”-stacked on the periphery. The purple circles represent sodium ions aligning themselves between phosphate groups, with salt bridge spacings of about 2.5 Å. But there’s a large “hole” in the middle of the structure, which is probably energetically inferior to the structure depicted in the next panel. (c) After the translation indicated by the yellow arrow in panel B, we have a more likely Form IV structure, with the “hole” in the middle now replaced by additional salt bridges. The bases have also been rotated slightly, to replace the “sandwich” stacking of panel B with the energetically- preferred “parallel-displaced” base stacking shown here.

Figure 9. Incorrect 2006 Form IV model. (a) Each of the 4 strands of the Wu tetraplex was rotated a full 180˚ about its phosphorus atom. (b) The axial phosphate core of the resultant structure was plausible, but the bases were stacked at the energetically-suboptimal spacing of 6.8 Å.
(>$100,000 US), I was not able to make a true molecular model, but only a graphic illustration of the general concept (Figure 9). Recently, however, the virtual modeling program Maestro (Schrödinger) has been made freeware for academic use, and, employing that software, I have now found that my 2006 model was flawed. It was based upon a full 180˚ axial rotation of each of the four strands of the Wu tetraplex [28] , and therefore had impossible 6.8 Å base stacking distances (since the bases of each of the 4 strands of the Wu tetraplex are thusly spaced).
At the time I first proposed this structure, I presumed that it could be perfected, but, when I attempted to do so with Maestro, I found that if the base stacking distance was corrected to 3.4 Å, the phosphate core became hopelessly packed with an impossibly-large number of atoms, resulting in severe Van der Waals violations. Conversely, if the phosphate core was plausibly modeled, the bases had to be stacked at distances much larger than 3.4 Å.
The solution to the problem was to limit the rotation of the DNA strands to about 90˚, as depicted above in Figure 8. The 90˚ rotation results in a structure that is perfectly stacked, whose salt bridges are properly positioned and of proper length, and which is totally devoid of Van der Waals violations. The final model fulfils all the requirements of a Form IV structure: it has no steric hindrances, no dependence upon base pairing, and it has almost exactly the same cross-sectional area as the Wu tetraplex, wherefore it can well-explain the 2.4-fold increase in s as the DNA goes from pH 12.3 to pH 13 [24] [29] .
What would be the predicted salt bridge length for this model? M. Harding, of the University of Edinburgh Centre for Translational and Chemical Biology, has compiled a massive database of metal-protein ligands: 17,342 of these are for sodium alone [30] . According to their sodium database, the average sodium coordinate number is about 5, which is consistent with the environment in a cylindrical structure whose axial core is packed with negatively-charged phosphate groups. The average sodium ligand length, for all 17,342 structures in the Edinburgh database, was 2.57 Å, wherefore I have spaced the DNA phosphate oxygen atoms, in the new Form IV model, at twice that distance, or about 5 Å.
An interactive Jmol model, and a downloadable PDB structure file for this Form IV model, may be found at https://notahelix.net.
3.4. What is the Structure of Form IV When the pH Is Restored to Neutrality?
The black circles, which comprise the upper curve at the top of Figure 1(a), show the neutralization data for Form IV fx174 DNA. Explaining this upper neutralization curve has been, by far, the most difficult part of the Form IV endeavor. The first part of the problem is that we cannot be absolutely certain about the reproducibility of the data itself, because this is the only Form IV neutralization study ever published. Whereas the data in the lower portion of the figure, i.e., the denaturation data (white circles), has been reproduced for other viral chromosomes and plasmids (Figure 10) [5] [8] , and whereas in each of these cases, the salient features of the Figure 1(a) lower curve are seen, the data at the top of Figure 1(a) (black circles) constitute the only neutralization study ever published.
This is most disconcerting with respect to the portion of the data highlighted by the vertical gray shading bar in the middle of Figure 1(a). The gray highlight has been added to focus our attention on the pH range 11.6 - 12.3, within which dramatic changes are taking place during the denaturation phase (white circles) of the pH titration. If we now direct our attention to the upper neutralization curve in that same range

Figure 10. Additional examples of pH vs. S titration curves. Both studies were limited to the denaturation phase of the Form IV cycle; that is, neither study included a neutralization curve. (a) The oncogenic virus polyoma [5] . (b). Penicillinase plasmid from S. aureus [8] .
of pH, we see what appears to be a small shoulder in the curve. Is this shoulder significant? If we were to move only the central data point in the shaded region, upwards by just a single s unit, the shoulder would vanish!
Before commenting further on the possible shoulder in the upper curve, we must address first the final data points in the neutralization phase of the experiment, at pH 7, which reveal a sedimentation coefficient of about 36 s (Figure 1(a)). This is a value which, complete neutralization notwithstanding, is still nearly twice the s value of the native chromosome (21 s). How can this be explained? For a chromosome in this size range, there is no known form of duplex DNA which has such an elevated sedimentation coefficient, but, at the same time, that sedimentation coefficient, 36 s, is also far too low to account for by means of a tetraplex structure.
If we are to explain this, by employing only structures which are known, then we must conclude that the chromosome, at pH 7, is still in the midst of a prolonged process of converting from the 4-stranded form seen at pH 13, to some sort of 2-stranded form, and that it has not yet completed that conversion. In other words, at pH 7, at the ionic strength of the Rush-Warner experiment (upper curve in Figure 1(a)), the chromosome must again have a hybrid structure, part tetraplex and part duplex (Figure 11)― as we saw previously in the denaturing chromosome in the pH range 12.3 - 12.8. In order for the neutralized Form IV chromosome to complete the transition back to 2-strandedness, as depicted in the electron micrographs in Figure 1(b), we must, in addition to lowering the pH to 7, also lower the ionic strength to that realm of ionic strength employed in the preparation of DNA samples for electron microscopy, namely 0.001 - 0.01 M salt. This is far lower than the ionic strength in the Rush-Warner experiment where, even at pH 7, the total ionic strength was still in the relatively high range of about 0.2 M.

Figure 11. A plausible explanation for the upper (neutralization) curve in Figure 1(a). At high pH (12.8-13) (drawing on the left), the structure is totally tetraplex, sedimenting at 50 s. Since the sedimentation coefficient does not return to the native 21 s at pH 7, but rather remains at the very elevated level of 36 s (middle drawing), we are more or less forced to conclude that some parts have reverted to a two-stranded, presumably superhelical conformation, while other parts remain in the compact tetraplex conformation. In order to get the chromosome to revert totally to the duplex form (drawing on the right), we must, in addition to neutralizing the solution, also greatly decrease the ionic strength; to the realm of ionic strengths employed in the preparation of samples for electron microscopy, i.e., 0.001 - 0.01 M.
If we examine this proposition, namely that when Form IV is neutralized back to pH 7, that it converts to a hybrid structure, part tetraplex and part duplex, we find that it is consistent with the proposition that at pH 13 there occurs the circular “drift” alluded to earlier, causing the two strands of DNA to be out of proper alignment with respect to base-pairing. If so, then when the pH is lowered, the proper native base-pairing cannot be re-established, wherefore there is relatively little energetic incentive to form a duplex structure. How then do the strands interact with one another?
There are only two likely answers that come to mind. The first possibility (Figure 12) is that when the two sides of the duplex circular structure, whose mutually-intercalated bases constitute the tetraplex, are no longer able to remain associated with one another as a 4-stranded structure, then they un-intercalate back to the original duplex circular form, and that the bases simply remain stacked, with no base-pairing at all (Figure 12(a)). The second possibility is that when the bases un-intercalate, they do engage in base-pairing, but only of a tautomeric variety (Figure 12(b)).

Figure 12. Neutralization of Form IV. Two competing models for the duplex form which, logically speaking, must progressively appear as the pH is lowered to neutrality. Due to the presumed “circular drift” alluded to in the text, it is a near-certainty that canonical Watson-Crick base pairing cannot re-appear under these circumstances. So what binds the strands together? (a) In this model, the 90˚ strand rotation portrayed in Figure 8 is reversed, so that the phosphate groups are once again peripheral, as in “traditional” Watson-Crick DNA. But the bases are portrayed as being organized into a single base stack, extending the length of the chromosome, with no base pairing at all. (b) In this model, base pairing is presumed to return, but, since only 25% of bases will be coincidentally aligned with the proper complementary base pair, the base-pairing here must be 75% tautomeric. There is a question as to whether either structure (i.e., “(a)” or “(b)”) is energetically feasible. Based upon the limited energetic information that is available, the answer must remain “Maybe”.
A review of tautomeric base-pairing [31] [32] reveals that any base can pair with any other base, although the energetics are vague. In solution studies of individual bases, the fraction of bases which are in tautomeric conformations, at any moment in time, is in the range 10−4 to 10−5. The DG for conversion of a normal DNA base to a tautomeric form is said to be [+5] to [+10] kcal/mol, whereas the DG for the formation of a base pair is [−5] to [−10] kcal/mol. These numbers obviously do not allow us to state persuasively that tautomeric base pairing, as the basis for a new structure, is likely, only that it’s well within the realm of possibility. In contemplating this possibility, we must bear in mind that in any random juxtaposition of DNA single strands, 1 out of every 4 base pairs will be coincidentally canonical, so that a basic framework for a tautomerically- base-paired structure automatically exists at all times. We must also bear in mind that in a faux-Watson-Crick tautomerically base-paired structure, the bases will also be stacked, and that base stacking is known to be considerably more important than base pairing in the stabilization of the double helix [24] . For these reasons, I am inclined to presume that the structure shown in Figure 12(b), which is base-paired and stacked, is preferable to that in Figure 12(a), which is stacked only.
In any event, the transition from a 4-stranded to an anomalous 2-stranded structure is clearly not a cooperative transition such as takes place during true DNA renaturation. The transition to either one of the 2-stranded hypothetical structures shown in Figure 12 provides much less of a free energy decrease than would a true renaturation to a fully- base-paired structure, wherefore, if we may again speak anthropomorphically, the chromosome converts back to the duplex form only “reluctantly”. In this respect, what we are seeing here seems to be a reversal of the denaturation events we saw in the pH range 12.3 - 12.8, which appeared to be a non-cooperative transition in the opposite sense, i.e., from a 2-stranded to a 4-stranded structure.
According to this point of view, during neutralization, the lowering of the pH from 13 back down to 7 affects the tetraplex structure in two ways: (1) the drop in ionic strength weakens the salt bridges which maintained the Form IV tetraplex structure at pH 13, and (2) the drop in pH gradually re-introduces the possibility of base-pairing, albeit tautomeric.
Stepping back a bit, and taking a broad view of the events in the upper neutralization curve (and simultaneously considering the low-ionic-strength, fully-duplex forms shown in the electron micrographs of Figure 1(b)), I believe we can confidently state that the neutralization process is one long, stepwise and continuous transition, beginning with the Form IV tetraplex structure depicted in Figure 8, and ending with one of the reconstituted duplex structures depicted in Figure 12. But the events between these extremes must necessarily remain murky.
The biggest question in my mind, which I cannot answer, is whether the intermediate points in the upper neutralization curve (Figure 1(a)) represent different stages in a single transition process, from the salt-bridge-based Form IV structure (Figure 8) to a 2-stranded structure (Figure 12), or, alternatively, whether the intermediate points in the upper curve represent a 2-phase transition, beginning first with a return to a Wu-tetraplex-like structure (only one stabilized by tautomeric base-pairing) somewhere around pH 12 (Figure 5), followed by a protracted transition from that tetraplex structure back to a 2-stranded structure. One could argue for either case.
In favor of including a Wu tetraplex intermediate in the neutralization process is that, in the lower denaturation data (Figure 1(a), white circles), the final conversion from the Wu intercalated tetraplex structure to the irreversibly-denatured Form IV structure is not seen until the pH exceeds 12.8. We might then assert (but with considerable uncertainty) that in the upper neutralization phase of this pH titration (black circles), that as the pH drops below 12.8, the strands must necessarily reverse the 90˚ axial rotation that―in the lower denaturation sequence of events―caused the chromosome to convert from the Wu tetraplex to the Form IV structure (Figure 8). We must keep in mind, however, that if, below pH 12.8, a Wu tetraplex-like structure does indeed re-appear, it must be one which is stabilized by weak tautomeric base pairing, which will not be favored by a free energy drop of anywhere near the magnitude we would see if the structure had canonical base pairing.
The electron micrographs (Figure 1(b)), however, might favor the alternative point of view, namely that the intermediate points in the upper neutralization curve represent an evolving transition between the Form IV structure only (Figure 8) and the duplex form, that is, that the Wu tetraplex never reappears at all during neutralization, but that all portions of the chromosome that are, at any given moment in time, in the tetraplex conformation, are in the Form IV tetraplex conformation, with axial phosphate groups bound together by sodium salt bridges (albeit salt bridges which are continually weakening as the ionic strength drops). In favor of this argument is the fact that DNA specimens prepared for electron microscopy, employing the type of methodology shown in Figure 1(b), are subjected to extremely low ionic strengths, in the range of 0.001 - 0.01 M sodium, suggesting that in the return from a 4-stranded to a 2-stranded form, the lowering of ionic strength is more important than the lowering of pH. This argument in turn might suggest that the structure from which the final electron micrographic duplex evolves is a form which is dependent upon high ionic strength, namely the Form IV structure depicted in Figure 8.
Regardless of which of these competing possibilities is the most likely, there is an additional broad generalization we can make. Insofar as we know anything about DNA helicity, it seems fairly clear that under physiological conditions of temperature, pH and ionic strength, DNA, once released from its native nucleoprotein environment, essentially always assumes a right-handed helical twist. Conversely, under conditions conducive to denaturation, such as the high pH conditions depicted by the denaturation data in Figure 1(a) above the nadir at pH 11.8, it seems equally clear that DNA “prefers” the left-handed helical conformation [33] . If so, then the sequence of events portrayed by the upper neutralization curve (black circles) entails a transition between a very-likely left-handed form at pH 13, to a right-handed form at pH 7. If so, then there must be a point in the upper curve where a transition in helical handedness causes a perturbation in the smoothness of the curve. Since, in the denaturation curve, the right-to-left helical transition takes place in the vicinity of pH 12, it would make sense to presume, as a first approximation, that the return trip during neutralization, i.e., the left-to-right helical reversion, would also take place in the same pH realm.
That is why I have superimposed a vertical gray shading bar on the Rush-Warner data in Figure 1(a), and also why I presume that the apparent shoulder in the upper neutralization curve is real, and not merely a random fluctuation of data points.
4. Discussion and Conclusions
When Form IV was first discovered in 1963 [4] , it generated a wave of intense interest, mainly because of its extreme compactness. Because separation of viral and plasmid DNA from cellular chromosomal DNA was a major problem in those days, it was fondly hoped that the compactness of Form IV would provide researchers with a simple and inexpensive way to purify these small circular DNA molecules, by massively increasing their buoyant density relative to that of the host cell DNA.
Insofar as the separation step, the researchers were amply rewarded. But Form IV, at first, seemed to be permanently denatured, and renaturing it proved to be a rather difficult task [6] [7] [34] [35] . Perhaps for this reason, the use of alkali denaturation as a purification procedure for small circular DNA was never perfected.
For about a decade, from the mid-1960s to the mid-1970s, at least three major groups labored to characterize Form IV: Jerome Vinograd’s group at CalTech, Robert Warner’s group at UC Irvine, and the group at the Dutch National Defense Research Organization under Jansz, Pouwels and their associates. Although these three groups collectively published dozens of papers between them, not one of those papers made any mention whatsoever of structure, and, to this day, the closest thing we have previously had to a “molecular model” for Form IV was the artist’s conceptual drawing shown in Figure 13.
In a world in which man has gone to the moon, split the atom down to the quark and beyond, and explored the bottom of the sea, surely we need to do better than this.
The current manuscript provides the first detailed molecular model ever proposed for Form IV. The starting point for the model is the Wu tetraplex [17] [18] , which has previously been successfully deployed to solve another long-standing molecular biological mystery, namely the structure of the protamine-DNA complex in sperm cells [19] [20] [21] . Protamine, as the name implies, is the very essence of nucleoprotein structure, being little more than a long string of positive charges, mostly from arginine residues. Conversely, DNA is a long string of negative charges from its phosphate groups. One might have naively thought that the solution to the puzzle of protamine-DNA structure would therefore be a “no brainer”, but, for over a half-century after the publication of the Watson-Crick structure, the puzzle was never solved. That’s because it’s impossible to align the negative charges on DNA with the positive charges on protamine, if the DNA is presumed to be in the Watson-Crick conformation. But as soon as the DNA structure is changed to the Wu tetraplex, the protamine-DNA structure virtually solves itself.
Likewise, in the case of Form IV, where no molecular model has ever been proposed or even hinted at, the application of the Wu structure once again leads quickly to a solution.
It is my opinion, for what it’s worth, that the protamine-DNA structure, which has not been challenged or criticized in any way since its publication, is well-nigh incontrovertible. The same, however, cannot be said for the Form IV structure proposed herein. I am confident that the assignment of the Wu mutually-intercalated tetraplex

Figure 13. Early Form IV “model” [5] . The drawing seems to imply an imploded, totally-collapsed structure without order of any sort.
structure to the pre-Form-IV conformation, between pH 12.3 - 12.8, will hold up to scrutiny, but the events beyond pH 12.8 are speculative. There is, for example, no precedent I know of for a nucleic acid structure with axial phosphate groups stabilized by salt bridges with the prevailing counterion. But the structure is entirely logical, and devoid of any steric hindrances whatsoever, and one might well ask “Why should such a structure not form, under the extreme conditions at which it appears?” That is to say, there is nothing barring it from forming.
That the primary “cement” for the proposed Form IV tetraplex is ionic, is supported by the observation that it is at the low ionic strength conditions of DNA which has been prepared for electron microscopy (0.001 - 0.01 M sodium), and only under those conditions, that we see a return of the Form IV tetraplex structure to a 100% duplex form. This would appear to clearly support a role for salt bridges in the maintenance of the Form IV structure at high pH and ionic strength.
Concerning the events in the neutralization phase of the data presented herein, as the pH is lowered from 13 back down to 7, the picture remains rather murky. It seems almost certain that the structure, in that pH range, is a hybrid structure, consisting of one of two tetraplex structures that we can imagine (i.e., the Form IV structure proposed herein, and/or the Wu mutually-intercalated tetraplex structure), gradually converting back to a two-stranded form. The nature of the two-stranded form is also obscure. It cannot possibly be canonically base-paired, yet it must logically be either stabilized by tautomeric faux-base-pairing (which sort of structure is not currently known to exist), or else by “pure” base stacking in the complete absence of base-pairing (which is also not known to exist) (Figure 12). And yet there is no reason to doubt that either one of these peculiar structures is entirely possible, requiring only a setting which provides a thermodynamic path for their formation. The alkali denaturation of the native chromosome may very well provide that path.
I am a theoretical biologist, having no laboratory facilities with which to verify the Form IV model by experimentation. Moreover, I believe myself to be, at this moment in time, the only scientist still alive and working who has even the slightest interest in Form IV, wherefore it may be a long time before the day comes that this structure will be experimentally proven. Hopefully this publication will bring that day closer.
Cite this paper
Biegeleisen, K. (2016) The Probable Structure of “Form IV” (Alkali-Denatured Circular DNA). Open Access Library Journal, 3: e3114. http://dx.doi.org/10.4236/oalib.1103114
References
- 1. Dulbecco, R. and Vogt, M. (1963) Evidence for a Ring Structure of Polyoma Virus DNA. Proceedings of the National Academy of Sciences of the United States of America, 50, 236- 243.
http://www.pnas.org/content/50/2/236.full.pdf
http://dx.doi.org/10.1073/pnas.50.2.236 - 2. Cairns, J. (1963) The Bacterial Chromosome and Its Manner of Replication as Seen by Autoradiography. Journal of Molecular Biology, 6, 208-213.
http://dx.doi.org/10.1016/S0022-2836(63)80070-4 - 3. Kleinschmidt, A.K., Burton, A. and Sinsheimer, R.L. (1963) Electron Microscopy of the Replicative Form of the DNA of the Bacteriophage ?X174. Science, 142, 961.
http://dx.doi.org/10.1126/science.142.3594.961 - 4. Weil, R. and Vinograd, J. (1963) The Cyclic Helix and Cyclic Coil Forms of Polyoma Viral DNA. Proceedings of the National Academy of Sciences of the United States of America, 50, 730-738.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC221253/pdf/pnas00238-0150.pdf
http://dx.doi.org/10.1073/pnas.50.4.730 - 5. Vinograd, J., Lebowitz, J., Radloff, R., Watson, R. and Laipis, P. (1965) The Twisted Circular Form of Polyoma Viral DNA. Proceedings of the National Academy of Sciences of the United States of America, 53, 1104-1111.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC301380/pdf/pnas00157-0216.pdf
http://dx.doi.org/10.1073/pnas.53.5.1104 - 6. Strider, W. (1971) Denatured Replicative Form and Complex DNA of X174: Isolation, Renaturation, and Sedimentation Properties. PhD Dissertation, Department of Biochemistry, New York University School of Medicine, New York.
- 7. Strider W. and Warner, R.C. (1971) Denatured Replicative Form and Complex DNA of X174: Isolation and Renaturation. Federation Proceedings, 30, 1053.
- 8. Rush, M.G. and Warner, R.C. (1970) Alkali Denaturation of Covalently Closed Circular Duplex Deoxyribonucleic Acid. Journal of Biological Chemistry, 245, 2704-2708.
http://www.jbc.org/content/245/10/2704.full.pdf+html - 9. Biegeleisen, K. (2006) The Double Non-Helix, Part I: The Science and History of Topologically Non-Helical DNA. Slides 127-234.
https://notahelix.net - 10. Grossman, L., Watson, R. and Vinograd, J. (1974) Restricted Uptake of Ethidium Bromide and Propidium Diiodide by Denatured Closed Circular DNA in Buoyant Cesium Chloride. Journal of Molecular Biology, 86, 271-283.
http://dx.doi.org/10.1016/0022-2836(74)90018-7 - 11. Keller, W. (1975) Determination of the Number of Superhelical Turns in Simian Virus 40 DNA by Gel Electrophoresis. Proceedings of the National Academy of Sciences of the United States of America, 72, 4876-4880.
http://www.pnas.org/content/72/12/4876.full.pdf
http://dx.doi.org/10.1073/pnas.72.12.4876 - 12. Shure, M., Pulleyblank, D.E. and Vinograd, J. (1977) The Problems of Eukaryotic and Prokaryotic DNA Packaging and in Vivo Conformation Posed by Superhelix Density Heterogeneity. Nucleic Acids Research, 4, 1183-1206.
http://nar.oxfordjournals.org/content/4/5/1183.full.pdf+html
http://dx.doi.org/10.1093/nar/4.5.1183 - 13. Crick, F.H.C., Wang, J.C. and Bauer, W.R. (1979) Is DNA Really a Double Helix? Journal of Molecular Biology, 129, 449-461.
http://profiles.nlm.nih.gov/ps/access/SCBCDD.pdf
http://dx.doi.org/10.1016/0022-2836(79)90506-0 - 14. Stanford University Biochemistry 201 (2000).
- 15. Bauer, W. and Vinograd, J. (1968) The Interaction of Closed Circular DNA with Intercalative Dyes. I. The Superhelix Density of SV40 DNA in the Presence and Absence of Dye. Journal of Molecular Biology, 33, 141-171.
http://dx.doi.org/10.1016/0022-2836(68)90286-6 - 16. Stettler, U.H., Weber, H., Koller, T. and Weissmann, C. (1979) Preparation and Characterization of form V DNA, the duplex DNA Resulting from Association of Complementary, Circular Single-Stranded DNA. Journal of Molecular Biology, 131, 21-40.
http://dx.doi.org/10.1016/0022-2836(79)90299-7 - 17. Wu, R. and Wu, T.T. (1996) A Novel Intact Circular dsDNA Supercoil. Bulletin of Mathematical Biology, 58, 1171-1185.
http://dx.doi.org/10.1007/BF02458388 - 18. Wu, T.T. (1969) Secondary Structures of DNA. Proceedings of the National Academy of Sciences of the United States of America, 63, 400-405.
http://www.pnas.org/content/63/2/400.full.pdf+html
http://dx.doi.org/10.1073/pnas.63.2.400 - 19. Biegeleisen, K. (2006) The Probable Structure of the Protamine-DNA Complex. Journal of Theoretical Biology, 241, 533-540.
http://dx.doi.org/10.1016/j.jtbi.2005.12.015 - 20. Biegeleisen, K. (2006) The Double Non-Helix, Part 2: The Probable Structure of the Protamine-DNA Complex.
https://notahelix.net - 21. Biegeleisen, K. (2005) Protein Data Bank, Accession Numbers 2AWR (Protamine-DNA Complex 1) and 2AWS (Protamine-DNA Complex 2).
http://www.rcsb.org/pdb/ - 22. Gehring, K., Leroy, J.L. and Gueron, M. (1993) A Tetrameric DNA Structure with Protonated Cytosine. Cytosine Base Pairs. Nature, 363, 561-565.
http://dx.doi.org/10.1038/363561a0 - 23. Biegeleisen, K. (2016) Form IV: The Final Puzzle Piece. Slides 93-108.
https://notahelix.net - 24. Biegeleisen, K. (2016) Form IV: The Final Puzzle Piece. Slides 195-212.
https://notahelix.net - 25. Yakovchuk, P., Protozanova, E. and Frank-Kamenetskii, M. (2006) Base-Stacking and Base-Pairing Contributions into Thermal Stability of the DNA Double Helix. Nucleic Acids Research, 34, 564-574.
http://dx.doi.org/10.1093/nar/gkj454 - 26. Biegeleisen, K. (2002) Topologically Non-Linked Circular Duplex DNA. Bulletin of Mathematical Biology, 64, 589-609.
http://dx.doi.org/10.1006/bulm.2002.0288 - 27. Biegeleisen, K. (2006) The Double Non-Helix, Part I: The Science and History of Topologically Non-Helical DNA. Slides 235-255.
https://notahelix.net - 28. Biegeleisen, K. (2016) Form IV: The Final Puzzle Piece. The Structure Is Developed in Slides 218-245. The Final Model Is Shown in Slides 239 and 244-245. A Jmol Model and a Downloadable PDB Structure File Are Provided.
https://notahelix.net - 29. Biegeleisen, K. (2016) Form IV: The Final Puzzle Piece. Slides 240-243.
https://notahelix.net - 30. Harding, M. Metal Coordination Sites in Proteins. Centre for Translational and Chemical Biology, University of Edinburgh, Edinburgh.
- 31. Topal, M. and Fresco, J. (1976) Complementary Base Pairing and the Origin of Substitution Mutations. Nature, 263, 285-289.
http://dx.doi.org/10.1038/263285a0 - 32. Biegeleisen, K. (2016) Form IV: The Final Puzzle Piece. Tautomeric Base Pairing Is Reviewed in Slides 109-125.
https://notahelix.net - 33. Ibid. The Preference of DNA for the Left-Handed (“Z”) Conformation, under Conditions Approaching Denaturation, Is Reviewed in Slides 49-59.
- 34. Strider, W., Camien, M. and Warner, R. (1981) Renaturation of Denatured, Covalently Closed Circular DNA. Journal of Biological Chemistry, 256, 7820-7829.
http://www.jbc.org/content/256/15/7820.full.pdf - 35. Biegeleisen, K. (2016) Form IV: The Final Puzzle Piece. Renaturation of Form IV Is Reviewed in Slides 23-44.
https://notahelix.net