R. LARABI ET AL.
Copyright © 2011 SciRes. OJPC
43
respectively and the 6-31G* to 6-311G* basis extension
increase slightly the activation barrier energy.
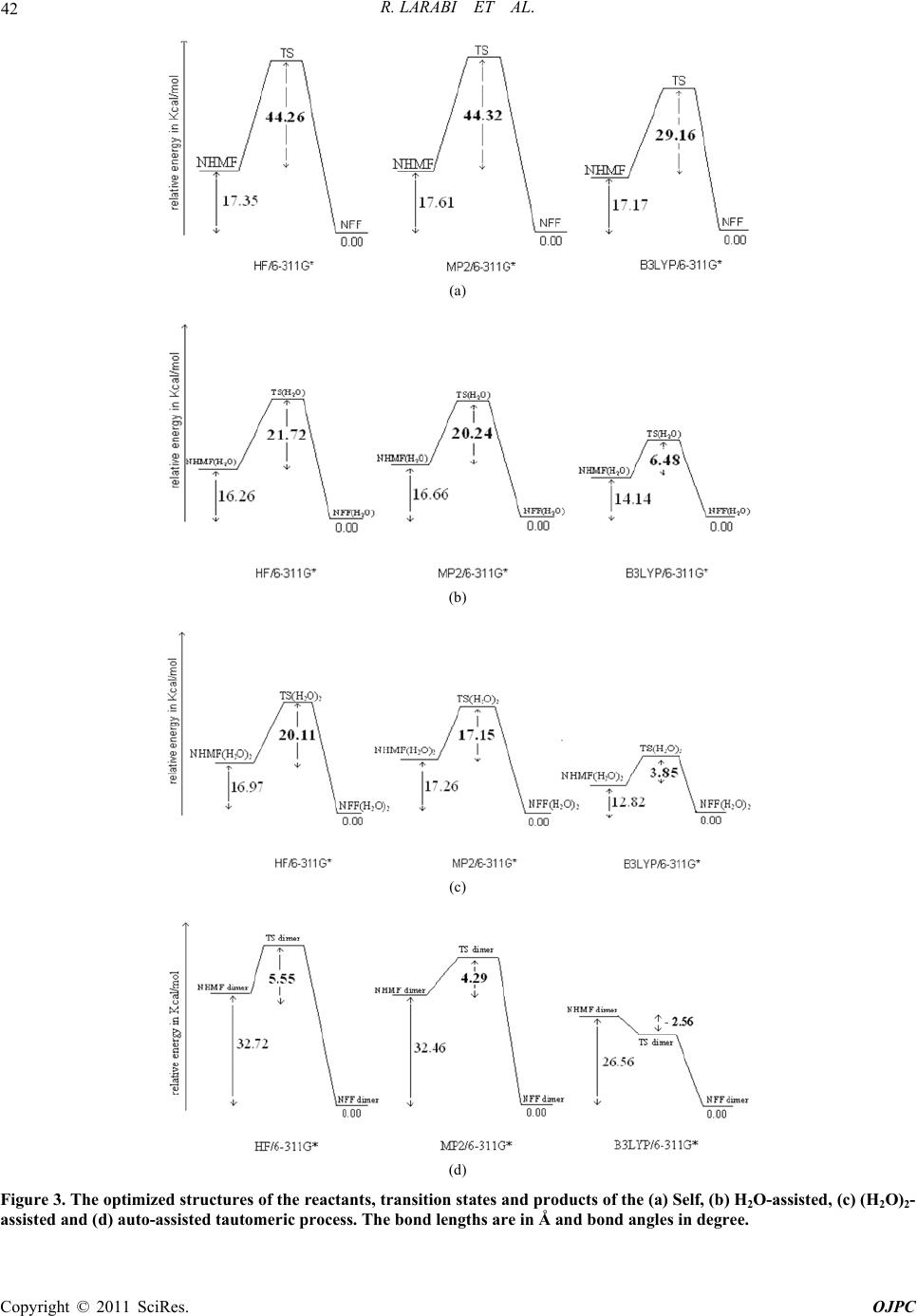
In another hand, the results, obtained at the different
level of theory (with ZPE correction), show that the
NHMF (H2O)2→NFF(H2O)2 is exothermic by 16.97, 17.26
and 12.82 kcal/mol at the HF, MP2 and B3LYP//6-311G*
level, respectively. These energies barriers are the same
with those obtained for the NHMF (H2O)→NFF(H2O)
reaction for the proton transfer when we use one water
molecule. The DFT/B3LYP level, conduce to the small-
est energy barrier, see 3.85 kcal/mol. This proves that the
second water molecule addition doesn’t have an effect on
all the different energetic value.
The NHMF-dimer→NFF-dimer reaction is exothermic
of 32.72, 32.46 and 26.56 kcal/mol. at the HF, MP2 and
DFT/B3LYP//6-311G* level, respectively. The transition
state, compared to the reactant one, is located at 5.55,
4.29 kcal/mol. at the HF, MP2//6-311G*, respectively
and –2.56 kcal/mol. at the DFT/B3LYP//6-311G* level.
The latter value is probably due to a spontaneous NHMF
NFF reaction. In the NHMF-dimer tautomerization, the
energy barrier decreases about 90% in comparison with
the self-NHMF tautomerization. According to this study,
we conclude that the NHMF→NFF reaction is au-
to-assisted by the NHMF-dimer proton transfer with the
smallest barrier and a good structural arrangement that
favoured the proton transfer mechanism.
4. Conclusions
The introduction of one or more water molecules has not
a great effect of the NFF geometrical parameters con-
trary to the NHMF ones. The water molecule restores the
planarity of the systems without affecting the no-conju-
gaison of the two C=N and C=O double bonds.
In all cases, the NFF molecules are more stable then
the NHMF ones.
All theoretical levels, in aqueous environments, tend
to decrease the energy barrier and the energy tautomeri-
zation compared to the gas phases. The HF and MP2
results promote the existence of the two NHMF and NFF
separate molecules in the gas phase and hydrated ones.
DFT leads a lower barrier energy compared with those
obtained at the HF and MP2 levels.
Thus this method provides the simultaneous existence
of the both molecules in the both environments. Finally,
the NHMF→NFF reaction is established on a self-as-
sisted NHMF dimmer with low barrier energies.
5. References
[1] M. Brahimi, Y. Belmiloud and D. Kheffache, “Har-
tree-Fock, Post Hartree-Fock and Density Functional
Theory Studies on Structure and Conformationa,” Jour-
nal of Molecular Structure: THEOCHEM, Vol. 759, No.
1-3, 2006, pp. 1-10. doi:10.1016/j.theochem.2005.10.017
[2] S. Petai, “The Chemistry of the Carbone-Nitrigene Dou-
ble Bond,” Interscience Publishers, London, New York,
1970, Chapter 1, p. 2.
[3] P. Bour, C. N. Tam, J. Sopkova and F. R. Trouw, “Meas-
urement and Ab Initio Modeling of the Inelastic Neutron
Scattering of Solid N-Methylformamide,” Journal of
Chemical Physics, Vol. 108, No. 1, 1998, p. 351-359.
doi:10.1063/1.475382
[4] V. R. Palakrishnan, G. Madrid, G. Cuevas and A. Thagler,
“Density Functional Studies of Molecular Structures of
N-Methyl Formamide, N,N-Dimethyl Formamide, and
N,N-Dimethyl Acetamide,” Proceedings of the Indian
National Science Academy: Chemical Science, Vol. 112,
2000, pp. 35.
[5] G. De Mare, Journal of Molecular Structure, Vol. 107,
1984, pp. 127-132.
[6] M. Nagaoka, Y. Okuno and T. Yamabe, “Chemical Reac-
tion Molecular Dynamics Simulation and the En-
ergy-Transfer Mechanism in the Proton-Transfer Reac-
tion of Formamidine in Aqueous Solution,” Journal of
the American Chemical Society, Vol. 113, 1991, pp. 769.
[7] A. Engdahl, B. Nelander and P. O. Astrand, “Complex
Formation between Water and Formamide,” Journal of
Chemical Physics, Vol. 99, No. 7, 1993, pp. 4894-4908.
doi:10.1063/1.466039
[8] X. C. Wang, J. Nichols, M. Feyereisen, et al., “Ab Initio
Quantum Chemistry Study of Formamide-Formamidic
Acid Tautomerization,” Journal of Physical Chemistry,
Vol. 95, No. 25, 1991, p. 10419-10424.
doi:10.1021/j100178a032
[9] J. D. Pranata and D. Geraldine, “Computational Investi-
gations of Reactive Intermediates in the Acid-Catalyzed
Proton Exchange in Formamide,” Journal of Physical
Chemistry, Vol. 99, No. 39, 1995, p. 14340-14346.
doi:10.1021/j100039a022
[10] R. L. Bell, D. L. Taveras, T. N. truong and J. simons, “A
Direct Ab Initio Dynamics Study of the Water-Assisted
Tautomerization of Formamide,” International Journal of
Quantum Chemistry, Vol. 63, No. 4, 1997, pp. 861-874.
[11] A.-P. Fu, H.-L. Li, D.-M. Du and Z.-Y. Zhou, “Theoreti-
cal Study on the Reaction Mechanism of Proton Transfer
In Formamide,” Chemical Physics Letters, Vol. 382, No.
3-4, 2003, pp. 332-337. doi:10.1016/j.cplett.2003.10.070
[12] Gaussian 03, Revision A.1, M. J. Frisch, G. W. Trucks, H.
B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheese-
man, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C.
Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone,
B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Pe-
tersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R.
Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,
O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P.
Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gom-
perts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cam-
mi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Moro-
kuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G.