Journal of Biomedical Science and Engineering
Vol.6 No.5(2013), Article ID:31888,11 pages DOI:10.4236/jbise.2013.65071
Long term survival and limited migration of genetically modified monocytes/macrophages grafted into the mouse brain*

1Centre de Recherche de l’Institut du Cerveau et de la Moelle Epinière, Equipe de Biotechnologie et Biothérapie, Hôpital Pitié- Salpêtrière, Paris, France
2CEA, Institute of Emerging Diseases and Innovative Therapies, Division of Immuno-Virology, UMR E1 CEA and University Paris South, Fontenay-aux Roses, France
3Centre d’Etudes d’Agents Pathogènes et Biotechnologies pour la Santé (CPBS), UMR 5236, Montpellier, France
4Laboratoire de Modélisation des Biothérapies, Groupe INSERM U986, MIRCen, CEA, Fontenay-aux-Roses, France
Email: chamsy.sarkis@newvectys.com, gabriel.gras@cea.fr, francoise.sanchez@cpbs.cnrs.fr, jacques.mallet@upmc.fr, #che.serguera@cea.fr
Copyright © 2013 Chamsy Sarkis et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 20 February 2013; revised 3 April 2013; accepted 7 May 2013
Keywords: Adenoviral Vector; Human Myeloid Cells; β-Glucuronidase; Ex Vivo Gene Therapy; Neurotransplantation
ABSTRACT
In mammals, myeloid progenitors infiltrate the developing central nervous system (CNS), through the immature blood-brain barrier (BBB), the ventricular layer or the pial surface migrate and give rise to resident microglia. In the mature brain, however, the BBB hampers such recruitment from the blood-stream and long-term establishment of blood borne myeloid cells in the CNS thus appears at best limited. Hematopoietic stem cell-derived microglia, nevertheless, represents a promising tool for the correction of genetic deficits in the brain. We thus investigated the fate of primary human monocytes, and monocyte-derived macrophages, following transplantation into the adult mouse brain overpassing the BBB. Furthermore, we documented the ability of such cells to deliver a lysosomal enzyme into the brain following genetic modification with a recombinant adenoviral vector carrying the human β-glucuronidase cDNA. When implanted into the mouse striatum, the engineered primary cells survived and expressed the transgene for as much as 8 months. Moreover, the donor cells could migrate out of the grafting site and settle along blood vessels or myelin tracts although at limited distance. Migrating donor cells down-regulated the expression of CD14 and HLA DR, suggesting the adoption of a deactivated microglia-like phenotype. Our observations establish the ability of circulating mononuclear phagocytes to integrate into the brain after transplantation and express a transgene on the long term. These cells might thus be employed for autologous transplantation for the delivery of secreted therapeutic proteins in the context of a wide range of brain affections.
1. BACKGROUND
Monocytes (Mo) belong to the hematopoietic myeloid lineage and circulate in the blood with a half-life of about 48 hours. They become tissue macrophages (MØ) as they leave the bloodstream and enter tissues [1], where they become resident for periods ranging from hours to years [2]. Tissue MØ are found in virtually all organs of mammals and display heterogeneous functional and phenotypic features according to their environment [3]. These cells are critical to tissue homeostasis as they scavenge cell debris and foreign particles and are, as well, central in initiating adaptive immunity [3].
Among all different organs, the CNS displays unique features such as an immune privilege environment and a particular BBB, which may explain the specificities of brain MØ populations, i.e. perivascular brain MØ and resident microglia. As these cells share many markers with Mo/MØ they are considered as the resident MØ of the brain [4]. Microglia are however a peculiar longlived population of MØ with specific characteristics that distinguishes them from other tissue MØ. In particular, gross renewal of resident microglia appears independent from bone marrow precursors in the adult life [5,6] even though blood derived cells may contribute to some microglia turnover, especially at the perivascular location [4], a property is used to intend for replacing resident microglia and complement genetic deficits in the brain [7].
Indeed, circulating Mo/MØ have been considered as possible vehicles for delivering therapeutic molecules through the organism, preventing in many cases the indication of hematopoietic stem cells transplantation (HSCT) and myeloablation. So far, MØ have been administered intravenously as therapeutic effectors in several models of diseases such as cancer [8], muscular damage [9], neuro AIDS [10] or lysosomal enzyme deficiencies [11,12]. In the CNS, grafts of MØ have been performed either to survey their fate and survival [13-15] or to evaluate a therapeutic contribution in spinal cord or brain models of injury [16,17] or neurodegenerative processes like Parkinson disease [18].
In adults, whether microglia cells are replaced by inner brain precursors or by circulating myeloid precursors still remains a controversial question [4,6]. In the mouse, following bone marrow transplantation [19] or intravenous mononuclear cell transfusion [11,12,20], donor Mo/MØ preferentially colonize the liver and the spleen and poorly enter the adult brain [19,21] in the absence of a lesion [22-24] or compared to newborn animals [11]. Such exclusion from the brain is due to the BBB that is not fully functional at birth and altered by inflammatory signals [4]. The use of Mo/MØ as vectors for therapeutic gene expression in the brain in place of hematopoietic stem cell transplantation is thus not obvious. On another hand, cerebral grafting of genetically modified Mo/MØ overpasses the BBB and might thus be an alternative to HSCT if the grafted cells establish locally on the long term with the expected therapeutic properties. To address this issue, we verified whether primary monocytes or macrophages could establish and settle within the CNS following cerebral injection. With that aim, purified human Mo and Mo-derived MØ were transduced with an adenoviral vector over-expressing the human lysosomal enzyme β-glucuronidase and were then grafted into the striatum of nude mice. We report that these cells efficiently engraft within the host brain, produce the transgene for extended periods of time and migrate out of the injection site even though at limited distance.
2. METHODS
2.1. Cells and Media
The culture medium was RPMI 1640, 1% L-glutamine, 1% penicillin/streptomycin (Life Technologies, Paisley, UK), 50 μM 2-Mercapto Ethanol (Sigma, St Louis, MO), 10% FCS (Dominique Dutscher, Brumath, France). Mo were obtained from Ficoll (Seromed, Biochrome, Berlin, Germany) separated PBMC of healthy volunteers, depleted of T and B-lymphocytes with M-450 Pan T/ CD2 and Pan B/CD19 Dynabeads (Dynal, Oslo, Norway) at 1:1 bead:cell ratio. The recovered population routinely contained >90% CD14+ cells. Cells were cultured in Costar plates (Cambridge, MA). Recombinant human MCSF (50 U/ml; R & D Systems, Minneapolis, MN) was used for Mo differentiation into MØ. Cultures were fed every 2 days by replacing 1:2 of the medium.
2.2. Structure of the Viral Vector, Cell Transduction and Flow Cytometric Cell Surface Marker Analysis
A second-generation adenoviral vector carrying the human β-glucuronidase cDNA under the control of the early CytoMegaloVirus promoter (Ad/CMV*β-glu) was generated as described [25]. The vector consisted in the CMV/β-glucuronidase.pA cassette in place of the E1 region, an E3-deleted background from the sub360 mutant, and a thermosensitive (ts) mutation in the E2A region (G to A conversion at position 1064 of the DBP cDNA generating a P to S amino acid change). Viruses were purified by three rounds of plaque purification at the permissive temperature of 32˚C. Stocks were devoid of detectable revertants as assayed by plating at 39˚C. The vector was amplified in 293 cells, purified by 2 successive CsCl gradient centrifugations and resuspended in PBS. The viral titer was 1.25 × 1012 particles/ml and 5 × 109 transducing units (TU)/ml. TU were assayed by infecting mucopolysacharidosis (MPS) VII fibroblasts with limiting dilutions and then staining for β-glucuronidase. Virus aliquots were stored at –80˚C in PBS, 10% glycerol (v/v) until use. For β-glucuronidase gene transduction, Mo or MØ, were seeded in 24-well tissue culture plates at a density of 5 × 105 cells/well, incubated with the Ad/CMV*β-glu vector at multiplicities of infection (M.O.I.) of 10, 50 or 100 in 400 μl medium. After 4 hours, supernatants were discarded and replaced by an equal volume of fresh medium. For analysis of β-glucuronidase expression, cells and supernatants were collected at various time points after infection and frozen until enzymatic assay. For grafting, freshly thawed Mo or previously M-CSF-differentiated MØ were incubated for 4 h with the Ad/CMV*βglu vector at a M.O.I. of 100. Supernatant was replaced by fresh serum-free medium. Eight hours after transduction, cells were harvested with 0.02% EDTA in PBS (10 min at 37˚C), resuspended in defined serum-free medium at 5 × 104 cells/μl, and kept on ice throughout the grafting session. Each cell population was evaluated for surface marker expression by flow cytometric analysis with PEor FITC-conjugated monoclonal antibodies (mAbs) to CD14 (BD Bioscience, Mountain View, CA) and HLA-DR (Dako, Glostrup, Denmark). The cells were incubated for 30 min at 4˚C in PBS, 2% FCS with the mAb or with irrelevant isotypematched mAb. After washing, the cells were resuspended in PBS 1% PFA (w/v). For each population, the percentage of cells expressing the respective phenotypic marker was assessed with a FACScalibur (BD Bioscience, Mountain View, CA).
2.3. Animals, Surgical Procedures and Cell Grafts
Nude male CD1 mice (Charles Rivers, Saint Aubain les Elbeuf, France) were housed and treated according to the European Community (86/609/EEC) guidelines. After approval by the local ethics committee, twelve 6-weekold mice (n = 6 for each cell type) were grafted bilaterally in both hemispheres with 5 × 104 cells, either unmodified (right striatum) or transduced with Ad/CMV*β- Glu (left striatum). Animals were first anesthetized with 1.5 ml/kg of a 1:1 mixture of ketamine (UVA, Ivry-surSeine, France) and Rompun (Bayer, Puteaux, France) by intraperitoneal injection. Using a 10 μl Hamilton syringe, 1 μl of cell suspension was injected stereotaxically into the striatum (0.25 μl/min) at the following coordinates: +4.7 anterior to bregma, +1.7 lateral to midline, –3.6 ventral to the dural surface. For each group of grafted cells, mice were euthanized 10 (n = 2), 30 (n = 3) or 240 (n = 1) days after grafting.
2.4. Fluorimetric Assay for β-Glucuronidase
Five days following Ad/CMV*β-glu transduction, cells were assayed for intracellular and secreted β-glucuronidase. Cell supernatants were collected after 24 h, centrifuged at 350 × g for 20 min at 4˚C and transferred into a new tube and frozen at –80˚C until use. Cells were trypsinized, washed in PBS, counted, centrifuged and resuspended in 100 μl cold lysis buffer (Tris-HCL pH 7.5 0.02 M; NaCl 0.05 M; Triton X-100 0.2%). Cell lysates were vortexed, kept on ice for 15 min, and then centrifuged at 12.000 × g for 20 min. Beta-glucuronidase activeity was assayed as follows: 10 μl of cell lysate or of supernatant were incubated with 100 μl of 10 mM 4-methylumbelliferyl-β-Dglucuronide (MUG, Sigma) in 0.1 M sodium acetate (pH 4.8) for 1 h at 37˚C. The reaction was stopped by adding 1.9 ml 0.32 M glycine, 0.2 M carbonate buffer, pH 10.5. Fluorescence was measured using the umbelliferone protocol of the Victor 1420 Multilabel counter (355 nm excitation, 460 nm emission) (Wallac, Turku, Finland). One unit β-glucuronidase activity was defined as 1 nmol 4-methylumbelliferone (4- MU) (Sigma) cleaved per hour, and fluorescence values were used to calculate units according to a calibretion curve of 4-MU fluorescence. Protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad Lab., Hercules, CA).
2.5. Histochemical and Immunohistochemical Analysis
Anesthetized animals were perfused intracardiacally with cold PBS and then with 4% PFA in PBS (w/v). Brains were removed, frozen in cold isopentane and cryopreserved in 15% sucrose. Twenty μm thick tissue sections were cut with a cryostat and used for all further histological analyses. For β-glucuronidase staining, tissue sections and cells in chambered slides were post-fixed in chloral-formal-acetone fixative solution, incubated in PBS at 65˚C for 90 min to inactivate murine β-glucuronidase and then washed twice with 0.05 M sodium acetate, pH 4.5, and incubated in 0.25 mM Naphtol-ASBi-β-D-Glucuronide (Sigma) in 0.05 M sodium acetate, pH 4.5, for 4 h at 4˚C. They were then incubated in 0.25 mM Naphtol-AS-Bi-β-D-Glucuronide (Sigma) in 0.05 M sodium acetate pH 5.2 with 1:500 2% hexazotized pararosaniline (Sigma) for 4 h at 37˚C (30). Transplanted cells were characterized by standard immunohistochemistry using, the following human-specific primary mAbs: anti-CD14 (BD Bioscience, 1:50), anti-CD68 (Dako, KP1 1:50), anti-human-adrenoleukodystrophy protein (hALDP) (Rabbit Polyclonal, 1:5000, a kind gift from N. Cartier, INSERM UMR745, University Paris-Descartes, Paris, France), and anti-HLA-DR (Dako, 1:50). Tissue sections on sister slides were thawed and fixed in 4% PFA in PBS for 20 min. Slides were incubated for 30 min in PBS, 10% goat serum and 0.2% Triton X-100, and then with the first antibody overnight at 4˚C. For immunofluorescence, sections were washed in PBS and incubated with the species-specific secondary antibody: FITC-conjugated goat anti-mouse IgG (Caltag, Tebu, Le Perray en Yvelines, France) or CY3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA).
2.6. Statistical Analysis
Bar graphs represent mean ± SD. Significance of difference between experimental variables was determined by unpaired two-tailed t-test. Differences were considered significant at p < 0.05.
2.7. In Situ Hybridization (ISH)
Human β-glucuronidase transcripts were detected in brain cryosections by ISH with a digoxygenin-11-UTP (Promega, Madison, WI) labeled cRNA probe directed against the human β-glucuronidase mRNA sequence, and consisting in the last 370 b.p. of the human cDNA [26]. Hybridization was performed overnight at 65˚C in 1x SSC, 50% formamide, 10% dextran sulfate, 1 mg/ml rRNA, 1× Denhardt’s solution. Sections were washed twice at 65˚C with 1× SSC, 50% formamide, 0.1% tween 20 and twice at room temperature in 1× TBS-T (Tris buffer saline, Tween 20). The digoxygenin-labeled hybrids were detected with an alkaline phosphatase-conjugated antidigoxigenin antibody (Roche Molecular Biochemicals, Mannheim, Germany).
3. RESULTS
3.1. Adenoviral Gene Transduction Slightly Shifts Mo and M Markers Expression
Here we tested the susceptibility of Mo and MØ to gene transduction by a 2nd generation adenovirus vector carrying the human β-glucuronidase cDNA (Ad/CMV*β- glu). To characterize each cell population, we used specific antibodies directed to well known phenotypic markers [27]. Non-transduced Mo and MØ were about 93% CD14+ and moderately expressed HLA-DR+ (Figure 1) but were CD1a– (not shown). After five days, cells transduced as just thawed Mo or as Mo differentiated MØ with M-CSF, displayed a 2 to 3 fold increase of HLA-DR expression and a 2 fold decrease of CD14 expression as compared to non transduced controls, while none expressed CD1a (not shown). This indicates that adenoviral transduction marginally altered expression of CD14 and HLA-DR in cells transduced as MO and MØ but did not induce their differentiation into dendritic cell.
In addition, transduced and control Mo cultured without cytokines, equally adhered to the plastic dishes and underwent spontaneous differentiation into round or spindle-shaped MØ with an immunophenotype similar to that of M-CSF differentiated MØ (not shown). Thus, the ability of Mo to differentiate into MØ either spontaneously or in response to M-CSF was not altered by Ad/ CMV*βglu transduction.
3.2. Transduced Mo and M Differently Express the Recombinant β-Glucuronidase in Vitro
We next determined whether transduction of Mo and MØ with Ad/CMV*β-glu led to increased expression and secretion of β-glucuronidase. To this end, freshly thawed Mo or in vitro-differentiated MØ were incubated with the vector at various M.O.I. (10, 50 and 100) and kept in culture for six days before β-glucuronidase activity was measured in cell lysates and culture supernatants. The
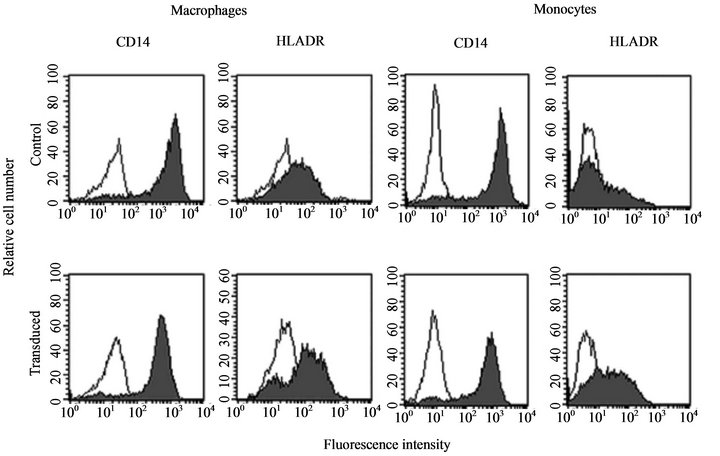
Figure 1. Immunophenotype of control and transduced monocytes and macrophages. Just thawed primary human monocytes (right panel) and primary human macrophages (monocytes differentiated with M-CSF) (left panel) were transduced (lower panel) or not (upper panel) with Ad/CMV*βglu at an M.O.I. of 100, put in culture with M-CSF for 5 additional days and stained with antibodies directed to CD14 and HLA-DR. FACS analysis shows that both monocytes and monocytes derived macrophages equally react to adenoviral transduction that induce a 2 fold decrease in CD14 expression and a 2 to 3 fold increase in HLA DR expression.
Intracellular β-glucuronidase activity significantly increased with M.O.I. in both cell types showing their susceptibility to adenovector transduction, but the levels of transgene expression were much lower in Mo than in MØ (Figure 2(a)). β-glucuronidase secretion also varied according to the cell type. At an M.O.I. of 100, Mo released low levels of enzyme that were only twice the ones of control Mo, whereas secretion considerably increased with M.O.I. in transduced MØ as compared to control MØ (31 fold at M.O.I. of 100, Figure 2(b)). This indicates that differentiation of Mo into MØ prior transduction, increases these cells ability to process the adenoviral vector and express the β-glucuronidase transgene.
3.3. Grafted Mo and M Establish in the Mouse Brain
Mo and MØ might be useful vehicles for ex vivo gene therapy in the brain, depending on their ability to establish long-term expression of a transgene in the target tissue. For this, cell availability is a critical parameter and, from this point of view, Mo as well as Mo-derived
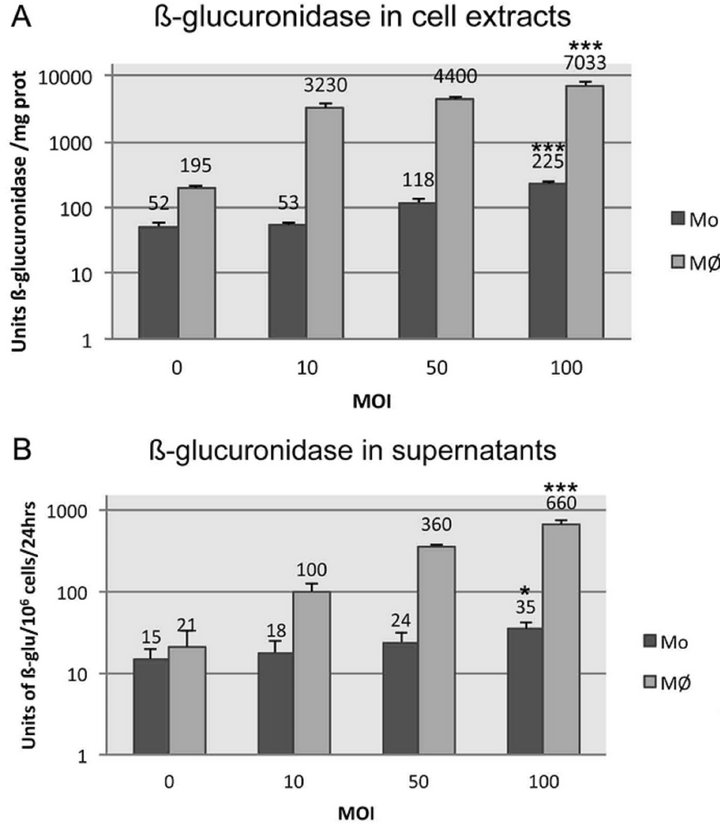
Figure 2. Beta-glucuronidase production and secretion by monocytes and macrophages transduced or not with Ad/CMV* βglu at M.O.I. of 10, 50 or 100. (A) Intracellular β-glucuronidase activity in units per mg of protein in cell lysates, five days after transduction; (B) β-glucuronidase activity released in supernatants in 24 h, five days after transduction. Mean values ± SD of triplicate experiments are shown. Statistical comparison of β-glucuronidase activity between non-transduced cells lysates or supernatants and that of cells transduced at M.O.I. 100 was performed by unpaired two-tailed t-test. Differences were considered significant at p < 0.05.
MØ may be suitable sources of autologous cells as they are abundant and accessible. In this experiment we assessed the survival and location of genetically modified Mo and MØ, grafted in the mouse striatum immediately after gene transduction, as well as transgene expression. To prevent cell rejection, we used athymic nude mice. Animals received a bilateral graft of transduced and nontransduced cells, in both hemispheres. The 12 recipient mice recovered well from transplantation and lived up to 240 days following surgery suggesting that the procedure was not pathogenic to nude mice.
To check for cell survival, location, and transgene expression, animals were euthanized 10, 30 and 240 days after transplantation, and brains were dissected and cut rostrally.
To assess the phenotype of donor cells, we used the same anti-CD14 and anti-HLA DR antibodies used for FACS phenotyping that specifically recognize human antigens expressed by these cells. In animals injected with PBS, no labelling was observed with these antibodies (not shown) while they allowed the detection of transplanted transduced and control cells. Ten days after grafting, Mo and MØ, either transduced or not, gathered at injection site, strongly expressed HLA-DR and CD14 (Figure 3(a)) but neither CD1a nor CD83, two markers of myeloid dendritic cells [27] (data not shown), indicating in either cases an activated MØ phenotype.
After 30 days, both transduced and control cells remaining in the grafting area, still strongly expressed HLA-DR (not shown). At close distance from the core of transplant though, numerous donor cells, transduced or not, were observed along blood vessels scattered at perivascular location (Figures 3(b) and (d)) displaying a weaker expression of CD14 and HLA-DR (Figure 3(b)). In the corpus callosum these markers could not be detected, whereas both human β-glucuronidase histochemistry and in situ hybridization indicated that donor cells had indeed migrated to this area (see below), strongly suggesting both CD14 and HLA-DR down modulation. We thus sought for donor cells in the corpus callosum using an anti-human CD68 antibody, recognizing myeloid human cells, and an anti-ALDP antibody, which specifically recognizes the human peroxisomal adrenoleukodystrophy protein (hALDP) expressed by myeloid cells [28]. In brains grafted with Mo or MØ, but not in mock brains, anti-CD68 and anti-hALDP antibodies indeed allowed detection of donor cells. These markers were co-localized in the same cells and the detection of the hALDP gave a characteristic punctuated staining (Figure 3(f)) consistent with the peroxisomal expression of this protein [28]. In hemispheres grafted with transduced cells, CD68- and hALDP-expressing cells were observed at the injection site or at close distance, associated to blood vessels (Figures 3(c) and (d)), but also
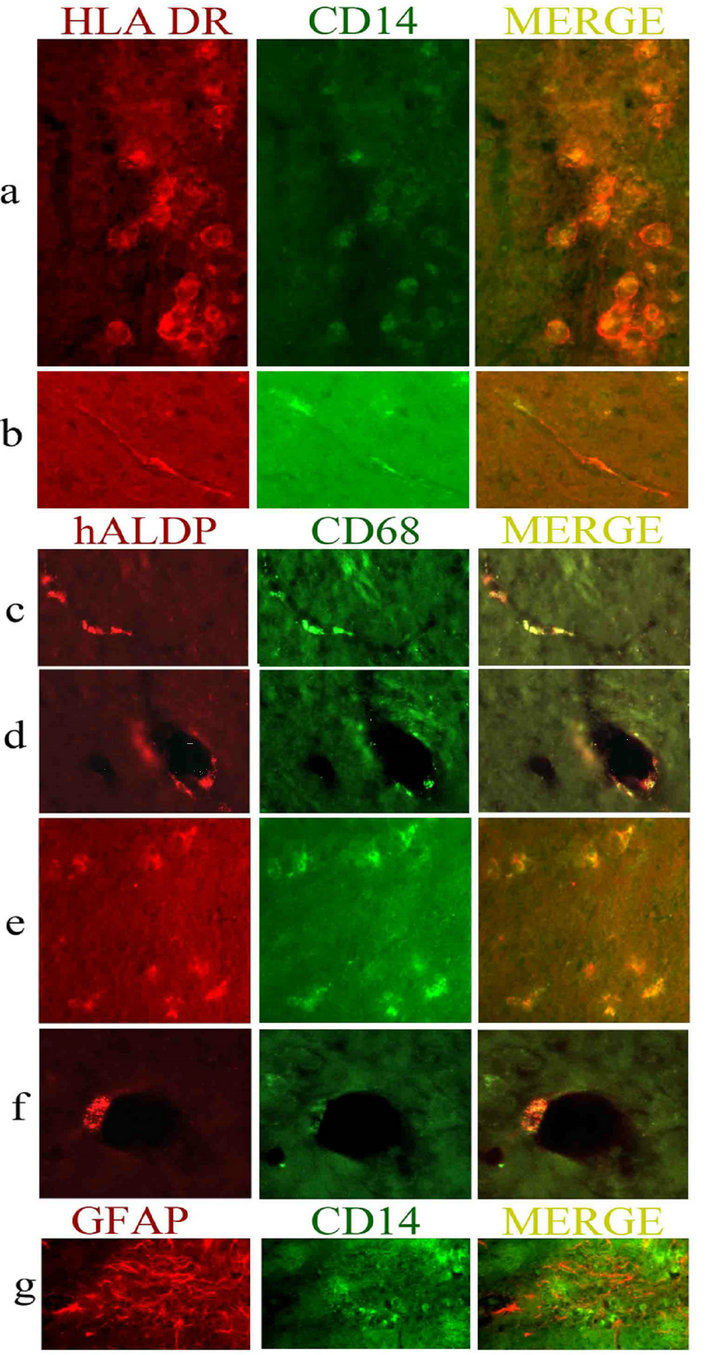
Figure 3. Phenotypic characterization and microscopy of transduced Mo and MØ, following grafting in the mouse striatum. (a) and (b) Immunohistochemical labelling of transduced Mo and MØ with HLA DR (red) and CD14 (green). (a) Mo at the site of injection 10 days after grafting, strongly express HLA DR and CD14 (magnification ×200); (b) Near the site of grafting in the striatum, a MØ expressing HLA-DR and low levels of CD14, migrating along a blood vessel 10 days after grafting (×400); (c) to (f) Immunohistochemical labelling of transduced Mo and MØ with hALDP (red) and CD68 (green); (c) and (d) Mo expressing hALDP and CD68 out of the grafting zone established or migrating along blood vessels: longitudinal (c) and transversal (d) views (×100) 30 days after grafting; (e) Donor Mo expressing CD68 and hALDP in the corpus callosum 30 days after grafting (×100); (f) Perivascular MØ expressing CD68 and hALDP, 30 days after grafting at high magnification (×400); (g) GFAP and CD14 expression at the site of grafting 10 days after injection (×100).
within the corpus callosum (Figure 3(e)), or located lining the ventricle or along myelin tracts within the internal capsula (not shown). On the contralateral hemisphere, non-transduced human cells expressing hALDP and CD- 68 were also detected at the site of injection within the striatum and at close distance of it (not shown) but were absent form the corpus callosum suggesting that transduced Mo and MØ have improved migrating capabilities.
Of 2 animals receiving grafts of Mo or MØ, kept for analysis at long term, that receiving Mo dyed prematurely 5 month after grafting and could not be analysed. Thus, eight months after grafting, in one animal grafted with human MØ, a few CD14/HLA-DR-positive cells, either transduced or not, were still present near the injection site in the parenchyma or along blood vessels. At distance from the injection site, few CD68/ALDP positive cells were still present at periventricular or perivascular locations, only in hemispheres grafted with transduced cells (not shown). Neither at any time after grafting, nor at any location in the recipient brains could we find cells co-expressing CD68 or ALD with glial markers (GFAP or CAII) or neuronal marker (NeuN) (not shown). However, 10 and 30 days after transplantation, GFAP immunostaining was up-regulated around the site of injection indicating local gliosis, which was equivalent for transduced and control cells (Figure 3(g)).
3.4. Genetically Modified Mo and M Similarly Express the Recombinant β-Glucuronidase in the Transplanted Brain
To detect human β-glucuronidase expression we used both enzymatic histochemistry and in situ hybridization (ISH). Brain sections were assayed by histochemistry for human β-glucuronidase activity following heat inactivation of endogenous murine β-glucuronidase that, in opposition to the human enzyme, is thermolabile [29]. Thus, to determine whether human β-glucuronidase expression and activity would display anatomical overlap we performed ISH in slides adjacent to those used for histochemistry, using a probe specific to the human transcript [26]. Histochemistry and ISH labelling for human β-glucuronidase were effective in brains receiving human cells (either transduced or not) but not in brain from a PBSinjected control animal, indicating that both techniques are reliable. As observed with cells phenotyping, histochemestry and ISH gave quite similar labelling when comparing Mo to MØ recipient brains related to staining intensity and cells shape and localization. Opposed to what we observed in vitro, where MØ expressed much more enzyme than Mo after adenoviral transduction (Figure 2), the intensity of staining of the human β-glucuronidase in vivo, assessed by histochemistry or ISH, was equally strong in brains receiving transduced Mo and MØ (Figure 4(a) for histochemistry and 4(g) for ISH). It was nevertheless weak in those grafted with non-transduced cells (Figures 4(c) and (h)) showing its ability to discriminate different expression levels. We also noticed that even though both techniques allowed
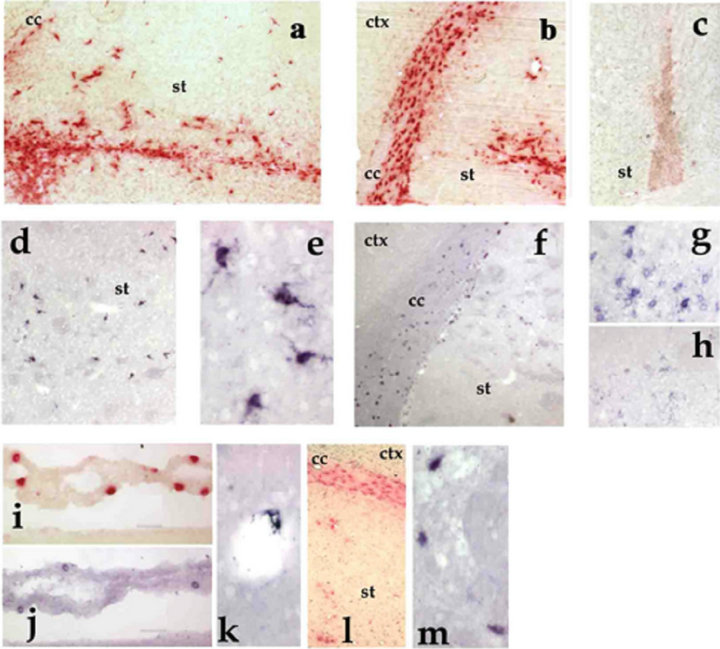
Figure 4. Fate of Mo and MØ, grafted into the mouse striatum as assessed by β-glucuronidase histochemical staining (red) and in situ hybridization of human β-glucuronidase mRNA (purple). From (a) to (f), pictures of brains grafted with transduced Mo; from (g) to (m), pictures of brains grafted with transduced MØ. For each type of cells, very similar histological views were obtained. (a) Ten days after transplantation of transduced Mo, the staining for human β-glucuronidase is limited to the striatum (st) around the site of injection (magnification ×40); (b) Thirty days after grafting of transduced Mo, most β-glucuronidase-positive cells are found in the corpus callosum (cc) (magnification ×40); (c) Ten days after grafting, non-transduced control Mo displayed a weaker staining for human β-glucuronidase activity than their transduced counterparts (magnification ×20); (d) Ten days after transplantation of transduced Mo, in situ hybridization to the human β-glucuronidase mRNA revealed sparse cells through the striatum in the area of grafting (magnification ×40); (e) Some of these cells displayed ramified microglial morphology (magnification ×200); (f) Thirty days after grafting of transduced Mo, human β-glucuronidase mRNA is mainly retrieved in cells located within the corpus callosum (cc) (magnification ×40); (g) Ten days after transplantation, transduced MØ strongly expressed human β-glucuronidase mRNA as revealed by in situ hybridization in the grafting area (magnification ×80); (h) In the hemisphere transplanted with control MØ (non-transduced), in situ hybridization signal in donor cells is much weaker (magnification ×80); (i) Cells expressing human β-glucuronidase within the choroid plexus 30 days after grafting of transduced MØ (×80); (j) Labelled cells with in situ hybridization in the choroid plexus 30 days after grafting of transduced MØ (×80); (k) Perivascular cell expressing the human β-glucuronidase mRNA 30 days after transplantation of transduced MØ (×100); (l) Human β-glucuronidase-expressing cells in the corpus callosum and in the vicinity of the ventricle 240 days after grafting of transduced MØ (magnification ×20); (m) Detection of cells expressing human β-glucuronidase mRNA in the corpus callosum 240 days after MØ transplantation (magnification ×100).
staining donor cells in overlapping areas, enzymatic detection revealed a greater number of positive bodies than ISH (compare Figures 4(a) to (d) and 4(b) to (f)). This is concordant with the fact that β-glucuronidase can be secreted and recaptured by distant cells through the mannose-6-phosphate receptor [30].
Ten and 30 days after transplantation, human β-glucuronidase activity was detected over a wide area of almost 2 mm in a radial axis surrounding the injection site. The detection of the human β-glucuronidase transcript instead was restricted to a shorter radius of about 0.2 mm. This indicates that the detection of β-glucuronidase activity at distance from the site of grafting was due to an extensive secretion of the recombinant enzyme through the parenchyma by the donor cells, as previously observed by grafting genetically modified astrocytes or fibroblasts [25, 30].
The anatomical locations of β-glucuronidase-expressing Mo and MØ evolved with time. Ten days after grafting, most β-glucuronidase-positive transduced cells were gathered at the injection site in the striatum (Figures 4(a) and (d)). At this time, when we counted the cells labelled with ISH on alternate slides of 2 animals of each group, we found that the estimated number of non-transduced Mo (1811 ± 149) or MØ (1705 ± 233) was not different to that of transduced Mo (1795 ± 70) or MØ (1873 ± 261).
After 30 days, the majority of the β-glucuronidasepositive, transduced cells were found in the corpus callosum enclosing the grafting area (Figures 4(b) and (f)) and those remaining in the striatum were found either gathered at the injection site or at short distance, associated to blood vessels (Figures 4(b) and (k)). Indeed, as we counted transduced cells in alternate slides of 3 animals per group we observed that the number of transduced Mo (1702 ± 241) was equivalent to that of transduced MØ (1588 ± 165) and most interestingly we found that at difference with the previous time of analysis, most of the cells counted were located in the corpus callosum Mo (74% ± 6%) and MØ (79% ± 10%) clearly indicating that they had migrated towards this new location between day 10 and 30 after grafting.
After 30 days, transduced cells expressing the human β-glucuronidase were also observed in the choroid plexus (Figures 4(i) and (j)). At that time, non-transduced cells were still present at the injection site but could not be detected in the corpus callosum or the choroid plexus. This could indicate that β-glucuronidase histochemistry was not sensitive enough to reveal untransduced cells that had reached this area. Indeed β-glucuronidase expression in tissue MØ is much stronger in inflammatory region but is down regulated otherwise [31]. A grater rate of activation induced by adenoviral transduction, as observed with an about 2 fold increase in HLA DR expression (Figure 1) could also explain a difference in migration between transduced and control cells. However, as cells migrating away from the injection site rather downregulated HLA DR expression, a more suitable explanation is that cells over-expressing the recombinant β- glucuronidase had increased migration capacity as compared to non-transduced cells. This observation is supported by the absence of labelling using antibodies directed to human antigens (i.e. CD68 or ALDP, see above) in any area distant from the injection site in hemispheres receiving non-transduced cells including the corpus callosum and the periventricular area. Moreover, this is coherent with several reports indicating that over-expression of β-glucuronidase is common in infiltrating microglia or gliomas, allowing extra-cellular matrix degradation and cell migration [32,33].
After 240 days, in one animal, β-glucuronidase-transduced MØ were still present in the brain parenchyma, although in reduced numbers, within the striatum as well as in the vicinity of the lateral ventricle, and in the corpus callosum, mostly associated with white fibre tracts (not shown). At this time, however, no untransduced cell could be detected using histochemestry or ISH, suggesting that detection of human β-glucuronidase was not sensitive enough to detect naïve cells.
Regarding cells morphology, 10 days after grafting, both grafted Mo and MØ displayed ramified morphology if located in the striatum (Figure 4(e)). At any other time or location, cells detected by ISH were round, and cell bodies appeared smaller if located in the corpus callosum than in the striatum.
The overlapping of immune labelling of strict human markers as HLA DR or hALDP with human β-glucuronidase immunohistochemistry and ISH staining over as much as 240 days strongly suggests that the grafted human cells were the actual bodies expressing the transgene rather than sparse engulfed debris of dead donor cells remaining functional for such a long period within host. Moreover in a previous comparable experiment involving human astrocytes transduced with the same vector grafted in the same area of the same mouse strain [25] an opposite pattern of human cell markers or transgene expression was observed, restricted for 2 months at the site of grafting. This further indicates that the location of transgene expression depends on the migrating ability of the carrier cells rather than on inner activity of host phagocytes of grafted cells.
4. DISCUSSION
We report that human Mo and Mo-derived MØ, genetically modified with an adenovirus vector allowing overexpression of the human lysosomal enzyme β-glucuronidase, behave similarly when grafted within the striatum of nude mice. These cells display a same propensity to leave the injection site and establish at close distance around blood vessels or associate to white fibre tracts where they sustain long-term transgene expression. Their migration pattern within the adult rodent striatum was modulated by the expression of β-glucuronidase and transduced cells migrated further than control cells, reaching the corpus callosum and the ventricle, indicating that over-expressing β-glucuronidase favoured a natural propensity of these cell for migration through heparan sulfates proteoglycans cleavage in the extracellular matrix [32-34]. Such migratory behaviour was similar to that of human bone marrow stromal cells [35], embryonic astrocytes [36], bulk of bone marrow cells [15] or mononuclear fraction of cord blood derived cells [15]. It however opposed to human adult astrocytes [25], rat amniotic epithelial cells [37] or mouse fibroblasts [30] that poorly migrate when implanted in the striatum. This mobility of transduced Mo and MØ suggests that migration from striatum towards typical locations (corpus callosum > perivascular spaces > periventricular areas) is determined by specific interactions of grafted cells with the tissue substratum. Most donor cells leaving the graft were associated with blood vessels, suggesting that they employed the outer walls of brain vasculature as a support for migration. The same was observed concerning activated parenchymal microglia following trauma [34] or monocytes infiltrating the inflamed brain [22,23,38]. The strong tropism of grafted transduced cells for the corpus callosum hints at a physiological perinatal process in mammals, when microglia precursors invade the CNS to locate in the supraventricular corpus callosum [39] which suggests a natural affinity of these cells for the corpus callosum or white matter. In our experiments however, the migration of grafted cells to an area adjacent to the needle tract in the corpus callosum, would rather be due to the mechanical lesion of the corpus callosum, inducing the release of chemokines such as MCP-1/ CCL2 and attraction of the grafted Mo/MØ [40,41].
Adenoviral transduction of Mo was barely effective in vitro, which contrasted with the efficient transduction of Mo-derived MØ. Moreover, even though in vitro transduced Mo adequately differentiated into MØ, they still expressed low transgene levels at this stage when kept in culture. In vivo, however, Mo grafted immediately after transduction, overexpressed β-glucuronidase to a level similar to that of transduced MØ as assessed by histochemistry and ISH. This suggests that the restriction observed in Mo transduction in vitro occurs at a post entry step, overpassed by a different maturation process within the CNS that is not recapitulated in vitro. In vivo differentiation of Mo into tissue macrophages is far more diverse and complex than that obtained in vitro [2,3] and the modulation of the transcription factor PU.1, that is central in hematopoietic cell differentiation, can shift the trafficking of an adenoviral vector within mononuclear cells from lysosomes to the nucleus [42], favouring expression over degradation. This finding opens interesting perspectives as it suggests that the in vitro differentiation step would not be useful for cell therapy protocols, preventing manipulation-induced cell loss.
An interesting point of our study is the longevity of grafted cells in the brain. In a previous report, primary human adult astrocytes transduced with the same adenoviral vector survived for only 2 months in the nude mouse brain [25]. Instead, transduced human Mo and MØ, showed improved aptitudes to remain in an inflammatory environment such as the site of injection but also at distance from it. The various growth factors and cytokines able to induce maturation of Mo into MØ (M-CSF, GM-CSF, TNF-α, IL-1β, IFN-γ, IL-6) may prevent their apoptosis in the injection site and thus support their initial survival [3].
In addition, we observed that transduced Mo and MØ were mobile, able to actively exit from the site of injection. Using immunohistochemistry, we observed that they expressed CD14 and HLA-DR when remaining at the site of injection. However, at all time points of analysis, the donor cells located at a distance from the injection site barely expressed HLA-DR and CD14 markers. This is in agreement with previous reports showing that Mo/MØ recruited in the CNS express activation markers only in the zone of inflammation [23,24]. In our experiments, the fact that Mo and MØ behave similarly after being grafted in the CNS, and underwent down regulation of their activation markers at distance from the injection site, suggests that they share a common susceptibility to the brain environment. This is in line with the concept of plastic and reversible MØ activation that we showed previously [43]. Thus, it is conceivable that the grafted cells are conditioned in the CNS to adopt resting phenotypes, irrespective of their previous differentiation at the time of grafting through mechanisms discussed elsewhere [4]. Our results further advocate for a deactivation of the grafted myeloid cells in the non-inflamed brain that does not rely on their ablation through cell death, preserving graft survival. Moreover, although grafted cells do undergo such deactivation by the CNS microenvironment, they nevertheless continue expressing the transgene for a long period of time as shown by present data. This makes them potential candidate for sustained delivery of secreted therapeutic proteins in a wide variety of disease affecting discrete areas of the brain, to prevent neuronal death in the course of neuro-degenerative diseases as Alzheimer or Parkinson diseases, to complement deficient enzymes as in lysosomal storage diseases or to modulate reperfusion or local immune response after brain ischemia or injury.
5. CONCLUSION
In agreement with previous reports, our results show that Mo/MØ have a significant potential for their integration in the CNS [14-16] and can ensure long lasting transgene expression [13] without inducing a particular inflammatory response [13-16]. Our data further show that Mo and MØ behave similarly and express the same levels of transgene after in vivo transplantation. Our results mostly contribute to validate the use of these cells, and especially Mo, within the framework of therapy in a wide range of brain affections of which course can be modified by secreted recombinant compounds. Monocytes are abundant and easily obtained from blood. Moreover, direct use of Mo instead of MØ would allow avoiding extensive cell manipulation before transduction, leading to a shorter ex vivo process and better survival and performances than in vitro differentiated cells. Blood Mo may thus represent an inestimable source of autologous cells for ex vivo gene therapy to target focal brain lesions, requiring no conditioning of the recipients and thus avoid putative side effects. Nevertheless, a critical issue still remains in the fact that Mo/MØ, although migrating further than other cell types tested to date, still do not allow expecting their use in diffuse brain pathology until their wide dissemination is made possible.
6. ACKNOWLEDGEMENTS
We thank Jean Claude Gluckman and François Lachapelle for helpful comments in the course of this study. This work was funded by the CNRS, The INSERM and Vaincre les Maladie Lysosomiales (vml-asso.org).

REFERENCES
- Kennedy, D.W. and Abkowitz, J.L. (1998) Mature monocytic cells enter tissues and engraft. Proceedings of the National Academy of Sciences of the United States of America, 95, 14944-14949. doi:10.1073/pnas.95.25.14944
- Naito, M., et al. (1996) Development, differentiation and phénotipique heterogeneity of murine macrophages. Journal of Leukocyte Biology, 59, 133-137.
- Murray, P.J. and Wynn, T.A. (2011) Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology, 11, 723-737. doi:10.1038/nri3073
- Ransohoff, R.M. and Perry, V.H. (2009) Microglial physiology: Unique stimuli, specialized responses. Annual Review of Immunology, 27, 119-145. doi:10.1146/annurev.immunol.021908.132528
- Ajami, B., et al. (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature Neuroscience, 14, 1142-1149. doi:10.1038/nn.2887
- Ajami, B., et al. (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature Neuroscience, 10, 1538-1543. doi:10.1038/nn2014
- Cartier, N., et al. (2009) Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science, 326, 818-823. doi:10.1126/science.1171242
- Faradji, A., et al. (1991) Phase I trial of intravenous infusion of ex-vivo-activated autologous blood-derived macrophages in patients with non-small-cell lung cancer: Toxicity and immunomodulatory effects. Cancer Immunology, Immunotherapy, 33, 319-326. doi:10.1007/BF01756597
- Parrish, E.P., et al. (1996) Targeting widespread sites of damage in dystrophic muscle: Engrafted macrophages as potential shuttles. Gene Therapy, 3, 13-20.
- Dou, H., et al. (2009) Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuro AIDS. Journal of Immunology, 183, 661-669. doi:10.4049/jimmunol.0900274
- Freeman, B.J., et al. (1999) Behavior and therapeutic efficacy of beta-glucuronidase-positive mononuclear phagocytes in a murine model of mucopolysaccharidosis type VII. Blood, 94, 2142-2150.
- Ohashi, T., et al. (2000) Reduction of lysosomal storage in murine mucopolysaccharidosis type VII by transplanttation of normal and genetically modified macrophages. Blood, 95, 3631-3633.
- Mordelet, E., et al. (2002) Brain engraftment of autologous macrophages transduced with a lentiviral flap vector: An approach to complement brain dysfunctions. Gene Therapy, 9, 46-52. doi:10.1038/sj.gt.3301591
- Oehmichen, M., et al. (1979) Features and distribution of intracerebrally injected peritoneal macrophages. Experimentelle Pathologie, 17, 71-76.
- Walczak, P., et al. (2004) Do hematopoietic cells exposed to a neurogenic environment mimic properties of endogenous neural precursors? Journal of Neuroscience Research, 76, 244-254. doi:10.1002/jnr.20042
- Franzen, R., et al. (1998) Effects of macrophage transplantation in the injured adult rat spinal cord: A combined immunocytochemical and biochemical study. Journal of Neuroscience Research, 51, 316-327. doi:10.1002/(SICI)1097-4547(19980201)51:3<316::AID-JNR5>3.0.CO;2-J
- Lazarov-Spiegler, O., et al. (1996) Transplantation of activated macrophages overcomes central nervous system regrowth failure. FASEB Journal, 10, 1296-1302.
- Brynskikh, A.M., et al. (2010) Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson’s disease. Nanomedicine (Lond), 5, 379-396.
- Krall, W.J., et al. (1994) Cells expressing human glucocerebrosidase from a retroviral vector repopulate macrophages and central nervous system microglia after murine marrow transplantation. Blood, 83, 2737-2748.
- Gorantla, S., et al. (2006) Quantitative magnetic resonance and SPECT imaging for macrophage tissue migration and nanoformulated drug delivery. Journal of Leukocyte Biology, 80, 1165-1174. doi:10.1189/jlb.0206110
- Mildner, A., et al. (2007) Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nature Neuroscience, 10, 1544-1553.
- D’Mello, C., Le, T. and Swain, M.G. (2009) Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. Journal of Neuroscience, 29, 2089- 2102. doi:10.1523/JNEUROSCI.3567-08.2009
- Djukic, M., et al. (2006) Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain, 129, 2394-2403. doi:10.1093/brain/awl206
- Flugel, A., et al. (2001) Transformation of donor-derived bone marrow precursors into host microglia during autoimmune CNS inflammation and during the retrograde response to axotomy. Journal of Neuroscience Research, 66, 74-82. doi:10.1002/jnr.1198
- Serguera, C., et al. (2001) Primary adult human astrocytes as an ex vivo vehicle for beta-glucuronidase delivery in the brain. Molecular Therapy, 3, 875-881. doi:10.1006/mthe.2001.0319
- Ghodsi, A., et al. (1998) Extensive beta-glucuronidase activity in murine central nervous system after adenovirus-mediated gene transfer to brain. Human Gene Therapy, 9, 2331-2340. doi:10.1089/hum.1998.9.16-2331
- Palucka, K.A., et al. (1998) Dendritic cells as the terminal stage of monocyte differentiation. Journal of Immunology, 160, 4587-4595.
- Benhamida, S., et al. (2003) Transduced CD34+ cells from adrenoleukodystrophy patients with HIV-derived vector mediate long-term engraftment of NOD/SCID mice. Molecular Therapy, 7, 317-324. doi:10.1016/S1525-0016(03)00002-9
- Frankel, H.A., Glaser, J.H. and Sly, W.S. (1977) Human beta-glucuronidase. I. Recognition and uptake by animal fibroblasts suggests animal models for enzyme replacement studies. Pediatric Research, 11, 811-816. doi:10.1203/00006450-197707000-00007
- Taylor, R.M. and Wolfe, J.H. (1997) Decreased lysosomal storage in the adult MPS VII mouse brain in the vicinity of grafts of retroviral vector corrected fibroblasts secreting high levels of β-glucuronidase. Nature Medicine, 3, 771-775. doi:10.1038/nm0797-771
- Laszlo, D.J., et al. (1993) Development of functional diversity in mouse macrophages. Mutual exclusion of two phenotypic states. American Journal of Pathology, 143, 587-597.
- Szaniawski, W. (1976) Localizations of beta-glucuronidase activity in sections and cultures of human gliomas. Acta Neuropathologica, 34, 47-54. doi:10.1007/BF00684943
- Takahashi, H., et al. (2008) Involvement of heparanase in migration of microglial cells. Biochimica et Biophysica Acta, 1780, 709-715. doi:10.1016/j.bbagen.2007.12.014
- Grossmann, R., et al. (2002) Juxtavascular microglia migrate along brain microvessels following activation during early postnatal development. Glia, 37, 229-240. doi:10.1002/glia.10031
- Azizi, S.A., et al. (1998) Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats—Similarities to astrocyte grafts. Proceedings of the National Academy of Sciences of the United States of America, 95, 3908-3913. doi:10.1073/pnas.95.7.3908
- Pundt, L.L., Kondoh, T. and Low, W.C. (1995) The fate of human glial cells following transplantation in normal rodents and rodent models of neurodegenerative disease. Brain Research, 695, 25-36. doi:10.1016/0006-8993(95)00753-D
- Kosuga, M., et al. (2001) Engraftment of genetically engineered amniotic epithelial cells corrects lysosomal storage in multiple areas of the brain in mucopolysaccharidosis type VII mice. Molecular Therapy, 3, 139-148. doi:10.1006/mthe.2000.0234
- Hess, D.C., et al. (2004) Hematopoietic origin of microglial and perivascular cells in brain. Experimental Neurology, 186, 134-144. doi:10.1016/j.expneurol.2003.11.005
- Cuadros, M.A. and Navascues, J. (1998) The origin and differentiation of microglial cells during development. Progress in Neurobiology, 56, 173-189. doi:10.1016/S0301-0082(98)00035-5
- Conductier, G., et al. (2010) The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. Journal of Neuroimmunology, 224, 93-100. sdoi:10.1016/j.jneuroim.2010.05.010
- Dzenko, K.A., et al. (2001) The chemokine receptor CCR2 mediates the binding and internalization of monocyte chemoattractant protein-1 along brain microvessels. Journal of Neuroscience, 21, 9214-9123.
- Carey, B., et al. (2007) PU.1 redirects adenovirus to lysosomes in alveolar macrophages, uncoupling internalization from infection. Journal of Immunology, 178, 2440- 2447.
- Porcheray, F., et al. (2005) Macrophage activation switching: An asset for the resolution of inflammation. Clinical & Experimental Immunology, 142, 481-489.
NOTES
*Competing interests: The authors declare that there exists no conflict of interest.
Authors’ contributions: CSe and CSa designed the experiments. CSe, CSa and FS conducted the experiments. FS, GG and JM provided resources and scientific input. CSe, CSa and GG wrote the manuscript.
#Corresponding author.