Journal of Cancer Therapy
Vol. 4 No. 4 (2013) , Article ID: 32574 , 15 pages DOI:10.4236/jct.2013.44098
Treatment with Paclitaxel Orotate and Carboxyamidotriazole Orotate in SC-Implanted OVCAR-5 Human Ovarian Tumor Xenografts
![]()
1SavviPharm Inc. and Tactical Therapeutics Inc., New York, USA; 2Cancer Therapeutics and Immunology Department, Southern Research Institute, Birmingham, USA; 3Pharmaceutical Sciences Research Institute, Samford University, Birmingham, USA.
Email: *karmali@aol.com, rashida@tacticaltherapeutics.com
Copyright © 2013 Rashida A. Karmali et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 24th, 2013; revised May 22nd, 2013; accepted May 29th, 2013
Keywords: Paclitaxel Orotate; Paclitaxel; Carboxyamidotriazole Orotate; Ovarian Cancer
ABSTRACT
Background: Paclitaxel (PTX) is approved for the treatment of refractory ovarian cancer and breast cancer, but is problematic due to severe, dose-dependent, potentially irreversible neurotoxicity. Alternative formulations using nanoparticles and liposomes have been developed to avoid solvent-related toxicity. These formulations allow improved delivery; however, toxicity, compensatory signaling, and drug resistance still pose challenges. Conversion of cytotoxic agents to their orotate compounds offers a potentially improved approach by increasing bioavailability and reducing toxicity. Orotate salts are neutral and acquire lipophilic properties, easing diffusion through lipid membranes. The orotate salt of PTX (PTXO) may yield an improved safety profile. Combination therapy with cytotoxic drugs, antiangiogenics and/or signal transduction pathway inhibitors has shown better efficacy than cytotoxic monotherapy. The combination of carboxyamidotriazole orotate (CTO, a calcium signal transduction pathway inhibitor) and PTX may be more effective than PTX alone at non-toxic doses. Materials and Methods: PTXO alone, and combinations of CTO with PTX and PTXO were first tested in female athymic NCr-nu/nu mice to evaluate tolerance of the combinations. The tolerated combinations, PTX monotherapy, and PTXO monotherapy were then tested to evaluate their antitumor activity in female athymic NCr-nu/nu mice with subcutaneously implanted OVCAR-5 human ovarian tumor. Antitumor activity was measured by median time to doubling, median tumor growth delay, and mean percent body weight loss. Results: CTO, PTX, and PTXO showed significant inhibition of growth of the human OVCAR-5 ovarian tumor xenografts. The combination of low PTX and CTO, or high PTXO monotherapy, had significant efficacy and it was less toxic than high PTX as measured by body weight loss. Conclusions: Low-dose CTO is effective and has low toxicity, suggesting the potential for maintenance therapy for ovarian cancer. PTXO offers efficacy and a strategy for minimizing body weight loss, and may improve outcomes for patients who demonstrate toxicity to PTX.
1. Introduction
Ovarian cancer is the 5th leading cause of cancer-related deaths among women in the United States, and about two-thirds of all patients are diagnosed with advanced disease, with a relative 5-year relative survival rate of 26.9% [1,2].
Paclitaxel (PTX) is a taxane that disrupts microtubule function and arrests mitosis and cell division [3]. Paclitaxel is approved for clinical use in the treatment of refractory ovarian cancer and breast cancer, and is a candidate for treatment of many other cancers [3].
Taxanes are highly hydrophobic [4]. Formulations of PTX contain Cremophor® EL (CrEL) as a part of the vehicle. CrEL comprises components that contribute to the severe toxicities observed in patients who are treated with PTX. Neurotoxicity manifests as dose-dependent sensory neuropathy, and occurs more frequently with longer infusions and doses above 175 mg/m2 [3]. Solventrelated toxicities are associated with sometimes irreversible neuropathies [5], hypersensitivity reactions [6-8], demyelination, possible interaction with PTX to cause myelosuppression, axonal swelling, complement activation, reduced cell penetration and increased systemic drug exposure [4,6]. CrEL must be delivered through specialized infusion sets with inline filters over 1 - 3 hours [4,9].
Alternative formulations of PTX, including albumin nanoparticles (nab, e.g., Abraxane®) and cationic liposomes (LP), have been developed to overcome these disadvantages. Nab-paclitaxel (Nab-PTX) is delivered as a suspension of albumin nanoparticles with an average size of 130 nm, eliminating the need for premedication or special infusion sets, with an infusion time of 30 minutes [4,7]. The nanoparticle has better efficacy, and the risk of hypersensitivity reactions is practically eliminated [7].
Overall tolerability is similar between nab-PTX and CrEL-PTX. In a Phase III study in metastatic breast cancer, grade 3 neuropathy was more common in nab-PTX, but improved rapidly to grade 1 - 2 within several weeks upon discontinuation [8]. The overall tolerability of nabPTX 260 mg/m2 was reported to be similar to that of CrEL-PTX 175 mg/m2 in a phase III trial [4]. Those results differed from the findings reported from phase III trial reported by Rugo et al. [10], which demonstrated that nab-PTX was unlikely to be superior to PTX for progression-free survival in metastatic breast cancer. The impact of prolonged recurrence, free and overall survival, as well as long-term toxicity, of a nab-PTX containing regimen, has not been well elucidated. There is still a need for a safer version of PTX despite the discovery of nab-PTX.
In a SCID mouse melanoma model, LP-PTX, but not CrEL-PTX, prevented melanoma growth and invasiveness, improved mouse survival, reduced vessel density at the tumor/dermis interface, and reduced endothelial cell mitosis to background levels [11]. While alternative PTX formulations are associated with fewer adverse reactions and can be delivered more quickly without specialized infusion sets, significant challenges remain. Among these issues, they are drug resistance and compensatory signaling, and toxicity.
In ovarian cancer cell lines with activated PI3K/AKT/ mTOR signaling, treatment with mTOR and AKT inhibitors resulted in compensatory upregulation of ERK signaling. PI3K/TOR/AKT and MEK/mERK signaling pathways may work together to promote tumor growth and survival and, in addition, foster resistance to therapy [2]. Combination therapy with chemotherapy and other agents such as antiangiogenic agents and signal transduction inhibitors are being explored to combat resistance and compensatory signaling.
Carboxyamidotriazole (5-amino-1(4-(4-chlorobenzoyl)- 3,5-dichlorobenzyl)-1,2,3-triazole-4-carbo-xamide, CAI) is a small molecule inhibitor with antiproliferative and antimetastatic effects related to inhibition of receptor operated calcium-channel-mediated influx [12,13]. Amongst its constellation of effects, CAI reduces VEGF production from tumor and endothelial cells [14-16].
Carboxyamidotriazole orotate (CTO) is the orotic acid salt of CAI. CTO possesses increased solubility compared to CAI [13]. CTO has reduced toxicity, increased oral bioavailability, and stronger efficacy compared to CAI [17]. CTO enters the bloodstream faster than CAI, and achieves higher plasma concentrations while maintaining a similar elimination half-life. PK data indicate that CTO may enable smaller dosages of CAI for inhibition of tumor cell proliferation, with reduced toxicity [16]. The combination of CTO and chemotherapy was found to produce significantly greater antitumor effects than chemotherapy alone in glioblastoma and colon xenograft mouse models [12].
Given the evidence that combination therapy with cytotoxic drugs and antiangiogenic and/or signal transduction pathway inhibitors demonstrates improved efficacy over cytotoxic therapy alone, we hypothesized that CTO alone, or the combination of CTO with PTX or paclitaxel orotate (PTXO), offered a potentially improved approach to treatment of ovarian cancer. This approach is also compared PTXO, a new salt of PTX, versus PTX to determine if PTXO has a safer profile and equal efficacy compared to PTX.
In this study, we first evaluated the tolerance of nontumored female athymic NCr-nu/nu mice to the combination treatment of PTXO plus CTO, and PTX plus CTO. We then compared antitumor activity of monotherapy with PTXO and PTX; the combination treatment of PTXO plus CTO; and PTX plus CTO when administered against subcutaneously (SC)-implanted human OVCAR- 5 ovarian tumor xenografts in female, athymic NCr-nu/nu mice.
2. Materials and Methods
2.1. Drug Formulation
2.1.1. Reagents
CTO (MW 580.76) was synthesized by Johnson Matthey Pharma Services (Devens, MA). PTXO (MW 992) was synthesized by Southern Research Institute. PTX was obtained from Hauser Laboratories (Boulder, CO). Polyethylene glycol (PEG 400, MW 400) and Cremophor® EL were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO). Deionized water (ASTM Type II) was purchased from LabChem, Inc. (Pittsburgh, PA). Ethanol (ethyl alcohol USP, 200 Proof) was purchased from Aaper Alcohol and Chemical Co. (Shelbyville, KY). Saline (Saline Solution 0.9%, for animal use only) was purchased from Phoenix Pharmaceutical, Inc. (St. Joseph, MO).
2.1.2. CTO Formulation
CTO was formulated once a week at a concentration of 51.3 mg/mL in 40% PEG 400 in deionized water. A portion of the resulting suspension was diluted further with 40% PEG 400 in deionized water to achieve a concentration of 34.2 mg/mL. CTO was administered to mice by exact individual animal’s body weight on each day of treatment. The injection volume was 0.1 mL/10g body weight.
2.1.3. Paclitaxel Orotate Formulation
A 2.32 mg/mL solution of PTXO was formulated on each day of treatment by adding 100% ethanol to the powder. An equal volume of CrEL was added and finally, saline was added to yield a concentration of 2.32 mg/mL in 12.5% CrEL/12.5% ethanol/75% saline. A portion of the 2.32 mg/mL solution was further diluted with the complete vehicle to 1.16 mg/mL. PTXO was administered to mice by exact individual animal’s body weight on each day of treatment. The injection volume was 0.1 mL/10g body weight.
2.1.4. Paclitaxel Formulation
An 8.0 mg/mL stock solution of PTX in 50% CrEL/50% ethanol was formulated on the first day of treatment. On each day of treatment, a portion of the 8.0 mg/mL stock solution was diluted with saline to yield a concentration of 2.0 mg/mL in 12.5% CrEL/12.5% ethanol/75% saline. A portion of the 2.0 mg/mL solution was further diluted with the complete vehicle to 1.0 mg/mL. Paclitaxel was administered to mice by exact individual animal’s body weight on each day of treatment. The injection volume was 0.1 mL/10g body weight.
2.2. Tumor Model
2.2.1. CTO/Paclitaxel Orotate/Paclitaxel Tolerance
Animals were non-tumored.
2.2.2. OVCAR-5 Ovarian Xenograft
Each mouse was implanted SC near the right flank with a 30 - 40 mg fragment of the OVCAR-5 human ovarian tumor from an in vivo passage using a 13 g needle. The day of tumor fragment implantation was designated as Day 0. Individual tumors of 150 animals grew to 75 - 198 mg in weight (75 - 198 mm3 in size) on Day 21 after tumor fragment implantation, the day of treatment initiation. Those animals selected with tumors in the proper size range were assigned to 15 treatment groups so that the mean tumor weights in all groups on Day 21 were as close to each other as possible (mean tumor weights ranged from 139 to 143 mg; median tumor weights were 126 or 162 mg).
2.3. Animal Care
Mice were purchased from Charles River Laboratories (Wilmington MA).
CTO/PTXO/PTX tolerance: Female 7-week-old athymic NCr-nu/nu mice were acclimated in the laboratories for 6 days prior to experimentation.
OVCAR-5 ovarian xenograft: Female 6-week-old athymic NCr-nu/nu mice were acclimated in the laboratories for 2 weeks prior to experimentation.
The animals were housed in microisolator cages, five per cage, in a 12-hour light/dark cycle. The animals received filtered Birmingham municipal water and sterilizable rodent diet (HarlanTeklad TD8656) ad libitum. Cages were changed twice weekly. The animals were observed daily and clinical signs were noted. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Southern Research. Animal laboratories of Southern Research are AAALAC accredited.
2.4. Drug Treatment
2.4.1. CTO/Paclitaxel Orotate/Paclitaxel Tolerance
The study consisted of six groups of five mice per group. PTXO was tested at the equivalent PTX doses (based on MW of PTX of 853.9 and MW of PTXO of 992).
PTXO and PTX were administered intravenously (IV) on Days 1, 3, 5, 8, 10, and 12 (Q2Dx3/2 weeks schedule), while CTO was administered by oral gavage (PO) on Days 1 - 14 (Q1Dx14 schedule).
Group 1 received PTXO 23.2 mg/kg; Group 2, 11.6 mg/kg. Group 3 received PTXO 23.2 mg/kg and CTO 513 mg/kg; Group 4, PTXO 11.6 mg/kg and CTO 342 mg/kg; Group 5 received PTX 20 mg/kg and CTO 513 mg/kg; Group 6, PTX 10 mg/kg and CTO 342 mg/kg.
2.4.2. OVCAR-5 Ovarian Xenograft and Treatments
The experiment consisted of 15 groups of 10 mice per group for a total of 150 mice. CTO was administered PO on Days 21 - 34 alone or in combination with PTXO or PTX, both of which were administered IV on Days 21, 23, 25, 28, 30, and 32. Animals in the control group were treated with the PTX and PTXO vehicle (12.5% cremophor EL/12.5% ethanol/75% saline) and the CTO vehicle (40% PEG 400 in deionized water). The following five treatments were designed for 14 treatment groups:
a) Two groups received CTO at doses of 513 mg/kg and 342 mg/kg.
b) Two groups received PTXO at doses of 23.2 mg/kg and 11.6 mg/kg.
c) Two groups received PTX at doses of 20 mg/kg and 10 mg/kg.
d) Four groups received combination treatment with PTXO and CTO:
i) PTXO 23.2 mg/kg and CTO 513 mg/kg;
ii) PTXO 11.6 mg/kg and CTO 513 mg/kg;
iii) PTXO 23.2 mg/kg and CTO 342 mg/kg;
iv) PTXO 11.6 mg/kg and CTO 342 mg/kg.
e) Four groups received combination treatment with PTX and CTO:
i) PTX 20 mg/kg and CTO 513 mg/kg;
ii) PTX 10 mg/kg and CTO 513 mg/kg;
iii) PTX 20 mg/kg and CTO 342 mg/kg;
iv) PTX 10 mg/kg and CTO 342 mg/kg.
2.5. Parameters Evaluated
2.5.1. CTO/Paclitaxel/Paclitaxel Orotate Tolerance
Change in mean body weight, mortality, and 22-day survival were evaluated.
2.5.2. OVCAR-5 Ovarian Xenograft
The SC tumors were measured and the animals were weighed twice a week starting on the first day of treatment. Tumor volume was determined by caliper measurements (mm) and using the formula for an ellipsoid sphere: Lx W2/2 = mm3, where Land W refer to the larger and smaller perpendicular dimensions collected at each measurement. This formula was also used to calculate tumor weight, assuming unit density (1 mm3 = 1 mg). The median time to reach two tumor mass doublings in each of the treated groups (T) and in the control group (C) was used in the calculation of the overall delays in the growth of the median tumors (T-C, days). Comparison of the median tumor weight in the treatment groups to the median tumor weight in the control group (T/C × 100%) on Day 35 (one day after the end of the treatment with CTO) and on Day 63 (the day of study termination) was used for an additional evaluation of the antitumor efficacy of the combination treatments.
2.6. Statistical Analysis
No statistical analysis was done for the CTO/paclitaxel orotate/paclitaxel tolerance experiment.
For the statistical analysis of the OVCAR-5 ovarian xenograft study, the individual animal’s time to reach two tumor mass doublings was used as the endpoint in a life tables analysis (Kaplan-Meier survival analysis followed by a log-rank test). The life tables analysis allows for the comparison of the growth data between the groups using the animals whose tumors did not reach the evaluation point, by censoring them. The individual animal’s tumor weight on Day 63 and the individual animal’s body weight on Day 35 were used as the endpoint in a Student’s t-test (or Mann-Whitney rank sum test) in order to statistically compare the growth data between the groups. A nonparametric test was used when the data set did not the pass the normality test. The difference between the groups was considered to be significant if the P value was equal to or less than 0.05.
3. Results
3.1. CTO/Paclitaxel Orotate/Paclitaxel Tolerance
Treatment for five of the six groups was tolerated without deaths. There was one moribund animal in the combination treatment group of PTXO 23.2 mg/kg plus CTO 513 mg/kg (at necropsy the animal was noted to have a possible blockage in the intestines). Percent mean body weight change in the six groups ranged from −6% to 13%.
The maximum tolerated dose (MTD) is defined as the dose that does not result in death, or produces no more than 20% mean body weight loss. Thus, the MTD of PTXO was above 23.2 mg/kg; for combination treatment with PTXO and CTO, the MTD of PTXO was 23.2 mg/kg, and of CTO, 513 mg/kg; and for combination treatment with PTX and CTO, the MTD of PTX was above 20 mg/kg, and of CTO, above 513 mg/kg.
A summary of the tolerance experimental results, including mean gross body weight, mean gross body weight change, and percent change in mean body weight are presented in Table 1. Overall, the combination of PTX plus CTO or PTXO plus CTO did not have an impact on body weight at different doses, indicating they were suitable to be used in the tumored animals.
3.2. OVCAR-5 Ovarian Xenografts
Control, vehicle-treated OVCAR-5 xenografts grew progressively in all 10 animals, reaching 1813 mg in weight on Day 63.
Twelve groups tolerated treatment without deaths. One animal died in each of the two remaining groups. In the PTXO 11.6 mg/kg + CTO 513 mg/kg group, no necropsy was possible. In the PTX 20 mg/kg + CTO 342 mg/kg group, necropsy revealed no gavage-related trauma.
3.2.1. Antitumor Activity of Treatment Groups versus Vehicle Controls
A summary of the OVCAR-5 experimental results, including median days to two doublings, median tumor growth delay, median T/C, and percent mean body weight loss, is presented in Table 2. Maximum median tumor weights on Days 35 and 63 are presented in Table 3. P values for median days to two doublings and tumor weights for all treatment groups compared to controls are presented in Table 4.
Statistically significant antitumor activity (as measured by time to two mass doublings and tumor weight) was observed for 12 of the 14 treatment groups, compared to controls:
a) CTO 342 mg/kg alone.

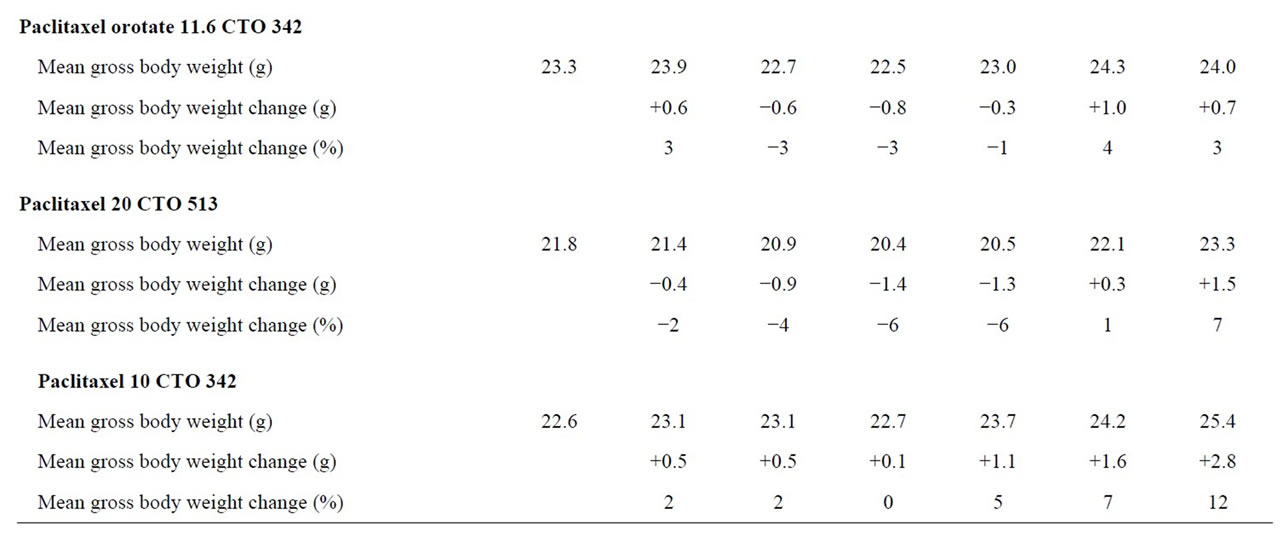
Table 1. Mean gross body weight percent change paclitaxel orotate, paclitaxel, CTO tolerance experiments.
b) PTXO 23.2 mg/kg and 11.6 mg/kg alone.
c) PTX 20 mg/kg and 10 mg/kg alone.
d) PTXO 23.2 mg/kg with CTO 513 mg/kg and 342 mg/kg.
e) PTXO 11.6 mg/kg with CTO 342 mg/kg.
f) PTX 20 mg/kg and 10 mg/kg with CTO 513 mg/kg and 342 mg/kg.
The response of the 14 treatment groups with OVCAR- 5 human ovarian tumor (as measured by tumor weight) is shown in Figures 1-4.
3.2.2. Antitumor Activity of Paclitaxel Orotate + CTO and Paclitaxel + CTO versus Paclitaxel Orotate and Paclitaxel
No statistically significant antitumor activity (as measured by time to two mass doublings and tumor weight) was observed for the following treatment groups when PTXO plus CTO was compared to PTXO alone, and PTX plus CTO was compared to PTX alone:
a) PTXO 23.2 mg/kg plus CTO 513 mg/kg or 342 mg/kg compared to PTXO 23.3 mg/kg.
b) PTXO 11.6 mg/kg plus CTO 513 mg/kg or 342 mg/kg compared to PTXO 11.6 mg/kg.
c) PTX 20 mg/kg plus CTO 513 mg/kg or 342 mg/kg compared to PTX 20 mg/kg.
d) PTX 10 mg/kg plus CTO 513 mg/kg or 342 mg/kg compared to PTX 10 mg/kg.
However, at the lower equivalent dose, PTX 10 mg/kg plus CTO 342 mg/kg showed a synergistic antitumor effect that was not observed at PTX 20 mg/kg plus CTO
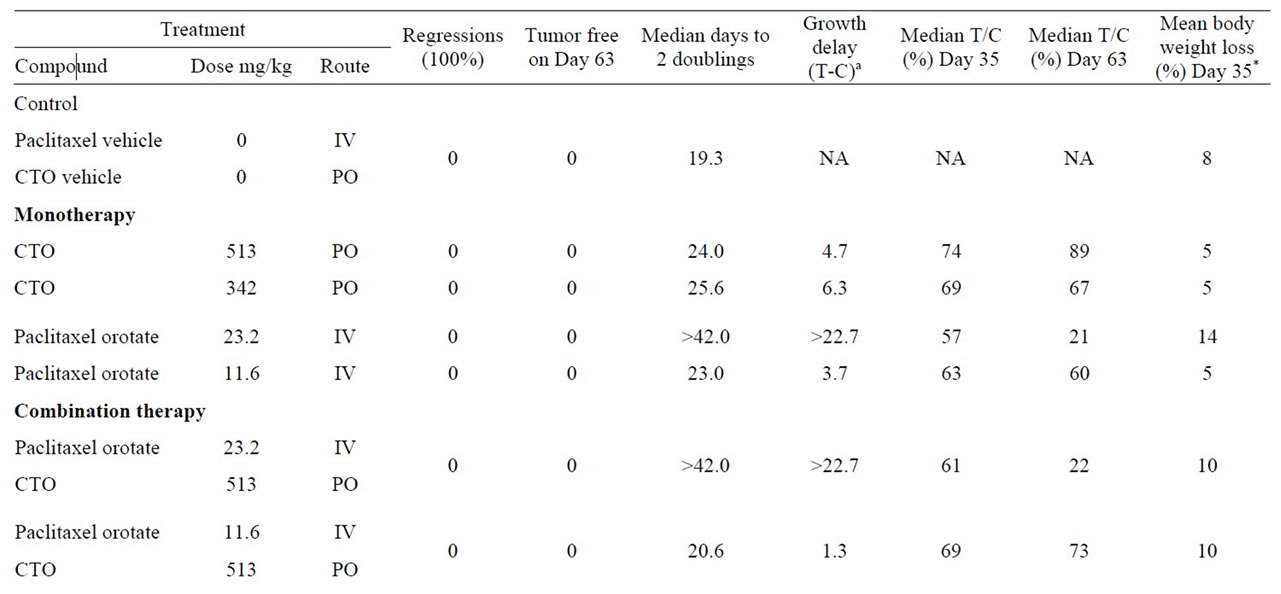
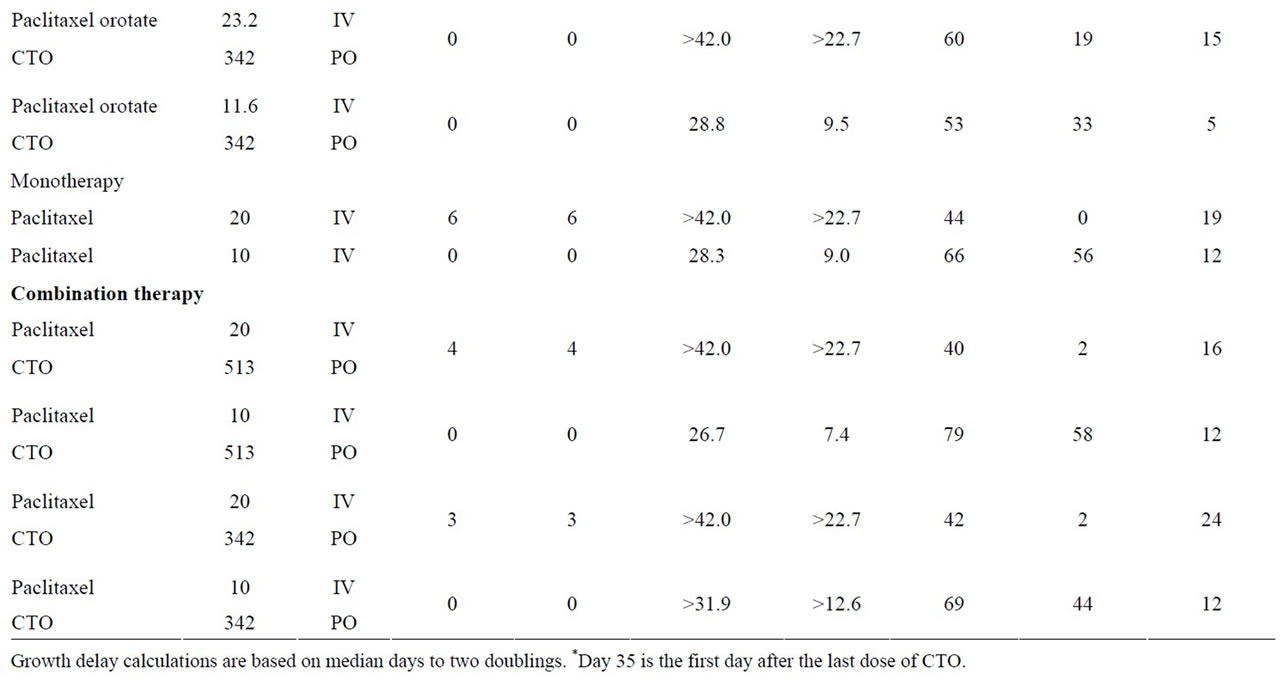
Table 2. Response of SC OVCAR-5 ovarian tumor to treatment with paclitaxel orotate, paclitaxel, CTO or CTO in combination with paclitaxel orotate or paclitaxel.
(Figure 4).
3.2.3. Antitumor Activity Paclitaxel versus Paclitaxel Orotate and Paclitaxel + CTO versus Paclitaxel Orotate + CTO
PTX was compared to the equivalent dose of PTXO, and PTX plus CTO 513 mg/kg or 342 mg/kg was compared to the equivalent dose of PTXO plus CTO 513 mg/kg or 342 mg/kg. Statistically significant antitumor activity as measured by tumor mass doubling time was observed only for PTX 10 mg/kg plus CTO 513 mg/kg compared to PTXO 11.6 mg/kg plus CTO 513 mg/kg (Table 5). Statistically significant antitumor activity as measured by tumor weight was observed only for the 20 mg/kg PTX groups (Table 5).
3.2.4. Body Weight Statistical Analysis
Animals treated with PTX 20 mg/kg had a statistically significant difference in body weights compared to controls. Those treated with PTXO 23.2 mg/kg and 11.6
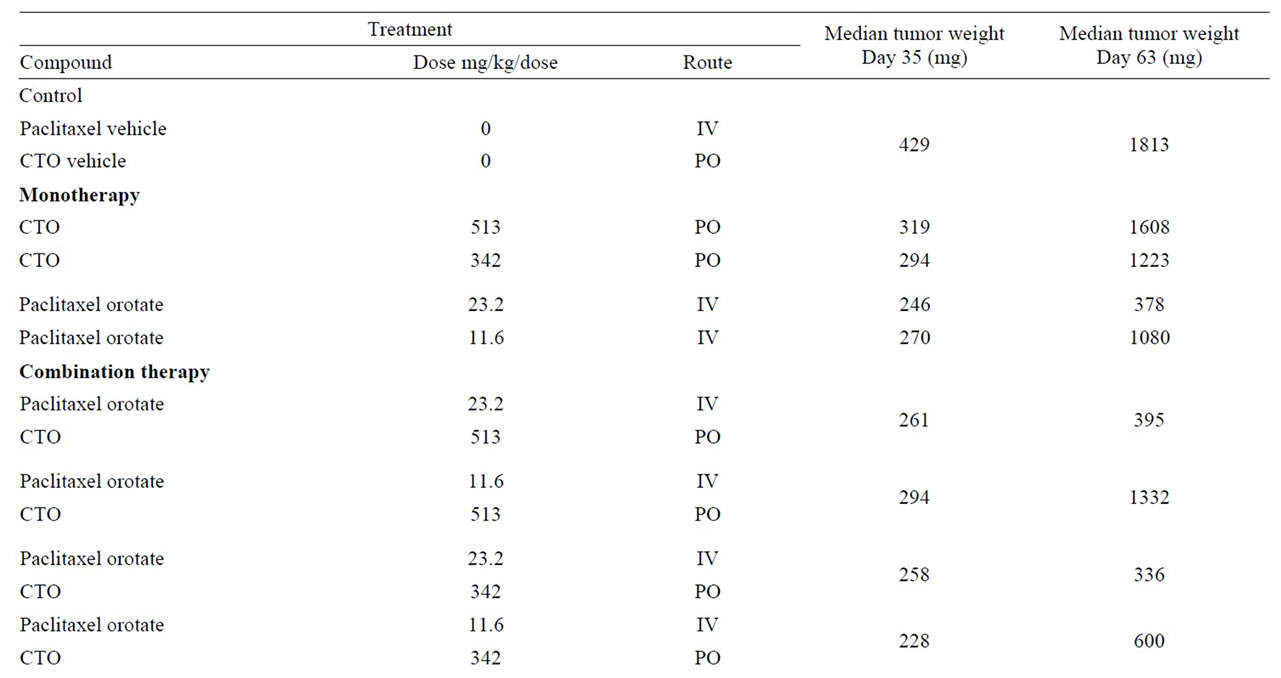
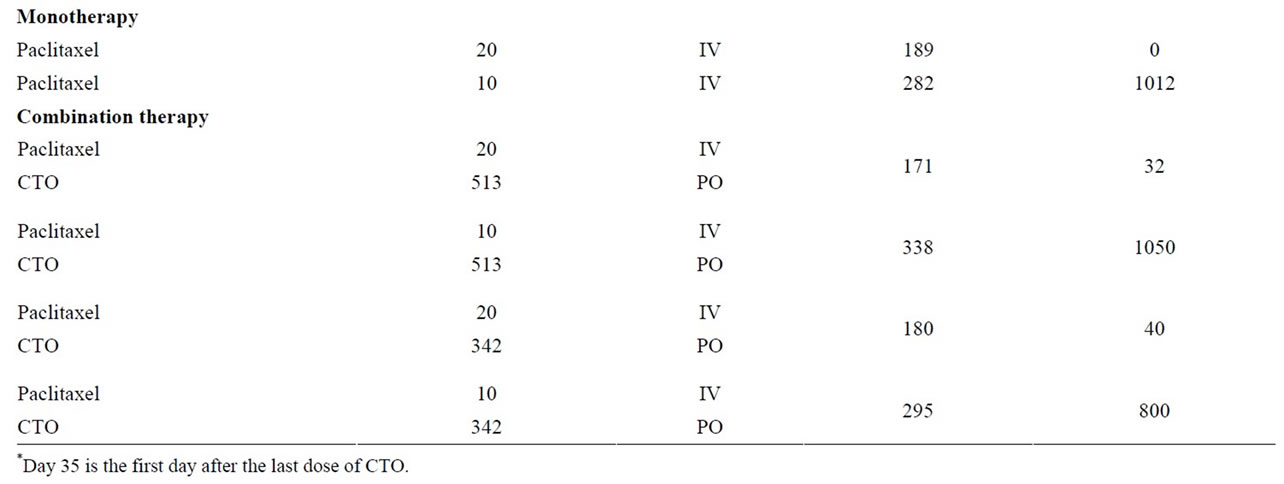
Table 3. Median tumor weights days 35 and 63.
mg/kg, and PTX 10 mg/kg, did not have statistically significant differences in body weight compared to controls. Nor did animals treated with PTX when compared to equivalent doses of PTXO (Table 6).
The effect of treatment with PTXO and PTX on mean body weight in animals implanted with OVCAR-5 human ovarian tumor is presented in Figure 5.
3.3. Hematological Analyses
PTXO and PTX administration to female athymic NCr-nu/nu mice resulted in statistically significant decreases in the mean value of leukocytes, neutrophils, lymphocytes and reticulocytes in comparison to the control group mean (data not shown). Comparison between groups of mice administered comparable dosages of PTXO or PTX did not show statistically significant differences, with the exception of eosinophils. In the high dose group, PTXO had a lower mean eosinophil count in comparison to the mice administered PTX.
4. Discussion
The goal of this study was to determine 1) if CTO alone, or the combination of CTO with PTX or PTXO, offered a potentially improved approach to treatment of ovarian cancer, and 2) if PTXO retained the antitumor activity and reduced toxicity compared with PTX. In this study, we compared antitumor activity of a) monotherapy with CTO; b) PTXO and PTX; c) the combination treatment


Table 4. P values time to tumor mass doubling and tumor weight of all treatment groups compared to control.

Figure 1. Response of SC implanted OVCAR-5 human ovarian tumor to treatment with CTO.
of PTXO plus CTO; and d) the combination treatment of PTX plus CTO when administered against subcutaneously (SC)-implanted human OVCAR-5 ovarian tumor xenografts in female, athymic NCr-nu/nu mice.
We found that oral administration of CTO 342 mg/kg resulted in a statistically significant growth delay of the human OVCAR-5 ovarian tumor xenografts when implanted SC in female athymic NCr-nu/nu mice, but CTO 513 mg/kg did not.
Intravenous PTXO at doses of 23.2 and 11.6 mg/kg and of PTX at doses of 20 and 10 mg/kg 3 times a week for 2 weeks resulted in a statistically significant inhibition of the growth of the human OVCAR-5 ovarian tumor xenografts. The highest antitumor activity was observed for PTX 20 mg/kg, followed by PTXO 23.2 mg/kg, PTX 10 mg/kg, and PTXO 11.6 mg/kg.
Treatment with PTX 20 mg/kg alone and in combination with CTO produced tumor-free animals on Day 63, while the equivalent dose of 23.2 mg/kg of PTXO alone and in combination with CTO did not.
However, the combination of PTX 10 mg/kg and CTO 342 mg/kg had significant efficacy and exhibited less toxicity given the lower body weight loss (12%) (Table 2). These findings suggest that a lower dose of PTX (10 mg/kg) combined with lower dose of CTO (342 mg/kg) may offer an alternative regimen in patients who exhibit serious toxicity to PTX 20 mg/kg. We have observed such dose dilution combinatorial effects of low CTO in other studies [13]. These results suggest a role for CTO in combination with PTX in treatment of ovarian cancer.
CTO is the orotate formulation of CAI. Treatment with single-agent CAI resulted in disease stabilization lasting

Figure 2. Response of SC implanted OVCAR-5 human ovarian tumor to treatment with paclitaxel orotate and paclitaxel.
6 months or more in 31% of patients with relapsed ovarian cancer [18], and was also observed in patients with melanoma and lung cancer. Treatment with the combination of CAI and paclitaxel resulted in partial responses in patients with melanoma, renal cell, fallopian tube, and ovarian cancers [14]. CAI had limited oral bioavailability, limited efficacy, and significant toxicity in clinical development [12]; however, patients responded even though CAI levels were not optimal. In the experiments presented here, low CTO showed significant efficacy and less toxicity indicated by lower body weight loss. Importantly, the addition of low CTO to low PTX in our OVCAR xenograft model resulted in increased antitumor activity with reduced toxicity, but was smaller in magnitude compared with high PTX with a higher toxicity. PTXO at the higher dose fell between high PTX and low PTX plus low CTO was regarding antitumor activity, but it had a safe toxicity profile.
The effect of PTXO in minimizing body weight loss was an unexpected result. PTXO 23.2 mg/kg alone had significant efficacy while exhibiting lower body weight loss (14%) compared to that in the PTX 20 mg/kg group (19%) (Table 2). The body weights on Day 35 in the groups treated with PTXO were not statistically different from control. Conversely, body weight was statistically significantly different between the PTX 20 mg/kg group and the control group (P = 0.01) (Table 6). These results suggest that PTXO is a safer version of PTX, and warrant further investigations. Rather than stopping PTX therapy altogether or reducing the PTX dose, using orotate compounds may offer safer alternatives for maintaining the antitumor therapy.
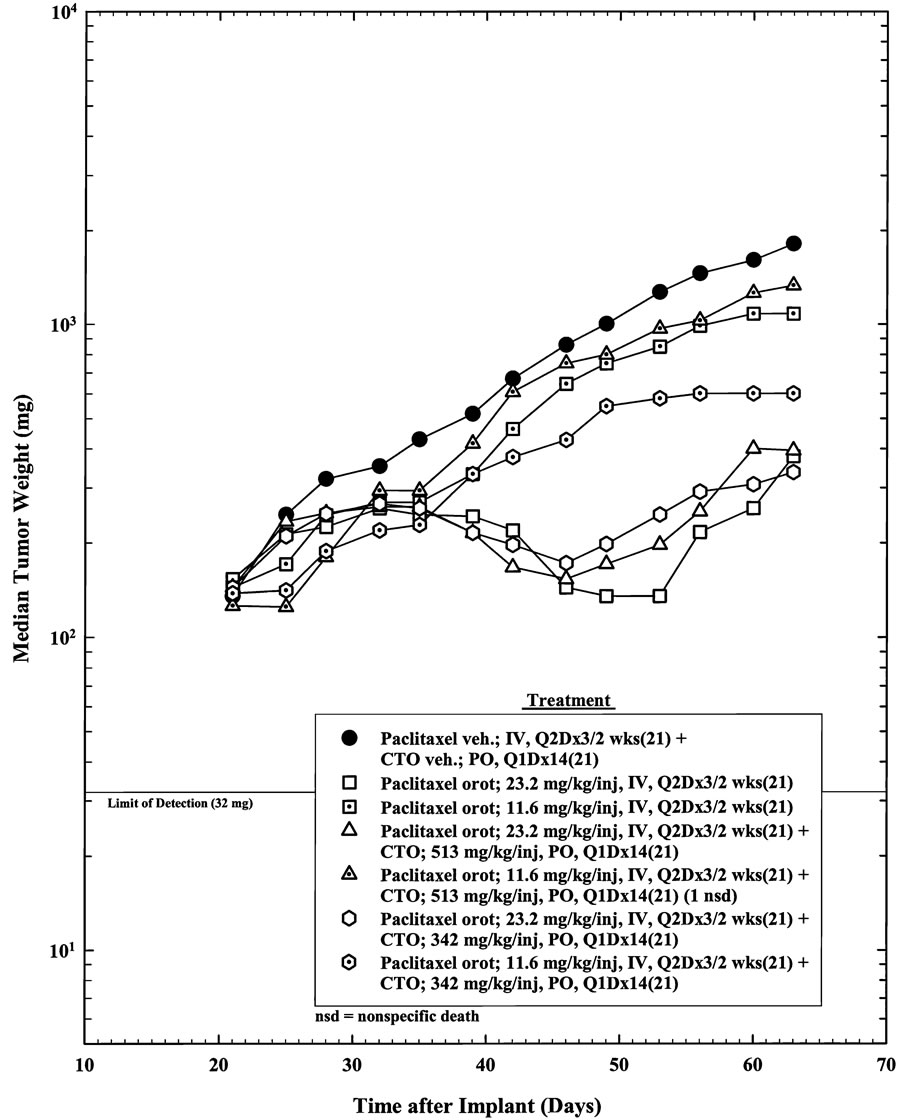
Figure 3. Response of SC implanted OVCAR-5 human ovarian tumor to combination treatment with paclitaxel orotate and CTO.
Body weight loss is a surrogate for the body’s reaction to cytotoxic agents, and may predict the quality of life and clinical outcome. Women who are diagnosed and treated with ovarian cancer are known to experience a variety of chemotherapy-related side effects that can affect quality of life. Ovarian cancer patients may experience cachexia as a result of advanced disease and therefore suffer body weight loss. The impact of changes in body weight during chemotherapy on survival has not been clear. Evidence about body weight changes, and specifically body weight loss due to side effects of cytotoxic agents, is mixed because of confounding variables. However, there is some evidence that a decrease in body weight during chemotherapy could have the potential to affect survival of cancer patients.
Hess and colleagues [19] found that change in body weight during primary chemotherapy was a strong prognostic factor for overall survival; that is, loss of body weight during primary therapy was an indicator of poor overall survival. Weight gain was an indicator of improved survival. A significant relationship was seen between median overall survival and weight change. The authors reported that the relative risk of death increased by 7% for each 5% decrease in body weight, and that strategies to minimize weight loss during primary chemotherapy should be developed to improve patient outcomes. LBM was a significant predictor of 5-FU toxicity in a study of colon cancer patients, an association not seen with body surface area (BSA). The differences seen in toxicities between patients were attributable to varia-

Figure 4. Response of SC implanted OVCAR-5 human ovarian tumor to combination treatment with paclitaxel and CTO.
tion in LBM [20]. Lean body mass was a better predictor of 5 FU pharmacokinetics than were standard anthropometric measures [21].
Comparison of the hematological parameters’ mean value between groups of mice administered equivalent doses of PTXO and PTX did not show statistically significant differences in leukocyte, erythrocyte, or platelet numbers, with the exception of eosinophils. In the high dose group (23.2 mg/kg for PTXO and 20 mg/kg for PTX), the mice administered PTXO had a lower mean eosinophil count in comparison to the mice administered paclitaxel. The significance of this result needs to be studied, as eosinophils are involved in inflammatory responses [22].
Lean body mass and weight change during chemotherapy have implications for toxicity and overall survival. The animal study results presented here show that low CTO doses have efficacy and low toxicity, suggesting CTO may have utility in maintenance therapy for ovarian cancer. Current formulations of PTX do not address the issue of preventing and/or reducing the body weight loss during chemotherapy, while maintaining the antitumor activity, as a strategy to reduce toxicity and adverse drug reactions. PTXO retained efficacy and minimized body weight loss during chemotherapy, and may have utility in the treatment of human ovarian cancer to improve patient outcomes. The effect of PTXO on body weight provides an important strategy to minimize body weight loss in prospective clinical studies in which various formulations of taxanes like PTX and docetaxel are used in chemotherapy to improve patient outcomes regardless of the formulation administered, i.e., in CrEL,
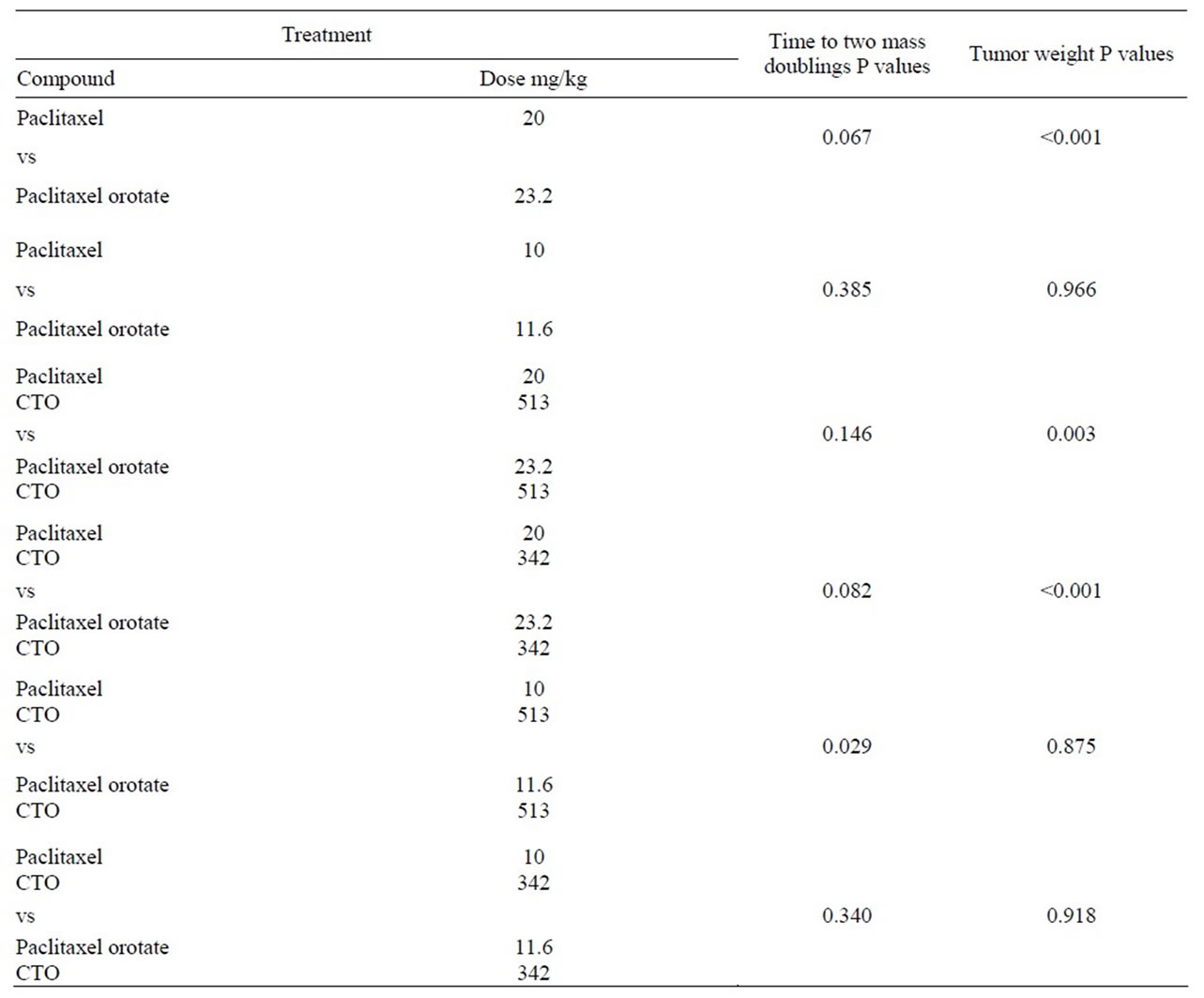
Table 5. Comparison of time to tumor mass doubling and tumor weight paclitaxel vs paclitaxel orotate.

Table 6. Body weight statistical analysis.
albumin nanoparticles, or liposomes.
Drug therapies that are used for the treatment of cancer can damage tissues with rapid cell turnover, such as the hematopoietic system, the gastrointestinal tract, and the genitourinary tract. Taxanes interfere with the metabolism and excretion of anthracyclines and potentiate anthracycline-induced cardiac toxicity [23]. Conversion of cytotoxic agents to their orotate compounds may yield alternative formulations with improved safety profiles. Orotic acid is a key intermediate in the biosynthesis pathway of pyrimidines and is found in cow’s milk. It has been used in clinical practice for conditions including pernicious anemia, neonatal jaundice, hyperuricemia and artherosclerosis [24]. Orotic acid bound to a mineral or drug becomes a neutral salt and acquires lipophilic properties, making diffusion through lipid membranes easier. Drug orotate derivatives undergo dissolution to release the drug as a charged molecule and free orotic acid, which in turn reduces drug-induced liver, heart, or other tissue toxicity. Chemical restructuring of pharmaceutical agents known for toxic side effects may allow them to retain tumor-inhibiting activity while inhibiting body weight loss during chemotherapy treatment.
5. Acknowledgements
Xenograft studies were carried out under contract with Southern Research Institute, Birmingham, AL. We thank

Figure 5. Effect of treatment with paclitaxel orotate and paclitaxel on mean body weight of female athymic nude mice implanted SC with OVCAR-5 ovarian tumor.
J. Abbott, who provided medical writing services on behalf of Tactical Therapeutics, Inc.
REFERENCES
- C. Santiskulvong, G. E. Konecny, M. Fekete, K. Y. Chen, A. Karam, et al., “Dual Targeting of Phosphoinositide 3-Kinase and Mammalian Target of Rapamycin Using NVP-BEZ235 as a Novel Therapeutic Approach in Human Ovarian Carcinoma,” Clinical Cancer Research, Vol. 15, No. 17, 2011, pp. 2373-2384. doi:10.1158/1078-0432.CCR-10-2289
- R. Wu, T. C. Hu, A. Rehemtulla, E. R. Fearon and K. R. Cho, “Preclinical Testing of PI3K/AKT/mTOR Signaling Inhibitors in a Mouse Model of Ovarian Endometrioid Adenocarcinoma,” Clinical Cancer Research, Vol. 12, No. 23, 2011, pp. 7359-7372. doi:10.1158/1078-0432.CCR-11-1388.
- E. Chu and V. T. DeVita, “Chemotherapeutic and Biologic Drugs,” In: E. Chu and V. T. DeVita, Eds., Physicians’ Cancer Chemotherapy Drug Manual 2010, Jones and Bartlett Publishers, Massachusetts, 2010, pp. 297- 301.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, et al., “Phase III Trial of Nanoparticle AlbuminBound Paclitaxel Compared with Polyethylated Castor Iil-Based Paclitaxel in Women with Breast Cancer,” Journal of Clinical Oncology, Vol. 23, No. 31, 2005, pp. 7794-7803. doi:10.1200/JCO.2005.04.937
- B. P. Schneider, F. Zhao, M. Wang, V. Stearns, S. Martino, et al., “Neuropathy Is Not Associated with Clinical Outcomes in Patients Receiving Adjuvant Taxane-Containing Therapy for Operable Breast Cancer,” Journal of Clinical Oncology, Vol. 30, No. 25, 2012, pp. 3051-3057. doi:10.1200/JCO.2011.39.8446
- M. Stenger, “Abraxane (Nanoparticle Albumin-Bound Paclitaxel) in Metastatic Breast Cancer,” Community Oncology, 2005, p. 214.
- V. Roy, B. R. LaPlant, G. G. Gross, C. L. Bane, F. M. Palmieri and North Central Cancer Treatment Group, “Phase II Trial of Weekly nab (Nanoparticle Albuminbound)-paclitaxel(nab-paclitaxel) (Abraxane) in Combination with Gemcitabine in Patients with Metastatic Breast Cancer (N0531),” Annals of Oncology, Vol. 20, No. 3, 2009, pp. 449-453. doi:10.1093/annonc/mdn661
- P. Vishnu and V. Roy, “Safety and Efficacy of Nab-Paclitaxel in the Treatment of Patients with Breast Cancer,” Breast Cancer (Auckl), Vol. 5, 2011, pp. 53-65. doi:10.4137/BCBCR.S5857
- J. L. Blum, “Clinical Implications of Using Nab Paclitaxel in Metastatic Breast Cancer,” Community Oncology, 2005, pp. 21-216.
- H. Rugo, W. T. Barry, A. Mareno-Aspitia, et al., “CALGB 40502/NCCTG N063H: Randomized Phase III Trial of Weekly Paclitaxel compared to Weekly Nanoparticle Albumin Bound Nab-Paclitaxel or Ixabepilone with or without Bevacizumab as First-Line Therapy for Locally Recurrent or Metastatic Breast Cancer,” 2012 ASCO Annual Meeting, Chicago, 1-5 June 2012, Abstract CRA1002.
- R. Kunstfeld, G. Wickenhauser, U. Michaelis, M. Teifel, W. Umek, et al., “Paclitaxel Encapsulated in Cationic Liposomes Diminishes Tumor Angiogenesis and Melanoma Growth in a ‘Humanized’ SCID Mouse Model,” Journal of Investigative Dermatology, Vol. 120, No. 3, 2003, pp. 476-482. doi:10.1046/j.1523-1747.2003.12057.x
- R. A. Karmali, Y. Y. Maxuitenko, G. S. Gorman and J. G. Page, “Carboxyamidotriazole Orotate and Cytotoxic Chemotherapy Have a Synergistic Effect on Tumor Inhibition in Glioblastoma and Colon Xenograft Mouse Models,” Cancer Therapy, Vol. 8, 2011, pp. 71-80.
- R. A. Karmali, Y. Y. Maxuitenko, G S. Gorman and Z. Qu, “Combinatorial Treatment with Carboxyamidotriazoleorotate and Temozolomide in SC-Implanted Human LOX IMVI Melanoma Xenografts,” Journal of Solid Tumors, Vol. 2, No. 5, 2012, pp. 1-16. doi:10.5430/jst.v2n5p13
- E. C. Kohn, E. Reed, G. A. Sarosy, L. Minasian, K. S. Bauer, et al., “A Phase I Trial of Carboxyamido-Triazole and Paclitaxel for Relapsed Solid Tumors: Potential Efficacy of the Combination and Demonstration of Pharmacokinetic Interaction,” Clinical Cancer Research, Vol. 7, No. 6, 2001, pp. 1600-1609.
- V. K. Oliver, A. M. Patton, S. Desai, D. Lorang, S. K. Libutti and E. C. Kohn, “Regulation of the Pro-Angiogenic Microenvironment by Carboxyamido-Triazole,” Journal of Cellular Physiology, Vol. 197, No. 1, 2003, pp. 139-148. doi:10.1002/jcp.10350
- G. J. Grover, J. Kelly, G. Moore, H. Jacoby, R. A. Karmali and G. S. Gorman, “Comparative Pharmacokinetic Profile of Carboxyamidotriazole and Carboxyamidotriazole-Orotate,” Cancer Therapy, Vol. 5, No. 2, 2007, pp. 437-442.
- C. Corrado, A. M. Flugy, S. Taverna, S. Raimondo, G. Guggino, et al., “Carboxyamidotriazole-Orotate Inhibits the Growth of Imatinib-Resistant Chronic Myeloid Leukaemia Cells and Modulates Exosomes-Stimulated Angiogenesis,” PLoS One, Vol. 7, No. 8, 2012, Article ID: e42310. doi:10.1371/journal.pone.0042310
- M. M. Hussain, H. Kotz, L. Minasian, A. Premkumar, G. Sarosy, et al., “Phase II Trial of Carboxyamidotriazole in Patients with Relapsed Epithelial Ovarian Cancer,” Journal of Clinical Oncology, Vol. 21, No. 23, 2003, pp. 4356-4363. doi:10.1200/JCO.2003.04.136
- L. M. Hess, R. Barakat, C. Tian, R. F. Ozols and D. S. Alberts, “Weight Change during Chemotherapy as a Potential Prognostic Factor for Stage III Epithelial Ovarian Carcinoma: A Gynecologic Oncology Group Study,” Gynecologic Oncology, Vol. 107, No. 2, 2007, pp. 260-265. doi:10.1016/j.ygyno.2007.06.010
- C. M. Prado, V. E. Baracos, L. J. McCargar, M. Mourtzakis, K. E. Mulder, et al., “Body Composition as an Independent Determinant of 5-Fluorouracil-Based Chemotherapy Toxicity,” Clinical Cancer Research, Vol. 12, No. 11, 2007, pp. 3264-3268. doi:10.1158/1078-0432.CCR-06-3067
- M. Gusella, S. Toso, E. Ferrazzi, M. Ferrari and R. Padrini, “Relationships between Body Composition Parameters and Fluorouracil Pharmacokinetics,” British Journal of Clinical Pharmacology, Vol. 54, No. 2, 2002, pp. 131- 139. doi:10.1046/j.1365-2125.2002.01598.x
- M. E. Rothenberg and S. P. Hogan, “The Eosinophil,” Annual Review of Immunology, Vol. 24, 2006, pp. 147- 174. doi:10.1146/annurev.immunol.24.021605.090720
- S. H. Giordano, D. J. Booser, J. L. Murray, N. K. Ibrahim, Z. U. Rahman, et al., “A Detailed Evaluation of Cardiac Toxicity: A Phase II Study of Doxorubicin and Oneor Three-Hour Infusion Paclitaxel in Patients with Metastatic Breast Cancer,” Clinical Cancer Research, Vol. 8, 2002, pp. 3360-3368.
- F. L. Rosenfeldt, “Metabolic Supplementation with Orotic Actic and Magnesium Orotate,” Cardiovascular Drugs and Therapy, Vol. 12, No. 2, 1998, pp. 147-152. doi:10.1023/A:1007732131887
NOTES
*Corresponding author.