Journal of Cancer Therapy
Vol.3 No.1(2012), Article ID:17216,10 pages DOI:10.4236/jct.2012.31007
Heat-Shocking of Murine Malignant Mesothelioma Cells Enhances Their Effectiveness as an Autologous Anti-Tumour Vaccine
![]()
1Queen Elizabeth II Medical Centre, School of Medicine & Pharmacology, University of Western Australia, Perth, Australia; 2National Research Centre for Asbestos Related Diseases, Western Australian Institute for Medical Research Inc., Nedlands, Australia; 3Institute for Molecular Bioscience, University of Queensland, Brisbane, Australia; 4School of Veterinary and Biomedical Sciences, Murdoch University, Murdoch, Australia.
Email: A.Currie@murdoch.edu.au, scott.fisher@uwa.edu.au
Received November 21st, 2011; revised December 23rd, 2011; accepted January 11th, 2012
Keywords: Hsp70; Immunotherapy; Adjuvant; Lung Cancer
ABSTRACT
Background: Malignant mesothelioma (MM) is a highly aggressive, incurable asbestos-induced cancer for which treatment options are limited. Surgical resection can reduce tumour burden, but patients ultimately succumb to disease due to reoccurrence of unresectable tumour, highlighting the need for new treatment modalities. In this study we describe the use of an easily translatable heat shock (HS) treated autologous tumour lysate vaccine and discus its potential application as an adjunct therapy for treating MM. Methods: Heat shocked autologous tumour lysate (HSL) vaccine was generated from AE17sOVA mesothelioma cells and tested for its ability to act as a protective or therapeutic vaccine in a murine tumour model. Vaccine efficacy was assessed by tumour growth/survival of vaccinated mice and FACS analysis used to assess DC maturation and trafficking from vaccine site to draining lymphnodes (dLN). Results: Mice vaccinated prior to tumour challenge with HS lysate induced protection in 40% of mice and caused a significant delay in tumour progression in remaining mice. Vaccine dose-response experiments showed that HS lysate was at least a log more efficient at retarding tumour growth and promoting survival than untreated lysate. HS and untreated lysate were equally effective at maturating DCs, but HS lysate improved trafficking of vaccine-site DCs to draining lymph nodes (dLN). Direct intratumoural injection of HS lysate significantly delayed tumour progression. Conclusions: HS treatment of tumour lysate improved vaccine immunogenicity, was associated with DC maturation, increased DC trafficking to dLNs and delayed tumour growth, particularly when administered intratumourally. Heat shocking autologous tumour cells is a simple and easily translatable approach to generate an immunogenic lysate vaccine with significant prophylactic and therapeutic effects. Coupling intratumoural HS vaccines with conventional therapies such as surgery may improve patient responses for otherwise refractive tumours.
1. Introduction
Tumour vaccine strategies in recent years have concentrated on the identification and isolation of specific antigens from a patients tumour and using the peptides either to stimulate exogenous DCs prior to adoptive transfer, or as a “typical” subcutaneous vaccine with adjuvant. Similar concepts have been adopted in the use of autologous heat shock protein (HSP)-peptide complexes isolated from patient tumour samples, with promising tumour-specific, CD8+ T lymphocyte observations and expansion of the natural killer (NK) cell population in immunized patients [1,2]. However, these methods of vaccine production are both time consuming and labour-intensive. Heat-shocking cells increases their immunogenicity [3-5]. Thus, an efficient and potentially enhanced immunogenic method of delivering HSP tumour vaccines may be to source autologous tumour cells via resection, and induce HSP production in the laboratory prior to vaccination at either a preadjuvant-conditioned site (to promote DC migration to the vaccination site) or in combination with another adjuvant.
HSP immunogenicity was originally observed due to the stimulation of a robust T cell response against tissues from which the HSPs had been sourced and purified [6]. Later, rejection of tumour was observed when directly injected with HSPs purified from autologous tumour cells in-vitro [7]. HSP released from necrotic cells are known to be efficiently endocytosed, mediated by receptors on antigen presenting cells (APCs) [8] and these HSP-peptide complexes are presented through the MHC class I antigen (Ag) presentation pathway. Aside from the crosspresentation capabilities of HSPs through various recaptors on APCs [9], they have also been attributed with an adjuvant effect that is independent of peptide and associated with stimulation of Toll like receptor 2 (TLR-2) or TLR-4 with the non-peptide bound HSP [10,11]. This response involves an upregulation of inflammatory cytokines including interleukin-12 (IL-12) and co-stimulatory molecules, with concomitant activation of NK cells and other immune system cells and co-stimulatory molecules [12-15].
In this study, we investigated the possibility of enhancing the immunogenicity of an autologous MM vaccine by enriching cell lysates with HSPs using a heatshocking protocol prior to lysis. We also examined the DC-activating properties and therapeutic potential of this heat-shocked MM vaccine.
2. Materials and Methods
2.1. Reagents and Mice
All reagents were purchased from Sigma (Castle Hill, NSW, Australia) unless stated otherwise. Female C57Bl/6 (H-2Kb) mice aged between 6 and 8 wks were obtained from the Animal Resources Centre (Murdoch, Western Australia) and maintained under standard SPF housing conditions. Animal experiments were carried out according to protocols approved by the University of Western Australia Animal Ethics Committee.
2.2. Tumour Cell Lines
The murine MM cell line AE17 was derived from the peritoneal cavity of C57Bl/6J mice injected with asbestos fibres (crocidolite) and stably transfected with the naturally occurring secreted form of OVA (AE17sOVA) under the control of the human β-actin promoter as described [16]. The AE17sOVA cell line was maintained in RPMI 1640 media (Invitrogen, Victoria, Australia) supplemented with 10% FCS (CSL, Victoria, Australia), 20mM HEPES (Life Technologies), pH 7.4, 48 mg/L gentamicin (Pharmacia and Upjohn, Western Australia, Australia), 60 mg/L benzylpenicillin (David Bulls Laboratory, Victoria, Australia) and 0.05 mM 2ME (Merck, West Point, PA). Supplemented with 400 µg/L of the neomycin analogue G418 (Geneticin: Invitrogen). Cells were cultured at 37˚C in a 5% CO2 atmosphere and passaged when 70% confluent.
2.3. Heat Treatment of Tumour Cells and Lysate Preparation
Monolayers of AE17sOVA cells in T175 flasks were incubated for 1 h at 43˚C using a water bath. The cells were then incubated in fresh media at 37˚C (5% CO2) for 3 hr. Cells were harvested with trypsin and centrifuged at 400 × g for 7 min, media was removed and the cells were washed × 2 with PBS. After a cell count using a haemocytometer, the cells were adjusted to, either 1 × 106, 1 × 107, or 1 × 108 cells/100 mL in PBS in cryovials and freezethawed in liquid N2/37˚C water bath for 5 cycles. Cryovials were then stored at –80˚C until use.
2.4. Hsp70 Quantification in Tumour Cell Lysate
The amount of inducible Hsp70 induced by heat-shocking of cultured AE17sOVA cells was quantified using an Hsp70 ELISA kit according to the manufacturer’s instructions (Stressgen Bioreagents, Vancouver, BC). Briefly, a cell pellet of 1 × 106 tumour cells was treated with extraction buffer in the presence of protease inhibitors and the cell suspension incubated on ice for 30 min. Extracts were centrifuged at 21,000 × g for 10 min/4˚C. The supernatant was removed and assayed by quantitative sandwich ELISA, which does not cross-react with other Hsp70 family members (e.g. Hsc70). Hsp70 levels were also visualised after western blot. Freeze-thawed lysates of AE17sOVA cells (untreated or heat-shocked), a positive control HeLa cell extract (BD Biosciences, New Jersey, USA), and recombinant Hsp70 (Stressgen Bioreagents) were resolved by SDS-PAGE on a 12% (w/v) acrylamide Tris-HCL pre-cast gel as per manufacturer’s instructions (Bio-Rad Laboratories, Australia). Proteins were transferred to nitrocellulose membrane before staining with mouse anti-human Hsp70 clone 7 (cross-reactive with mouse, BD Biosciences, New Jersey USA) followed by rabbit-anti-mouse IgG-HRP secondary antibody (Invitrogen Pty Ltd, Carlsbad CA) and ECL substrate (Pierce, Thermo Scientific, Rockford IL) prior to film exposure & development.
2.5. Protective and Therapeutic Vaccine Experiments
For single vaccination experiments, mice received 100 ml of untreated or heat-shocked tumour lysate (102 to 108 cells/dose depending on experiment) in the left flank by subcutaneous (s.c.) injection. For multiple vaccination experiments mice received 3 doses of either untreated or heat-shocked lysate vaccine (107 cells/dose) in the right flank in one week intervals. Mice injected with saline alone were used as controls throughout. All mice were challenged 14 days after the last dose of vaccine by s.c. injection in the right flank with 1 × 106 viable AE17- sOVA tumour cells, unless otherwise stated. Subsequent tumour growth was monitored by taking two perpendicular diameters using microcalipers. Mice were sacrificed when tumour dimension reached 100 ± 5 mm2. For therapeutic treatments, mice were first inoculated with 1 × 106 viable AE17-sOVA tumour cells in the right flank on day 0 and solid tumours allowed to establish for 10 days (reaching approximately 4 mm2). Tumour-bearing mice then received a single 100 ml injection of heatshocked lysate vaccine (107 cells/dose) either in the opposing flank or directly into the tumour itself, and tumour growth monitored as above.
2.6. DC Culture and Stimulation
Bone-marrow-derived DC (BMDC) was generated using recombinant murine granulocyte/macrophage-colony stimulating factor (GM-CSF; Prospec-Tany, Rehovot, Israel) as described [17]. After 10 days in culture, BMDC were exposed for 24 h to either; saline, lipopolysaccharide (LPS; 100 ng/ml), heat-shocked or untreated tumour lysates (equivalent to 5 × 106 tumour cells) in Teflon 24-well inserts (Savillex, Mn, USA) at 1 × 106 BMDC per well. BMDC were then washed twice with PBS and stained with FITC-anti-mouse CD11c (eBioscience, San Diego, CA) along with either; PE-conjugated anti-mouse CD80 or CD86 (eBioscience), or MHC class II (I-Ab; clone TIB120, prepared in house). Appropriate FITC and PE-conjugated IgG isotypes were used as controls. Cells were acquired on a FACScalibure flow cytometer and data analysed using CellQuest and FlowJo software.
2.7. In Vivo Tracking of Vaccine-Site DC Migration
Mice (5 per group) were vaccinated s.c. with either heatshocked AE17sOVA lysate or untreated lysate (107 tumour cells/dose), or with saline as a control. Eight hours later, mice received 25 mM of cell tracker blue (Invitrogen) s.c. in 100 mL saline into the same site. After a further 12 h, mice were sacrificed and the inguinal and axillary lymph nodes draining the injection site along with the contralateral non-draining lymph nodes were then taken for analysis. A cell suspension was prepared as described [18] and stained with PE-anti-mouse CD11c, TriColor-anti-mouse CD8a, and APC-streptavidin/biotinanti-mouse CD11b and biotin-anti-mouse Dec205, costained with FITC-streptavidin (all from eBioscience). Appropriate fluorophore-conjugated IgG isotypes were used as controls. Cells were acquired and analysed by FACS as above.
2.8. Statistical Analysis
Student’s t test was used to measure significance between two individual groups, Log rank analysis was performed on survival curves. All analysis was performed using Graph Pad Prism Software (Graph Pad Software Inc., CA, USA) and a P value < 0.05 considered signifycant.
3. Results
3.1. Heat-Shocking Induces Hsp70 Expression in AE17sOVA Cells
The susceptibility of the ovalbumin-transfected malignnant mesothelioma cell line (AE17sOVA) to heat-induced stress was examined by measuring the levels of Hsp70 by western blot and ELISA in the untreated and heatshocked lysates. Hsp70 was undetectable by ELISA in the untreated lysate and was only observed at low levels by western blot (Figure 1(a)). Heat-shocking for 1 h at 43˚C induced the expression of Hsp70 in AE17sOVA to a level of 250 ng/106 cells, similar to that observed in human mesothelioma cell lysate (p. 42; Figure 1(b)). Heatshocking of a Balb/c-restricted mesothelioma cell line,
 (a)
(a)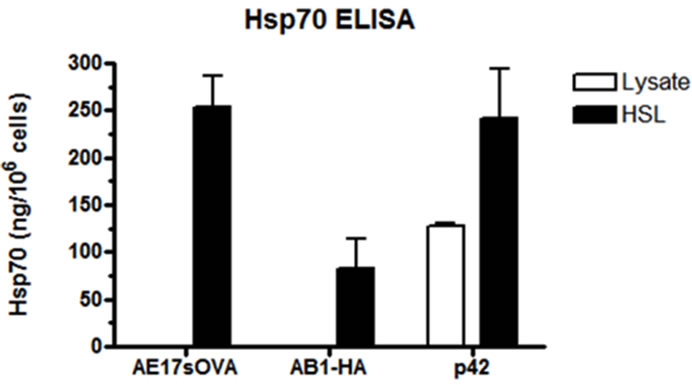 (b)
(b)
Figure 1. Induction of Hsp70 expression in AE17sOVA following heat-shock. Monolayers of AE17sOVA cells were incubated at 37˚C (untreated) or heat shocked at 43˚C for 1 hour in a water bath prior to recovery in fresh media for 3 hrs at 37˚C. (a) Hsp70 specific western blot analysis demonstrating an increase in Hsp70 expression in cell lysates following heat shock treatment relative to untreated controls; (b) Hsp70 specific ELISA demonstrating that the level of Hsp70 protein expression increased upon heat shock treatment. Similar levels of Hsp70 was observed between AE17sOVA and human mesothelioma cell lysates, but not AB1-HA (Balb/c mesothelioma) cell lysate following heat shock treatment.
AB1-HA [19], also induced detectable Hsp70 expression but at 3-fold lower levels than AE17sOVA.
3.2. Heat-Shocked AE17sOVA Cell Lysate is More Effective at Delaying and Preventing Tumour Progression
To determine the effectiveness of both untreated and heatshocked tumour cell lysate vaccines at protecting against tumour growth, mice were vaccinated once, subcutaneously with increasing doses of either vaccine and then challenged 14 days later with viable AE17sOVA tumour cells. Both vaccines formulations were partially protective (20% - 40% long-term survival) at the highest dose tested (108 cells/dose; Table 1). However, only the heatshocked cell lysate vaccine was still protective when a 10-fold lower dose was used, although no protection was observed for doses lower than this. Both vaccines also significantly delayed tumour growth (~2-fold) in unprotected mice when using 108 cells/dose. Here again, the heat-shocked cell lysate vaccine was approximately a log-fold more effective than the untreated cell lysate with significant delays in tumour growth observed using as little as 106 cells/dose (Table 1). Vaccination with purified ovalbumin in incomplete Freund’s adjuvant (IFA) had no effect on survival or growth of AE17sOVA (Table 1) despite promoting robust OVA-specific CD8 responses (data not shown). Interestingly, vaccination of BALB/c mice with heat-shocked AB1-HA lysate (107cells/dose) had no effect on survival or tumour growth after AB1-HA challenge (data not shown), consistent with the lower Hsp70 response observed in this line (Figure 1(b)).
3.3. Multiple Vaccinations Do Not Improve Resistance to Challenge
To ascertain if a prime-boost strategy would be more effective at promoting survival against AE17sOVA, mice received 3 vaccine doses (107 cells/dose s.c.) one week apart, prior to tumour challenge. Multiple vaccinations with heat-shocked cell lysate had no added benefit on survival over a single vaccination, and multiple vaccinetions with untreated cell lysate were still unable to promote survival (Figure 2(a)). Emergence of tumour in unprotected mice receiving multiple doses of heat-shocked cell lysate was also significantly delayed (~20 days; Figure 2(b)). However, unlike tumour kinetics in the singledose experiments, once tumours did emerge they appeared to grow more rapidly than in saline or untreated cell lysate vaccinated mice (Figure 2(b)).
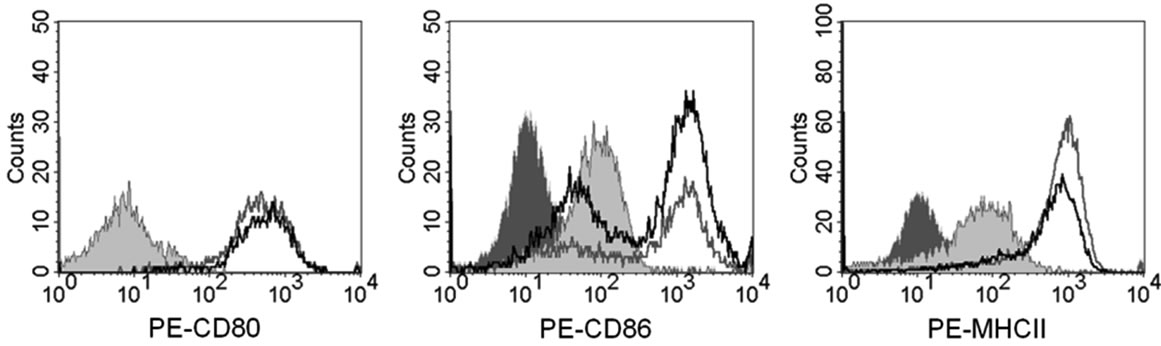
3.4. Both Heat-Shocked and Non Heat-Shocked Lysate Vaccines Induce Maturation of BMDC
To determine if the increased potency of HSL vaccination was due to adjuvant effects on DCs we measured the level of maturation induced by HSL and lysate on BMDC in-vitro, BMDCs were stimulated 5 to 1 with equivalent tumour cells for 24 hr and then stained for MHC and co-stimulatory molecules. Unstimulated BMDC had relatively low levels of CD80, CD86 and MHC
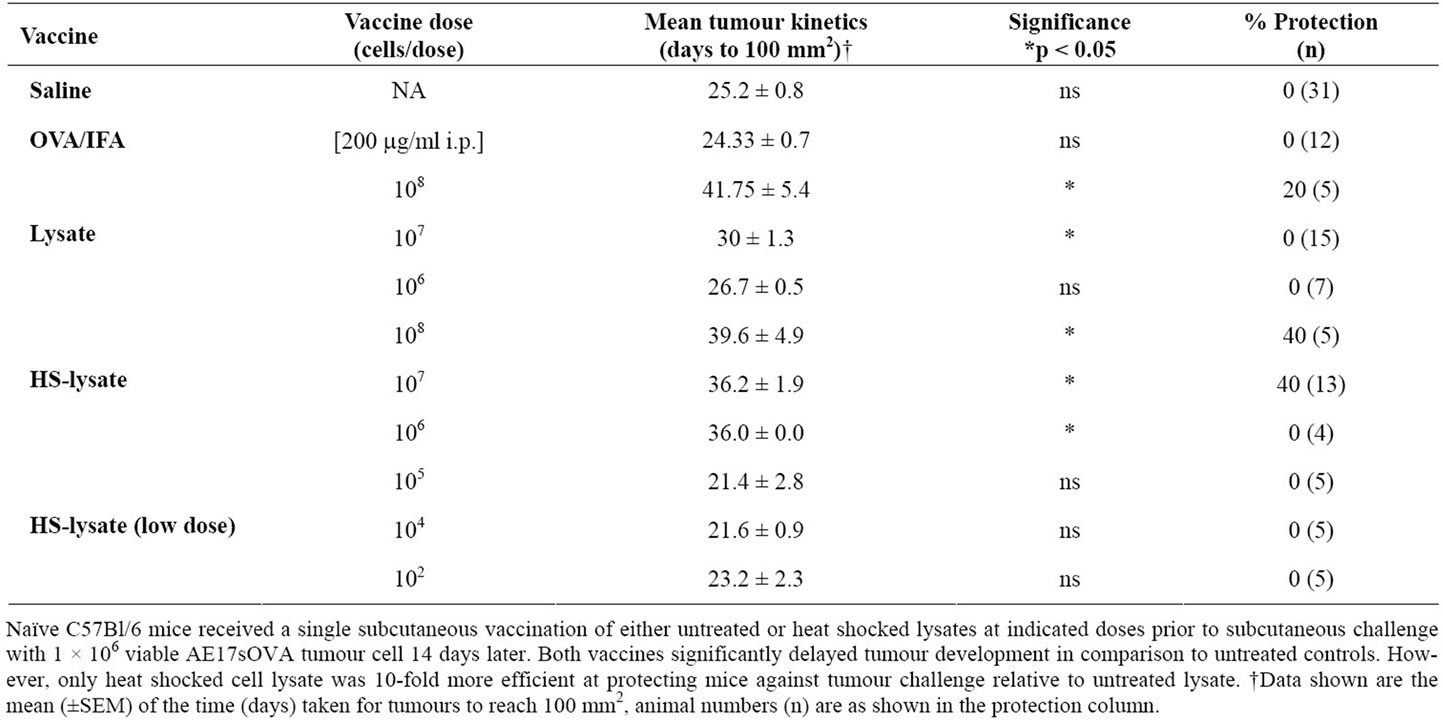
Table 1. Effect of dose in the efficacy of lysate and heat shocked lysate vaccines.
 (a)
(a) (b)
(b)
Figure 2. Multiple HSL vaccination does not improve resistance to tumour challenge. (a) Naïve C57Bl/6 mice were vaccinated subcutaneously with three doses of either untreated or heat-shocked cell lysates on days 0, 7 and 14 (1 × 107 cells/dose) prior to subcutaneous challenge with viable AE17sOVA tumour cells on day 28. No additional survival benefit was observed for mice receiving multiple vaccinations versus those that received single vaccination; (b) Mice that ultimately failed to respond heat shocked lysate vaccination demonstrated significantly delayed tumour growth relative to saline and untreated lysate controls; although once tumour began to develop it did so at a much faster rate relative to control mice.
class II (Figure 3(a)). Stimulation with lysate caused a marked upregulation in expression of all three surface markers. Stimulation with HSL also induced upregulation of CD80, CD86 and MHC class II but responses were not higher than with normal lysate.
3.5. Heat-Shocked AE17sOVA Lysate Induces Local DCs to Preferentially Migrate to Draining Lymph Node
We next investigated the kinetics involved in DC trafficking to lymph nodes, subsequent to exposure to HSL. Mice were vaccinated s.c. with 1 × 107 HSL followed 8 h later by treatment of the injection site with cell tracker blue dye in order identify migrating DCs. HSL-vaccinated mice showed a four-fold increase in the proportion of cells trafficking from the vaccine-site in the draining lymph node compared to those treated with lysate, and over a seven-fold increase compared to saline treated mice (Figure 3(b)). Trafficking cells were CD11c+ and CD8– (Figure 3(b)) and expressed CD11b and Dec205 (data not shown) consistent with a migratory DC profile [20]. Together, these data indicate that HSL and lysate vaccines can induce DC activation but that HSL treatment promotes superior recruitment of activated DCs in the draining lymphnodes.
3.6. Prophylactic Vaccination Promotes Long Term Immunological Protection
We next tested whether HSL vaccination could promote long term immunological protection (i.e. memory) against AE17sOVA tumour. Surviving mice from single vaccination experiments (Table 1, n = 4) were subcutaneously rechallenged on the contra-lateral flank with 1 × 106 viable tumour cells, 100 days after the initial tumour challenge (i.e. d14 + 100). On the same day, naïve control mice were inoculated with 1 × 106 AE17sOVA cells, having received saline vaccination 14 days earlier. Control mice succumbed to tumour growth by day 30, post tumour challenge, while 100% of HSL vaccinated mice survived tumour rechallenge with no visible tumours (Figure 4).
3.7. Therapeutic Intratumoural Injection of HSL Vaccine Delays Tumour Growth, but Does Not Protect against Tumour Challenge
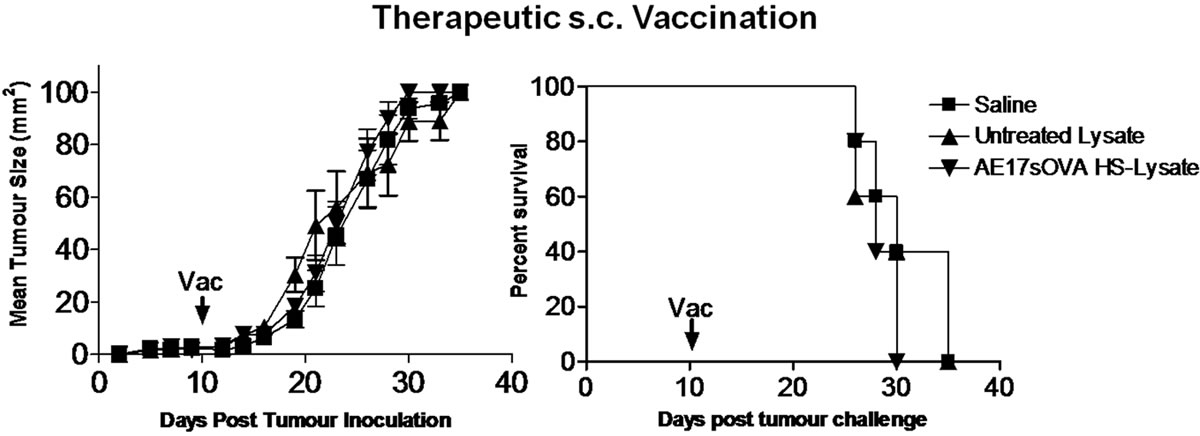
To determine whether AE17sOVA HSL vaccination could protect against tumour challenge in a therapeutic setting, groups of 5 mice were first inoculated s.c. on day 0 with 1 × 106 viable AE17sOVA cells and then treated on day 10 with either AE17sOVA HSL or untreated AE17s OVA lysate equivalent to 1 × 107 cells, or with saline (control). Tumour growth was consistent between all groups with all mice succumbing to tumours by day 35 when vaccines were administered subcutaneously (Figure 5(a)). Interestingly, when vaccines were administered intratumorally (i.t.), we observed a significant delay in tumour growth (p < 0.05; ≥1.3-fold) in HSL vaccinated mice relative to other groups. However, therapeutic i.t. vaccination failed to eliminate AE17sOVA tumour development (Figure 5(b)).
4. Discussion
Aggressive surgery with adjunct chemotherapy or radiotherapy is one treatment regimen used for malignant
 (a)
(a)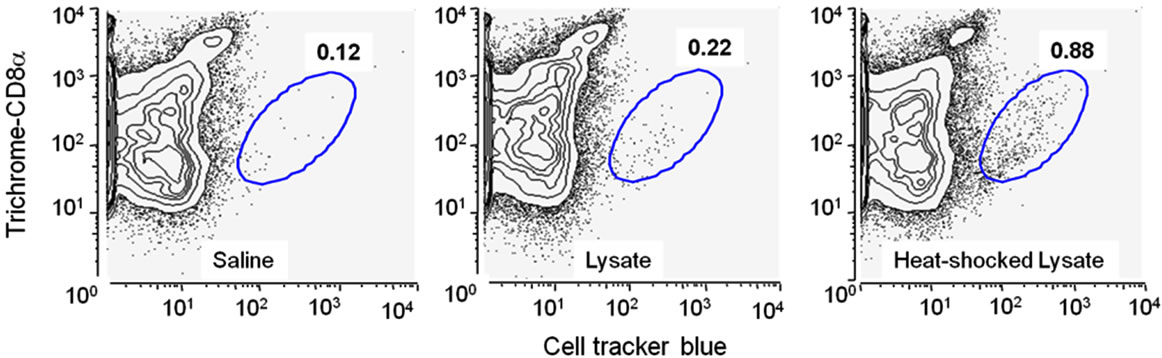 (b)
(b)
Figure 3. Heat shocked lysate promotes equivalent level of maturation as lysate-stimulated BMDC. (a) Bone marrow derived dendritic cells were stimulated in vitro with heat-shocked or non-heat-shocked AE17sOVA lysate (equivalent to 5:1 tumor cells/DC) of left unstimulated for 24 hrs prior to staining for surface marker expression of CD11c and the DC maturation markers CD80, CD86 and MHC class II. Dark grey shaded histogram = isotype control; light grey shaded histogram = unstimulated; grey line = untreated lysate; black line = heat shock treated lysate; (b) Vaccine-induced DC trafficking to lymphnodes. Mice were injected with either heat-shocked lysate or untreated lysate (both 107 cells/injection) for 8 h. Cell Tracker Blue dye was then added to the injection site and after a further 12 h the number of “dye” containing CD11c+ DCs in the draining lymph node determined by flow cytometry after first pooling lymphnodes from 5 mice per group.
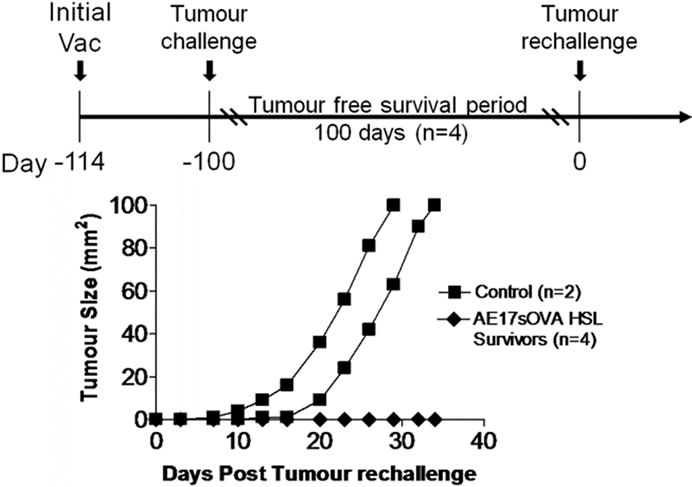
Figure 4. Prophylactic vaccination promotes long term immunological protection against tumour rechallenge. Surviving mice from single vaccination experiments (Table 1, n = 4) were rechallenged with 1 × 106 viable AE17sOVA cells (s.c) on the contra-lateral flank 100 days after initial tumour challenge. Control mice (n = 2) received tumour challenge 14 days post saline vaccination. 100% of HSL vaccinated mice survived tumour rechallenge while all control mice succumb to tumour by day 30.
mesothelioma with the intent of prolonging life, especially in younger patients. Yet they are seldom curative. Most MM patients experience a high recurrence rate of local disease and succumb within 9 to 12 months of diagnosis [21] highlighting the need for improved adjuvant therapies that will target residual or metastatic disease and improve survival after surgery. Immunotherapies, such as tumour vaccines have emerged as a therapeutic option for the management of cancer patients. These vaccines have shown most potential when targeted toward relatively immunogenic cancers with known tumour antigens such as melanoma and prostate cancer [22, 23]. However, despite these limited clinical successes, cancer vaccines to date have not lived up to their potential as many cancers, such as MM, are poorly immunogenic and their tumour associated antigens (TAAs) remain uncharacterised. In the absence of known tumour antigens, the use of autologous tumour lysates have provided an alternative vaccine approach, as by definition they should contain all relevant TAAs within them (reviewed in [24]). Hyperthermic treatment of tumour lysates can enhance their immunogenic potential due to the
 (a)
(a)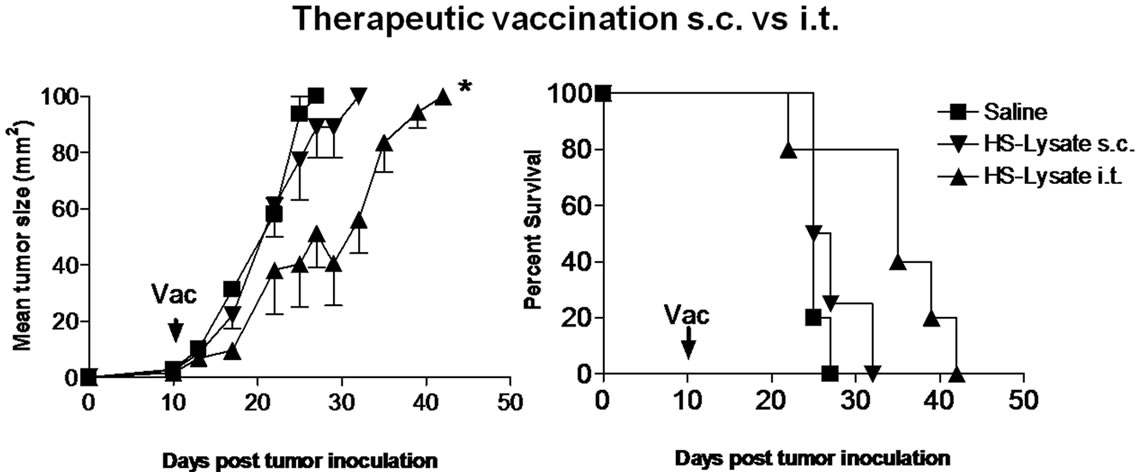 (b)
(b)
Figure 5. Therapeutic vaccination fails to protect against tumour rechallenge. Naïve mice were inoculated subcutaneously with 1 × 106 viable AE17sOVA cells 10 days prior to receiving saline control, untreated lysate or heat shock lysate via subcutaneous (s.c) or intratumoral (i.t) vaccination. (a) Consistent tumour growth was observed between all groups with all subcutaneously vaccinated mice succumbing to tumour by day 35; (b) Intratumoral therapeutic vaccination with heat shocked lysate significantly delayed tumour development relative to control or subcutaneous vaccination groups, but failed to protect against overall tumour development.
ability of heat shock proteins to promote cross presentation of antigenic peptides [3-5], a necessary step for inducing anti-tumour immunity. In contrast to previous studies that utilised HSP-peptide complexes purified from tumour lysates [1,2,25], we investigated whether a simple heat-shock protocol could be used to enrich MM tumour cells with HSPs prior to lysis and to test whether HS treatment enhanced the immunogenicity of the resulting tumour lysate vaccine.
Our data demonstrate that HS treatment of tumour cells results in increased Hsp70 expression equivalent to that observed in human mesothelioma cells. When lysates made from these cells were used as vaccines, we observed significantly improved protection and delayed tumour development. These results are consistent with previous reports demonstrating the anti-tumour properties of tumour derived HSP-peptide complexes [1,2,5]. Interestingly, both HSL and untreated lysate were able to induce similar levels of BMDC maturation following vaccination, although HSL showed enhanced recruitment of local DCs to the draining lymph node (dLN) compared to untreated lysate. Cross presentation of tumour antigen by DCs to T cells is essential for the development of antitumor immunity and the increased DCs trafficking to the dLN plus the ability of HSPs to more efficiently chaperon antigenic peptides for presentation [5,25] may explain the enhanced immunogenicity of the HSL vaccine. This has been exploited in a recent study using DC vaccines fused with purified HSP complexes to enhance DC vaccination efficacy [26]. However, these protocols are generally labour intensive, and require substantial time commitments and cost associated with preparation of vaccine components. Our data demonstrates that a simple, inexpensive method of enriching HSP in tumour lysates is comparable to more costly and time consuming autologous lysate preparation methods. Importantly, we have previously demonstrated the feasibility of this preparation method for treating MM [27].
In the prophylactic setting, a single s.c dose of the HSL vaccine was sufficient to induce long term immunological memory and protect against subsequent tumour rechallenge. However, as with many tumour immuno-therapies the s.c HSL vaccine failed to protect if administered therapeutically. Importantly, when HSL was used as a therapeutic vaccine administered intratumourally it was able to significantly delay tumour growth. Thus directly delivered HSL vaccines could boost local anti-tumor responses, at possibly the most relevant site. We observed a similar result in an earlier study using our MM model following i.t delivery of interleukin-2 (IL-2) plus anti-CD40 combined adjuvant immunotherapy [28]. Here, i.t. delivered IL-2 + anti-CD40, induced local inflammation that prompted tumour destruction and release of autologous TAA, which in turn promoted the development of a systemic anti-tumour immune response. These results demonstrate the potential for locally administered adjuvant immunotherapies to manipulate the tumour milieu such that the tumour can now become its own vaccine.
This raises the possibility of using HS treated tumour lysates to complement current treatment options for the management of solid malignancies like MM. Thermal treatment modalities such as radio frequency ablation (RFA), microwave ablation (MVA) and cryoablation have been used to treat solid malignancies, including lung cancers [29-31]. RFA induces hyperthermia within the tumour resulting in necrotic cell death and the release of endogenous adjuvants such as Hsp70 [32], which can then promote a primary anti-tumour response. Indeed, the combination of HSL pulsed autologous DC vaccine with RFA was recently shown to abrogate tumour recurrence [33], demonstrating the practical advantage of using HS treatment of autologous vaccines as an adjuvant therapy in combination with standard treatment modalities to target residual disease.
5. Conclusion
In conclusion, we have shown that heat shocking of autologous tumour cells is a simple and translatable approach to generate an immunogenic lysate vaccine with significant prophylactic and therapeutic effects when delivered intratumourally. Coupling intratumoural autologous heat shocked vaccines with conventional therapies such as chemotherapy and surgery may improve patient responses for otherwise refractive tumours such as malignant mesothelioma.
6. Acknowledgements
The authors have no conflict of interest to report. This work was supported by the Dust Diseases Board, NSW, Australia. A.J. Currie was kindly supported by the Raine Medical Research Foundation, Western Australia.
REFERENCES
- S. Janetzki, D. Palla, V. Rosenhauer, H. Lochs, J. J. Lewis and P. K. Srivastava, “Immunization of cancer patients with autologous cancer-derived heat shock protein gp96 preparations: A Pilot Study,” International Journal of Cancer, Vol. 88, No. 2, 2000, pp. 232-238. doi:10.1002/1097-0215(20001015)88:2<232::AID-IJC14>3.0.CO;2-8
- V. Mazzaferro, J. Coppa, M. G. Carrabba, L. Rivoltini, M. Schiavo, E. Regalia, L. Mariani, T. Camerini, A. Marchiano, S. Andreola, R. Camerini, M. Corsi, J. J. Lewis, P. K. Srivastava and G. Parmiani, “Vaccination with Autologous Tumor-Derived Heat-Shock Protein gp96 after Liver Resection for Metastatic Colorectal Cancer,” Clinical Cancer Research, Vol. 9, No. 9, 2003, pp. 3235-3245.
- H. Bendz, S. C. Ruhland, M. J. Pandya, O. Hainzl, S. Riegelsberger, C. Brauchle, M. P. Mayer, J. Buchner, R. D. Issels and E. Noessner, “Human Heat Shock Protein 70 Enhances Tumor Antigen Presentation through Complex Formation and Intracellular Antigen Delivery without Innate Immune Signaling,” Journal of Biological Chemistry, Vol. ,282 No. 43, 2007, pp. 31688-31702. doi:10.1074/jbc.M704129200
- R. J. Binder, N. E. Blachere and P. K. Srivastava, “Heat Shock Protein-Chaperoned Peptides but Not Free Peptides Introduced into the Cytosol Are Presented Efficiently by Major Histocompatibility Complex I Molecules,” Journal of Biological Chemistry, Vol. 267, No. 20, 2001, pp. 17163-17171. doi:10.1074/jbc.M011547200
- R. J. Binder and P. K. Srivastava, “Peptides Chaperoned by Heat-Shock Proteins Are a Necessary and Sufficient Source of Antigen in the Cross-Priming of CD8+ T Cells,” Nature Immunology, Vol. 6, No. 6, 2005, pp. 593-599. doi:10.1038/ni1201
- P. K. Srivastava, “Peptide-Binding Heat Shock Proteins in the Endoplasmic Reticulum: Role in Immune Response to Cancer and in Antigen Presentation,” Advance in Cancer Research, Vol. 62, 1993, pp. 153-177.
- P. K. Srivastava, A. Menoret, S. Basu, R. J. Binder and K. L. McQuade, “Heat Shock Proteins Come of Age: Primitive Functions Acquire New Roles in an Adaptive World,” Immunity, Vol. 8, No. 6, 1998, pp. 657. doi:10.1016/S1074-7613(00)80570-1
- B. Berwin, R. C. Reed and C. V. Nicchitta, “Virally Induced Lytic Cell Death Elicits the Release of Immunogenic GRP94/gp96,” Journal of Biological Chemistry, Vol. 276, No. 24, 2001, pp. 21083-21088. doi:10.1074/jbc.M101836200
- P. Srivastava, “Interaction of Heat Shock Proteins with Peptides and Antigen Presenting Cells: Chaperoning of the Innate and Adaptive Immune Responses,” Annuls Reviews of Immunology, Vol. 20, 2002, pp. 395-425.
- A. Asea, M. Rehli, E. Kabingu, J. A. Boch, O. Bare, P. E. Auron, M. A. Stevenson and S. K. Calderwood, “Novel Signal Transduction Pathway Utilized by Extracellular HSP70: Role of Toll-Like Receptor (TLR2) and TLR4,” Journal of Biological Chemistry, Vol. 277, No. 17, 2002, pp. 15028-15034. doi:10.1074/jbc.M200497200
- R. M. Vabulas, P. Ahmad-Nejad, C. da Costa, T. Miethke, C. J. Kirschning, H. Hacker and H. Wagner, “Endocytosed HSP60s Use Toll-Like Receptor 2 (TLR2) and TLR4 to Activate the Toll/Interleukin-1 Receptor Signaling Pathway in Innate Immune Cells,” Journal of Biological Chemistry, Vol. 276, No. 33, 2001, pp. 31332-31339. doi:10.1074/jbc.M103217200
- A. Asea, S. K. Kraeft, E. A. Kurt-Jones, M. A. Stevenson, L. B. Chen, R. W. Finberg, G. C. Koo and S. K. Calderwood, “HSP70 Stimulates Cytokine Production through a CD14-Dependant Pathway, Demonstrating Its Dual Role as a Chaperone and Cytokine,” Nature Medicine, Vol. 6, No. 4, 2000, pp. 435-442. doi:10.1038/74697
- S. Basu, R. J. Binder, R. Suto, K. M. Anderson and P. K. Srivastava, “Necrotic but Not Apoptotic Cell Death Releases Heat Shock Proteins, Which Deliver a Partial Maturation Signal to Dendritic Cells and Activate the NFKappa B Pathway,” International Immunology, Vol. 12, No. 11, 2000, pp. 1539-1546. doi:10.1093/intimm/12.11.1539
- C. Massa, C. Melani and M. P. Colombo, “Chaperon and Adjuvant Activity of hsp70: Different Natural Killer Requirement for Cross-Priming of Chaperoned and Bystander Antigens,” Cancer Research, Vol. 65, No. 17, 2005, pp. 7942-7949.
- G. Multhoff, L. Mizzen, C. C. Winchester, C. M. Milner, S. Wenk, G. Eissner, H. H. Kampinga, B. Laumbacher and J. Johnson, “Heat Shock Protein 70 (Hsp70) Stimulates Proliferation and Cytolytic Activity of natural Killer Cells,” Experimental Hematology, Vol. 27, No. 11, 1999, pp. 1627-1636. doi:10.1016/S0301-472X(99)00104-6
- C. Jackaman, C. S. Bundell, B. F. Kinnear, A. M. Smith, P. Filion, D. van Hagen, B. W. Robinson and D. J. Nelson, “IL-2 Intratumoral Immunotherapy Enhances CD8+ T Cells That Mediate Destruction of Tumor Cells and Tumor-Associated Vasculature: A Novel Mechanism for IL-2,” Journal of Immunology, Vol. 171, No. 10, 2003, pp. 5051-5063.
- K. Inaba, M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu and R. M. Steinman, “Generation of Large Numbers of Dendritic Cells from Mouse Bone Marrow Cultures Supplemented with Granulocyte/Macrophage Colony-Stimulating Factor,” Journal of Experimental Medicine, Vol. 176, No. 6, 1992, pp. 1693-1702. doi:10.1084/jem.176.6.1693
- S. Henri, D. Vremec, A. Kamath, J. Waithman, S. Williams, C. Benoist, K. Burnham, S. Saeland, E. Handman and K. Shortman, “The Dendritic Cell Populations of Mouse Lymph Nodes,” Journal of Immunology, Vol. 167, No. 2, 2001, pp. 741-748.
- A. L. Marzo, R. A. Lake, B. W. Robinson and B. Scott, “T-Cell Receptor Transgenic Analysis of Tumor-Specific CD8 and CD4 Responses in the Eradication of Solid Tumors,” Cancer Research, Vol. 59, No. 5, 1999, pp. 1071- 1079.
- R. S. Allan, J. Waithman, S. Bedoui, C. M. Jones, J. A. Villadangos, Y. Zhan, A. M. Lew, K. Shortman, W. R. Heath and F. R. Carbone, “Migratory Dendritic Cells Transfer Antigen to a Lymph Node-Resident Dendritic Cell Population for Efficient CTL Priming,” Immunity, Vol. 25, No. 1, 2006, pp. 153-162. doi:10.1016/j.immuni.2006.04.017
- B. W. Robinson and R. A. Lake, “Advances in Malignant Mesothelioma,” The New England Journal of Medicine, Vol. 353, No. 15, 2005, pp. 1591-1603. doi:10.1056/NEJMra050152
- C. Jandus, D. Speiser and P. Romero, “Recent Advances and Hurdles in Melanoma Immunotherapy,” Pigment Cell & Melanoma Research, Vol. 22, No. 6, 2009, pp. 711- 723.
- J. Rotow, S. R. Gameiro, R. A. Madan, J. L. Gulley, J. Schlom and J. W. Hodge, “Vaccines as Monotherapy and in Combination Therapy for Prostate Cancer,” Clinical and Translational Science, Vol. 3, No. 3, 2010 pp. 116- 122. doi:10.1111/j.1752-8062.2010.00186.x
- T. D. de Gruijl, A. J. van den Eertwegh, H. M. Pinedo and R. J. Scheper, “Whole-Cell Cancer Vaccination: From Autologous to Allogeneic Tumorand Dendritic Cell-Based Vaccines,” Cancer Immunology, Immunotherapy, Vol. 57, No. 10, 2008, pp. 1569-1577. doi:10.1007/s00262-008-0536-z
- P. K. Srivastava, M. K. Callahan and M. M. Mauri, “Treating Human Cancers with Heat Shock Protein-Peptide Complexes: The Road Ahead,” Expert Opinion on Biological Therapy, Vol. 9, No. 2, 2009, pp. 179-186. doi:10.1517/14712590802633918
- J. Gong, Y. Zhang, J. Durfee, D. Weng, C. Liu, S. Koido, B. Song, V. Apostolopoulos and S. K. Calderwood, “A Heat Shock Protein 70-Based Vaccine with Enhanced Immunogenicity for Clinical Use,” Journal of Immunology, Vol. 184, No. 1, 2010, pp. 488-496. doi:10.4049/jimmunol.0902255
- A. Powell, J. Creaney, S. Broomfield, I. Van Bruggen and B. Robinson, “Recombinant GM-CSF plus Autologous Tumor Cells as a Vaccine for Patients with Mesothelioma,” Lung Cancer, Vol. 52, No. 2, 2006, pp. 189-197. doi:10.1016/j.lungcan.2006.01.007
- C. Jackaman, A. M. Lew, Y. Zhan, J. E. Allan, B. Koloska, P. T. Graham, B. W. Robinson and D. J. Nelson, “Deliberately Provoking Local Inflammation Drives Tumors to Become Their Own Protective Vaccine Site,” International Immunology, Vol. 20, No. 11, 2008, pp. 1467- 1479.
- E. Liapi and J. F. Geschwind, “Transcatheter and Ablative Therapeutic Approaches for Solid Malignancies,” Journal of Clinical Oncology, Vol. 25, No. 8, 2007, pp. 978-986. doi:10.1200/JCO.2006.09.8657
- R. A. McTaggart and D. E. Dupuy, “Thermal Ablation of Lung Tumors,” Techniques in Vascular & Interventional Radiology, Vol. 10, No. 2, 2007, pp. 102-113. doi:10.1053/j.tvir.2007.09.004
- B. B. Pua, R. H. Thornton and S. B. Solomon, “Ablation of Pulmonary Malignancy: Current Status,” Journal of Vascular and Interventional Radiology, Vol. 21, Supplement 8, 2010, pp. S223-S232.
- W. L. Yang, D. G. Nair, R. Makizumi, G. Gallos, X. Ye, R. R. Sharma and T. S. Ravikumar, “Heat Shock Protein 70 Is Induced in Mouse Human Colon Tumor Xenografts after Sublethal Radiofrequency Ablation,” Annuals of Surgical Oncology, Vol. 11, No. 4, 2004, pp. 399-406.
- Q. Liu, B. Zhai, W. Yang, L. X. Yu, W. Dong, Y. Q. He, L. Chen, L. Tang, Y. Lin, D. D. Huang, H. P. Wu, M. C. Wu, H. X. Yan and H. Y. Wang, “Abrogation of Local Cancer Recurrence after Radiofrequency Ablation by Dendritic Cell-Based Hyperthermic Tumor Vaccine,” Molecular Therapy, Vol. 17, No. 12, 2009, pp. 2049-2057. doi:10.1038/mt.2009.221