Health
Vol. 4 No. 11A (2012) , Article ID: 24996 , 13 pages DOI:10.4236/health.2012.431176
Unilateral 6-OHDA th-fgfr1(tk-) mouse model supports the role of FGFs in Parkinson’s disease and the effects of nicotine and L-DOPA on spontaneous motor impairments
![]()
1Department of Pathology and Anatomical Sciences, State University of New York, Buffalo, USA; *Corresponding Author: mks4@buffalo.edu
2Department of Biochemistry and Neuroscience, Lake Erie College of Osteopathic Medicine, Erie, USA
3Targacept Inc., Winston-Salem, USA
Received 3 October 2012; revised 6 November 2012; accepted 12 November 2012
Keywords: Parkinson’s Disease; Fibroblast Growth Factor (FGF) Signaling; Transgenic Mouse; Nicotine; L-DOPA
ABSTRACT
In the developing and adult brain, neurotrophic growth factors support the growth and protection of dopaminergic neuronal systems. Recently, links between impaired neurotrophin support of dopamine (DA) neurons has been described in Parkinson’s Disease (PD). Fibroblast growth factor (FGF) has a unique association with DA neurons in that FGF signaling is vitally important for the development and protection of adult DA neurons. We assessed the role of substantia nigra (SN)-expressed FGFs in the nigrostriatal dopaminergic system using a transgenic mouse, th-fgfr1(tk-). In these mice, generated by expression of dominant negative FGFR1(TK-) from the tyrosine hydroxylase (TH) gene promoter, reduced FGF signaling results in smaller and less dense adult nigrostriatal DA neurons, similar to what is observed in PD. With unilateral 6-hydroxydopamine (6-OHDA) lesions, th-fgfr1(tk-) mice exhibited extensive unilateral nigrostriatal damage with robust spontaneous (non-drug induced) asymmetrical turning and a decreased latency to remain on the accelerating rotarod. L-DOPA remains the gold standard for PD therapy despite debilitating hyperkinetic and dyskinetic side effects. The nicotinic acetylcholine system has recently been targeted as an alternative system to combat PD motor symptoms. Nicotine effectively stimulates dopaminergic transmission in the nigrostriatal pathway and mediates movement. Using unilaterally lesioned th-fgfr1(tk-) mice, long term (11 day) oral administration of nicotine increased spontaneous bidirectional turning and increased the latency before falling from the accelerating rotarod. In a separate analysis, L-DOPA treatment reversed directionality of rotation and further deepened motor discoordination, suggesting activation of hypersensitive postsynaptic DA receptors in the denervated striata. These results in a transgenic model of PD provide insights into the cellular mechanisms underlying L-DOPA and nicotinic therapies and offer further evidence of nicotine’s capacity to facilitate movement and enhance motor coordination in PD.
1. INTRODUCTION
The central pathological feature of Parkinson’s Disease (PD) is a degeneration of nigrostriatal dopamine (DA) neurons that project from the substantia nigra pars compacta (SNc) to the dorsolateral striatum. The loss of DA innervation leads to progressive dysfunction of basal ganglia motor circuits, resulting in movement impairments such as bradykinesia, resting tremor and altered motor coordination [1].
Over the past several years it has become increasingly evident that the loss of DA neurons in PD may be due to a combination of environmental insults, such as excessive levels of reactive oxygen species (ROS), combined with varying degrees of genetic/metabolic susceptibility. Brain neurons, and particularly DA neurons, are subjected to high levels of oxidative stress from oxygen and glucose consumption and breakdown, which contributes to their selective degeneration in PD and related neurodegenerative disorders [2,3]. Healthy neurons have various protective mechanisms against ROS-induced cell death, and the decline or absence of these systems may leave neurons more vulnerable to deterioration [4].
Neurotophic factors are small proteins that aid in the maturation and protection of developing neurons as well as maintenance and survival of adult neurons [5]. The antioxidant effects of many neurotrophic factors have been shown to protect and enhance the vitality of DA neurons including basic fibroblast growth factor (FGF-2) [6], glial cell line-derived neurotrophic factor (GDNF) [7, 8], epidermal growth factor (EGF) [9], brain-derived neurotrophic factor (BDNF) [10], conserved dopamine neurotrophic factor (CDNF) [11] and mesencephalic astrocyte-derived neurotrophic factor (MANF) [12]. Reports show decreased levels of FGF, BDNF, NGF and GDNF in the nigrostriatal DA system and cerebrospinal fluid of PD patients [13-17] and genetic links between CDNF [18], BDNF [19] and FGF [20,21] and PD have been described.
FGF distinguishes itself from other neurotrophic factors due to its critical role in supporting developing and adult DA systems [22,23]. FGF-2 promotes the growth and survival of cultured DA neurons [24-26], protects against neurotoxin-induced cell death [27-32] and cografting of DA neurons with FGF-2 increases neuronal survival [17,33,34]. It was reported that FGF signaling increases DA levels in calbindin-negative SN neurons, those at greatest risk in PD patients [35]. In human PD patients there was a reduction in the percentage of FGF- 2 immunoreactive DA neurons relative to healthy subjects (12.7% FGF-2 positive DA neurons versus 82% FGF-2 positive DA neurons in control brains) [17]. Timmer and colleagues (2007) reported that in adult FGF-2 knockout mice with 6-hydroxydopamine (6-OHDA) lesions, significantly fewer DA neurons survived compared to non-lesioned controls [36].
To further assess the role of substantia nigra (SN)- expressed FGFs and its homologues in PD, a transgenic mouse model expressing fgfr1(tk-) receptor from the tyrosine hydroxylase gene promoter was generated [37]. Phenotypically, th-fgfr1(tk-) mice have smaller and lessdense midbrain DA neurons, resembling the DA neuronal hypoplasia observed in adult PD patients [38,39]. These mice display behavioral abnormalities associated with dysfunctional DA release such as impaired sensorimotor gating (reduced prepulse inhibition of the startle response), abnormal social interaction and cognitive dysfunction [37,40,41].
Here we demonstrate that unilateral 6-OHDA lesions to the medial forebrain bundle (MFB) in th-fgfr1(tk-) mice generates a robust unilateral PD model. Relative to control mice, th-fgfr1(tk-) mice made a higher percentage of contralateral rotations with apomorphine administration and showed an extensive unilateral reduction of hydroxylase (TH) immunoreactivity in nigrostriatal DA neurons and loss of striatal DA and DA metabolites. Furthermore, unilaterally lesioned th-fgfr1(tk-) mice exhibited consistent spontaneous (drug free) motor impairments—ipsilateral rotation preference in the novel Cylinder Turning Test and a reduced latency to remain on the accelerating rotarod.
L-3,4-dihydroxyphenylalanine (L-DOPA), the gold standard of PD therapy, effectively, but nonspecifically, increases movement by enhancing DA content lost from deterioration of DA terminals of the nigrostriatal system. However, chronic administration of L-DOPA leads to motor abnormalities such as abnormal involuntary movements (AIMs) or “dyskinesias” [42,43], which may reflect activation of hyper responsiveness of postsynaptic DA targets [44].
The nicotinic acetylcholine system is currently being investigated as a potential therapeutic target for PD. Nicotine improves dopaminergic neuronal integrity [45] and stimulates movement and other DA-mediated functions impaired in PD [46,47]. There is a strong anatomical overlap [48-50] and dynamic functional relationship between the DA and nicotinic acetylcholine systems in the SNc and the striatum [51-54]. Chronic nicotine administration improved motor functions in advanced PD patients [55,56] and in some animal models [57-59]. However, there are conflicting results in human and animal studies, perhaps due to differences in methods of administration and behavioral testing paradigms [60].
Here we tested the effects of nicotine on spontaneous motor asymmetries and impaired motor coordination in unilaterally lesioned th-fgfr1(tk-) mice. Long-term (11 day) oral administration of nicotine improved 6-OHDAinduced impairments on the accelerating rotarod and increased bidirectional rotation movement. In a separate analysis, acute L-DOPA reversed the direction of unilateral turning and further deepened motor discoordination on the accelerating rotarod. These results provide further evidence that targeting the nicotinic acetylcholine system may be advantageous to facilitate coordinated movement in PD and illustrate different synaptic mechanisms between nicotine and L-DOPA in a unilaterally lesioned transgenic mouse.
2. MATERIALS AND METHODS
2.1. Animals
Homozygous transgenic th-fgfr1(tk-) mice were described in Klejobr et al. (2006). These mice express fgfr1(tk-) fused to rat TH gene promoter (4.5 kb). The progenies were screened for the presence of the transgene by PCR amplification of tail DNA. Mice homozygous for th-fgfr1(tk-) transgene and control mice (transgene free) lines were derived from the same parental BCF1 (C57BL/10J/C3H/HeJ) mouse strain. The lines showed stable behavioral differences in all generations investigated [37]. Mice used in all specific experiments were selected from multiple litters. Mice (males and females) were singly housed throughout the course of the experiments and for at least four weeks before testing. The housing facility was maintained on a light:dark cycle of 12:12 h and animals had free access to food and water in the home cage.
All experiments were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and with approval from the University at Buffalo IACUC.
2.2. Drugs
Nicotine and L-DOPA were obtained from RBI/Sigma (St. Louis, MO). Nicotine was dissolved in 1% sucrose solution at a concentration of 0.0125 mg/mL (3.78 mg/kg/day). Animals were administered either nicotine in 1% sucrose solution or 1% sucrose solution (control) in their drinking bottles. RO 4-4602 (Benserazide Hoffman-La Roche) was dissolved in phosphate buffered saline (PBS) at a concentration of 50 mg/kg and injected intraperitonealy 5 minutes before L-DOPA or saline administration. L-DOPA (25 mg/kg) or saline was injected subcutaneously. All injections were given at a volume of 100 µl/30g of body weight. Apomorphine (RBI/Sigma St. Louis, MO) was injected s.c. in th-fgfr1(tk-) and control mice at a dose of 0.5 mg/kg 30 minutes before the start of the Rotometer Test.6-hydroxydopamine hydrobromide (6-OHDA) was obtained from Regis Technologies, Inc. 6-OHDA (3.5 mg/mL) dissolved in 0.9% NaCl and 0.2% ascorbic acid solution was stereotaxically injected (2 µL per animal) into the right medial forebrain bundle (AP – 1.2, ML + 1.1, DV + 4.8 mm) a band of fibers that connects the SNc with the striatum. Mice were anesthetized with isoflurane (2% in O2) and surgeries were performed in a Kopf Model 900 Small Animal Stereotaxic frame modified for mice (Kopf Model 921 mouse adaptor).
2.3. Dopamine and Metabolites Assay
The mice (8 th-fgfr1(tk-) and 5 controls) were approximately 8 months of age at time of analysis. The brains were removed and frozen at –80 degrees Celsius and the left and right striatum were isolated using punching needles [61]. For each mg of tissue collected, 19 µL of 100 mM perchloric acid containing 100 µM metabisulfite and 50 nM DHBA as an internal standard was added. The samples were injected through a Suppelco Discovery C18 reverse phase 15 cm column with a 2 cm guard column. Electrochemical HPLC detection was done with a BAS LC-4C with redundant data collection using a strip chart recorder, an HP integrator and a LabView A-D converter and software. Custom written computer softsware by author M. Parry was used for analysis. All three methods gave the same relative areas for all peaks with the computer method giving the most accuracy.
2.4. Immunohistochemistry
Two weeks after 6-OHDA surgeries, mice were deeply anesthetized then perfused transcardially with saline followed by 4% paraformaldehyde. Brain cryostat sections (40 um) were immunostained with anti-TH specific antibody (1000:1; Chemicon, cat# AB151), followed by biotinylated anti-rabbit secondary antibody (100:1), then by treatment with 3% H2O2 and then avidin-HRThe staining was completed with diaminobenzidine (DAB) and the sections were mounted on slides in sequential order and coverslipped for examination using a light microscope. Fluorescent microscopic images (12-bit) were acquired with a XILLIX Microimager cooled CCD camera (Qimaging, Vancouver, BC, Canada) on a Nikon FXA fluorescent microscope (Nikon, New York, NY, USA). All images acquired were within the linear range of the camera.
2.5. Behavioral Tests
54 th-fgfr1(tk-) mice between the ages of 4 - 5 months were tested in the Cylinder Turning Test before 6-OHDA lesions. Latency remaining on the rotarod was also tested in a separate group of 87 th-fgfr1(tk-) mice of approximately the same age before 6-OHDA administration. All animals were tested approximately 4 weeks later after 6-OHDA to determine effects of the lesions.
Cylinder Turning Test: This test measures spontaneous rotational movement, defined as circular movement up to a body length in distance, which included spinning in place. Mice were placed individually in the center of a Plexiglas cylinder (20 cm diameter, 20 cm height) and video recorded with an infrared camera (Sony) under dim light for 3 min. Testing took place during the dark cycle and mice were allowed at least 10 min to acclimate to the room prior to testing. The amount of time spent on rotation movement towards the lesion (ipsilateral) and away from the lesion (contralateral) were quantified. Unlike rotammetry, the mice are not attached to a tethering device or restrained in any way and are able to move freely throughout the test and without the use of drugs to evoke movement. The duration behaviosr were tracked either from the recording or live during the test using Noldus Pocket Observer (v5.0) by an experimenter who was blind to the treatment condition.
Rotarod Test: The apparatus (Rotarod, San Diego Instruments, San Diego, CA, USA) consists of a continuously accelerating spinning rod (5 cm diameter) and a foam pad 20 cm underneath to brace the fall of the animals. Each mouse was loaded facing against the rod’s direction of movement. The mice were allowed at least 10 min to acclimate to the room prior to testing, which took place during the dark cycle but with the lights turned on. Each mouse was trained to perform this task for 5 consecutive days at the start of the experiment in order to attenuate any practice-related improvements in performance or inherent differences among physical or learning capabilities among the subjects. Training and testing days were identical, consisting of 5 consecutive trials for each mouse. The rotarod beam accelerated at a rate of 0.33 rpm/s with a maximum speed of 40 rpm achieved at 120 seconds. Each trial continued until the mouse fell off the rod or 120 seconds had elapsed. The number of seconds each mouse was able to stay on the rod was recorded The average of the 4 top performances out of the 5 test day trials served as the dependent variable for the test. The Rotarod Test examines global motor coordination in PD models [62], which may be dependent on coordinated action of both intact and nonlesioned nigrostriatal pathways.
Rotometer Test: Apomorphine-induced rotational movement was compared between control (n = 4) and thfgfr1(tk-) (n = 4) mice using individual metal bowls with a diameter of 20 cm. Mice were attached by a specially adapted harness affixed to a light and flexible tether. The tether is secured to an automated rotary encoder that monitors the angular direction of the subject (RotaCount-8, Columbus Instruments, Columbus, OH). Full and partial ipsilateral and contralateral turns were counted by the software. Experiments were conducted 2 weeks after 6-OHDA lesions. Mice were given 15 minutes to acclimate to the apparatus and then injected with apomorphine (s.c. 0.5 mg/kg). Counting of rotations began 30 minutes after injections. Rotations were counted for one hour and the percentage of full (360˚) and partial (30˚ - 360˚) contralateral rotations were compared between control and transgenic mice.
2.6. Experimental Design
Nicotine Experiment: After initial post lesion behavioral analyses, mice were divided into two groups with half receiving nicotine (0.0125 mg/ml in a 1% sucrose solution) in the first drug session while the other half receiving a control, 1% sucrose-only solution (Figure 1). These treatments were reversed in the second treatment session, beginning 4 weeks after the end of the first.
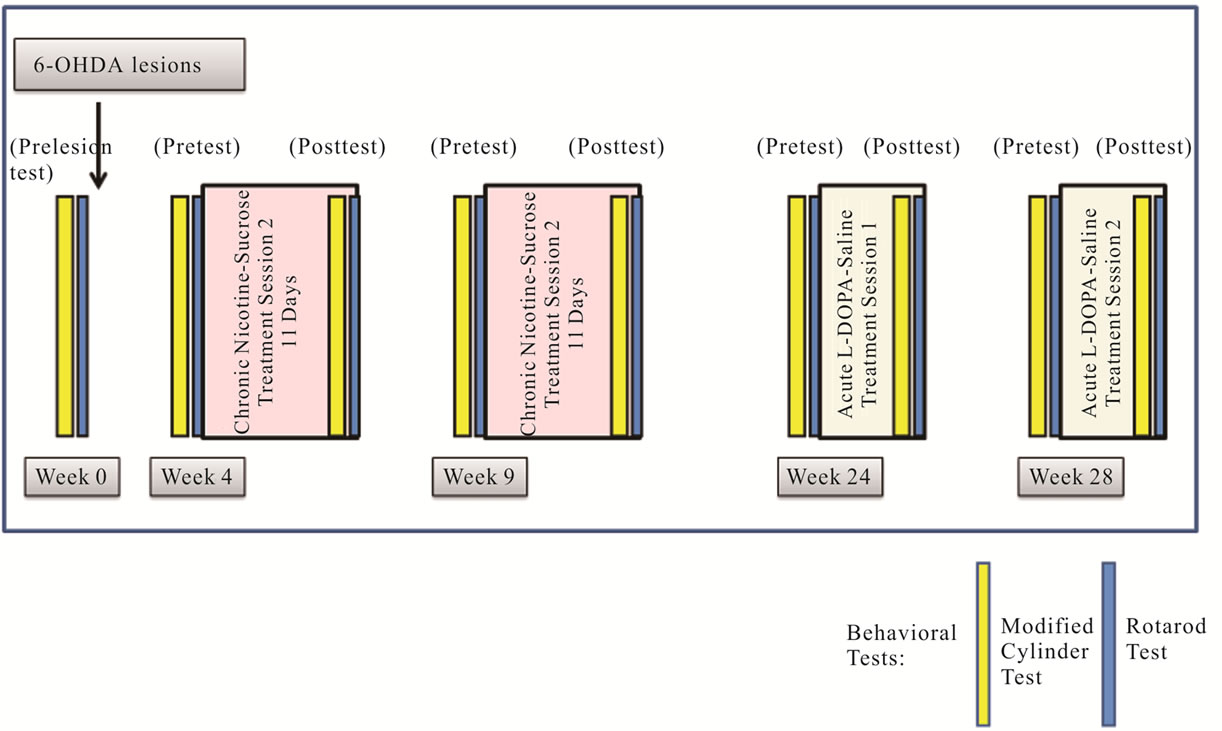
Figure 1. Experimental Design. Nicotine was administered to th-fgfr1(tk-) mice in 1% sucrose drinking solution (3.78 mg/kg/day) for 11 days starting 4 weeks post 6-OHDA lesions. Mice were tested for behaviors in the two days prior to treatment (pretest) and during the last two days of nicotine/sucrose administration (post test). For L-DOPA experiments, animals were either injected once with L-DOPA (25 mg/kg s.c.) preceeded by Benserazide (50 mg/kg s.c.) or equal volumes of saline. Behavioral tests were run one and two hours after L-DOPA or saline injections.
Mice received drinking solutions beginning the first morning of each drug treatment session. Solutions were changed daily and consumption volume was measured. Mice on average consumed 5.19 ± 0.31 mL of nicotine/ sucrose solution and 5.69 ± 0.34 mL of sucrose-only solution (no significant difference). The weights of the mice were measured daily and there were no treatment group differences in body weight and no change of more than 10% in any animal in the experiment. For all analyses, there were no significant differences in performance with sucrose or nicotine between drug sessions, so scores from both sessions were combined. Two days prior to the start of each drug session, all mice were pre-tested on the Cylinder Turning Test followed by the Rotarod Test the next day. This sequence was repeated during the final two days of each drug session (post test). All tests were conducted during the dark cycle at approximately the same time each day.
Since it is known that 6-OHDA produces DA neuronal lesions of variable size and the MFB is a difficult structure to target in mice, it was expected that not all lesioned animals would exhibit spontaneous unilateral turning. This was also observed in the present study, as 28 out of 54 of 6-OHDA injected mice showed 65% or higher post lesion ipsiversive turning preference in the Cylinder Turning Test. Thus, to account for mice that had not been sufficiently lesioned to display spontaneous motor asymmetry, only animals that exhibited a 65% or higher ipsiversive turning were analyzed for the effects of nicotine. Animals meeting this criterion after surgeries test tended to maintain this strong ipsilateral preference throughout the experiment.
L-DOPA Experiment: Approximately four months after the nicotine experiments (7 months after 6-OHDA lesions), the remaining mice (n = 52) were pre-tested in the Cylinder Turning. Sufficiently lesioned animls (65 + % ipsileteral turning preference) were randomly divided into two groups. Half received L-DOPA and the other half received saline injections. For saline administration, injections were made at the same time, using the same volume and using the same method of administration (i.for L-DOPA and s.c. for RO-4-4602) as the drugs. One hour later the mice were tested in the Cylinder Turning Test immediately followed by the Rotarod Test. After a break of one week, the experiment was repeated with treatments reversed. The mice were also pre tested before the second treatment session, and pre test scores were compared between the first and second session to determine any carry-over effects of L-DOPA. Since no effects were observed, data from both sessions were combined.
2.7. Statistics
The effects of 6-OHDA lesions on each behavior were analyzed using One Way ANOVAs comparing pre and post lesion results. For each behavior, potential carryover effects of either nicotine and sucrose (or L-DOPA and saline) treatments were examined by comparing performances between the first pre drug test (pre test one) and the second pre drug test (pre test two). Two way ANOVAs were conducted using pre test sequence number (1 or 2) and treatment experience (nicotine or sucrose, L-DOPA or saline) as independent variables and each behavioral score (ipsilateral, contralateral and total rotation movement or latency remaining on the rotarod) as the dependent variables for each analysis. No significant main effects of pre test or treatment experience or interaction effects were found in either the nicotine or L-DOPA experiments, therefore scores from both treatment sessions were combined for analyses of drug effects. To analyze drug effects on rotation directionality, changes in turning movement as a result of sucrose or nicotine treatment (or L-DOPA verus saline treatment) were compared separately for time spent making ipsilateral, contralateral and total movement using One Way ANOVAs. The pre test values (conducted two days prior to the start of drug treatment) were considered baseline values and changes from this baseline as a result of drug administration were compared between treatments for each type of movement using One Way ANOVAs. One Way ANOVAs were also used to compare control and th-fgfr1(tk-) mice in the Rotometer Test and for levels of DA, DOPAC and HVA.
3. RESULTS
3.1. Changes in the Nigrostriatal System
Previous studies of FGF-2 KO mice showed an increased susceptibility of DA neurons to 6-OHDA [36]. To assess this possibility in th-fgfr1(tk) mice, we first analyzed rotational movement after administration of apomorphine in the Rotometer Test. Apomorphine induces contralateral turning by stimulating supersensitive D1 and D2 receptors on the deneravated side [63-66]. However, this supersenstivity is only observed with severe depletion of DA terminals [65,67,68]. In a group of 8 [4 th-fgfr1(tk) and 4 nontransgenic] mice with unilateral 6-OHDA lesions, transgenic mice made a significantly higher percentage of full and partial counterclockwise turns than control mice (Figure 2). This indicates DA postsynaptic hypersensitivity in the transgenic mice and a greater extent of uniltateral DA deafferentation.
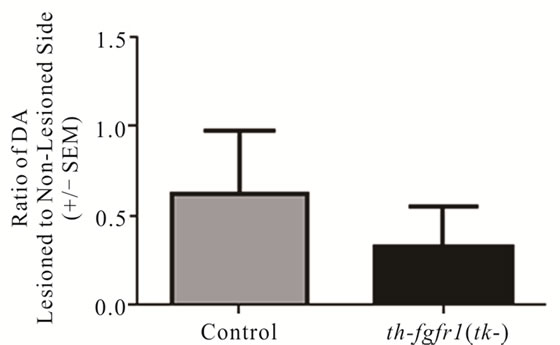
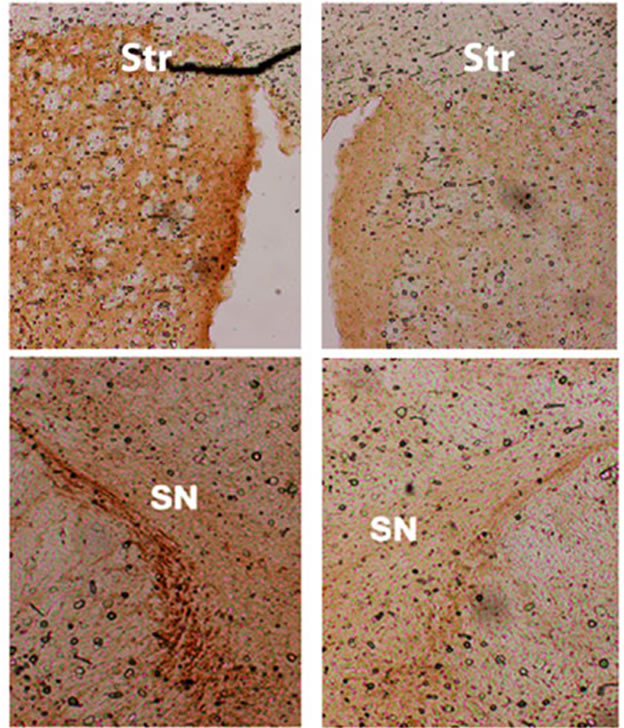
In a separate (drug-naïve) group of mice [5 controls and 8 th-fgfr1(tk-)], the lesioned striata were analyzed for DA and DA metabolite (DOPAC and HVA) content by HPLC-EC. Changes in striatal tissue DA and DA metabolite levels have been shown to correlate well with the number of surviving terminals assessed with TH immunostaining or by measuring DA uptake sites [69]. In th-fgfr1(tk-) mice, the loss of striatal DA as well as DA metabolites DOPAC and HVA appeared greater than in the control mice (Figure 3), However, statistical significance was not obtained, likely due to high variances which was observed previously after administration of 6-OHDA [70]. Immunoflourescence of TH-expressing neurons in the striatum and SN (Figure 4) verified a heavy loss of TH-immunostained terminals in the striatum (caudate) and TH expressing neurons of the SNc in th-fgfr1(tk-) mice.
3.2. Spontaneous Unilateral Rotation and Impaired Motor Coordination in Unilateral 6-OHDA th-fgfr1(tk-) Mice
To further characterize nigrostriatal impairments, behavioral assessments of spontaneous (drug-naïve) movement were carried out before and after 6-OHDA lesions in th-fgfr1(tk-) mice. Four weeks after 6-OHDA administration the mice were tested for spontaneous rotational asymmetries in the Cylinder Turning Test. Ipsilateral turning significantly increased (from 19.37 ± 1.05 pre lesion to 27.20 ± 2.75 seconds; p = 0.02) and contralateral turning decreased (from 21.06 ± 1.34 seconds pre lesion to 9.21 ± 1.04 seconds; p < 0.001), while total movement did not change (Figure 5). A second spontaneous behavior, the latency before falling from the accelerating rotarod, was also tested in a separate group of mice. The rotarod test assesses global motor coordination in PD models [62] and may be dependent on coordinated action of both intact and nonlesioned nigrostriatal pathways. The 6-OHDA lesions significantly reduced time remaining on the rotarod beam (65.16 ± 2.10 s pre lesion, 51.60 ± 1.87 s post lesion; p < 0.001). Impaired rotaord performance has also been reported in previous unilateral 6-OHDA models [66,71].
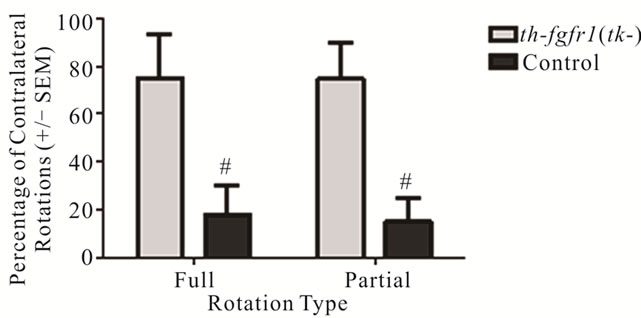
Figure 2. Rotation behavior in 6-OHDA-lesioned control and th-fgfr1(tk-) mice with administration of apomorphine. Percentage of full (360˚) and partial (30˚ - 360˚) contralateral rotations made during a one-hour test session beginning 30 minutes after administration of apomorphine (0.5 mg/kg, s.c.) in 8 mice (4 between th-fgfr1(tk-) and 4 controls). #Significant difference between th-fgfr1(tk-) and control, one way ANOVA (p < 0.05).
 (a)
(a)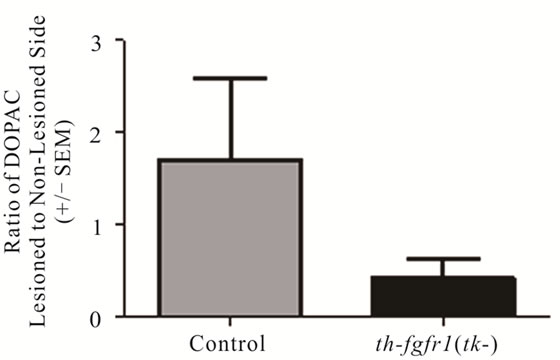 (b)
(b)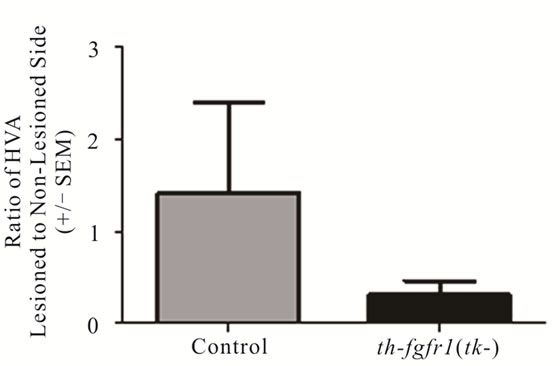 (c)
(c)
Figure 3. Dopamine in the striatum of 6-OHDA-lesioned control and th-fgfr1(tk-) mice. Ratio of DA on the lesioned-to-non-lesioned side of the brain. There were no significant differences in all mice (n = 8 thfgfr1(tk-) and n = 5 controls). In mice with strong lesions (greater than 50% DA depletion) (n = 5 thfgfr1(tk-) and n = 4 controls), the DA ratio was significantly reduced in th-fgfr1(tk) mice. #Significant difference between th-fgfr1(tk) and control mice, one way ANOVA (p < 0.05).
3.3. The Effect of Nicotine and L-DOPA on Spontaneous Motor Impairments
After the spontaneous motor impairments were established, nicotine was administered to the lesioned mice in 11 day treatment sessions (see Figure 1 for experimental design). Relative to sucrose-only treatment, nicotine increased time spent on ipsilateral (–5.46 ± 3.11 s with sucrose; +3.65 ± 2.62 s with nicotine; p = 0.032) and con-
Non-lesioned side Lesioned side
Figure 4. Immunostaining for tyrosine hydroxylase (TH) in the striatum (caudate) and substantia nigra pars compacta (SNc) of th-fgfr1(tk-) mice. TH-immunopositive DA terminals and cell bodies in 6-OHDA-lesioned th-fgfr1(tk-) mice.
tralateral (+0.82 ± 0.85 s with sucrose; +3.87 ± 1.11 s with nicotine; p = 0.032) rotations (Figure 6), indicating a general facilitation of movement. Nicotine also significantly increased the latency to fall from the accelerating rotarod relative to sucrose treatment (8.48 ± 1.76 s improvement from pre to post test with nicotine, 3.99 ± 1.34 s with sucrose; p = 0.043) (Figure 6).
The effects of L-DOPA on spontaneous motor impairments were analyzed beginning 3 months after the conclusion of nicotine experiments and 7 months after 6-OHDA lesions. Animals were first pre tested in the Cylinder Turning Test to isolate mice that maintained asymmetrical rotation (65 + % ipsilateral turning—see Materials and Methods). In these mice, L-DOPA decreased time spent engaging in ipsilateral movement (L-DOPA: 5.39 ± 1.81 s, saline: 30.18 ± 7.36 s; p < 0.01) and increased time spent on contralateral movement (L-DOPA: 68.93 ± 12.04 s, saline: 10.89 ± 2.04 s; p < 0.001) relative to saline (Figure 7). These results are consistent with previous models of unilateral PD in which a reversal of ipsilateral rotation is observed following L-DOPA administration, apparently reflecting DA hypersensitivity of denervated striatal neurons [72,73]. In the Rotarod Test, latency to fall from the accelarating rotarod beam after L-DOPA was significantly reduced relative to saline treatment (L-DOPA: 54.39 ± 8.63, saline: 78.92 ± 9.41; p = 0.035) (Figure 7).
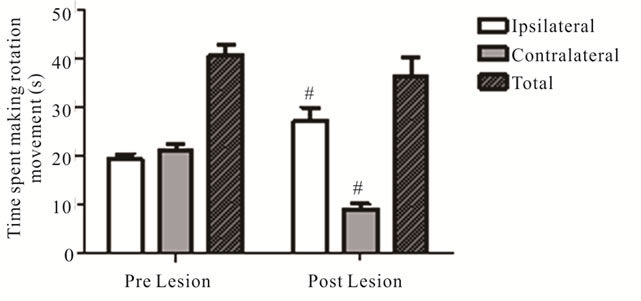
Figure 5. Effect of 6-OHDA Lesions on Spontaneous Rotational Movement in the Cylinder Turning Test. Rotation movement in the Cylinder Turning test assessed before and 4 weeks after 6-OHDA (n = 54). #Significant difference in time spent on movement from pre to post 6-OHDA lesion, one way ANOVA (p < 0.05).
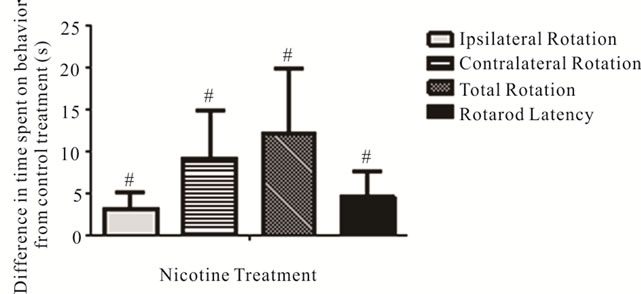
Figure 6. Effects of nicotine on spontaneous rotation movement in the Cylinder Turning Test and time remaining on the accelerating rotarod. The changes in time spent on rotation movements and latency before falling from the accelerating rotarod with nicotine treatment (values shown relative to sucrose-only control solution). #Significant difference from control, one way ANOVA (p < 0.05).
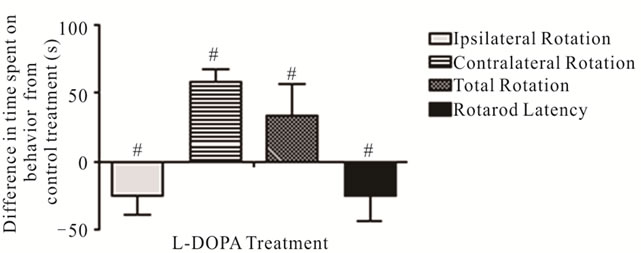
Figure 7. Effects of nicotine on spontaneous rotation movement in the Cylinder Turning Test and time remaining on the accelerating rotarod. The changes in time spent on rotation movements and latency before falling from the accelerating rotarod with L-DOPA treatment (values shown relative to saline control administration). #Significant difference from control, one way ANOVA (p < 0.05).
4. DISCUSSION
There is increasing evidence that PD may be elicited by a failure of endogenous neuroprotective mechanisms, such as impaired neurotrophin support of DA neurons. Here we show that mice with a deficiency of FGF signaling in nigrostriatal DA neurons [37] are highly susceptible to damage from oxidative stress incurred by administration of neurotoxin 6-OHDA. The unilaterally lesioned th-fgfr1(tk-) mice exhibited increased contralateral turning with administration of apomorphine relative to control mice (Figure 2). It was shown that apomorphine-induced contralateral turning correlates strongly with the extent of nigrostriatal damage [66] and animals with severe depletion of DA will markedly exhibit this behavior [65]. Analysis of depletion of DA biomarkers in the striatum showed a trend towards increased loss of DA and DA metabolite content in the transgenic mice relative to controls (Figures 3(a)-(c)) and TH immunostaining in the striatum and SNc further substantiated strong unilateral nigrostriatal damage in th-fgfr1(tk-) mice (Figure 4). These results are in agreement with previous findings of high susceptibility to 6-OHDA in mice with impaired FGF signaling [36].
Although about 80% - 90% of patients suffering from PD acquire what is termed the “sporadic” or non- (known) genetic origin of the disease, several genetic causes have been identified in recent years, including autosomal dominant genes alpha-synuclein [74] and LRRK2 [75] and autosomal recessive genes parkin [76] and DJ-1 [77] and recently FGF20 [20]. Given this diversity, it is likely that these genes influence common mechanisms affecting the development, maintenance and protection of DA neurons. We postulate that such mechanisms involve signaling by FGF and related receptors as intermediary in diverse biological pathways as found in the recently established “Integrative Nuclear FGFR1 Signaling” (INFS) pathway [78,79]. Central to the INFS mechanism, which is activated by FGFs and other diverse stimuli, is nuclear translocation of newly synthesized FGFR1 (along with its ligand) and direct modulation of several homeostatic gene programs by FGFR1 [80-84]. INFS may link signaling pathways in which PD-linked mutations have been reported.
Recently a genetic link between PD and another member of the FGF-2 family, FGF20, has been established. FGF-20 polymorphisms and haplotypes were shown to strongly influence risk of PD in diverse populations [85]. FGF20 is located on chromosome 8 and has been identified as a risk factor for PD [20,21]. Studies by Ohmachi and colleagues showed that the neurotrophic activity of FGF20, similar to FGF-2, is mediated through interactions with FGFR1. FGF-20 is preferentially expressed in the SNc and striatum of the rat brain and enhances the survival of cultured midbrain DA neurons [86].
An important role of FGF receptor signaling to the survival of DA neurons was shown using transfections of episomal dominant negative fgfr1(tk-) DNA directly into the SN in and cultured neurons [87]. In vivo transfection of FGFR1(TK-) into the adult rat midbrain using polyethylenimine resulted in a loss of TH+ neurons in the SNc and depletion of striatal DA detected at 30 but not at 10 days [88]. Inhibition of FGF20 and FGF-2 signaling in DA neurons by their dominant negative receptor fgfr1(tk-) is likely to affect DA transmission by reducing expression of the rate limiting enzyme, TH. In cultured adrenal medullary cells transfected with fgfr1 (tk-), TH gene activity was diminished by fgfr1(tk-) interference with the INFS mechanism, an effect which in SN DA neurons would likely reduce in vivo DA transmission.
Behavioral analysis of th-fgfr1(tk-) mice with unilateral 6-OHDA lesions revealed a spontaneous ipsilateral turning preference in the drug-free Cylinder Turning Test. The increase of movement towards the lesioned side indicates a predominant use of intact (non lesioend) DA neurons. Previously in 6-OHDA lesioned th-fgfr1(tk-) mice administration of amphetamine increased (data not shown), consistent with other unilateral PD models. Given the non-vesicular source of amphetamine-released DA and the unclear relevance to sensory-motor processing by the nigrostriatal DA system, our analyses focused on spontaneous (non-drug induced) motor impairments. The baseline unilateral rotation preference suggests “steady-state” unbalanced DA-ergic tone. In addition the lesioned mice exhibited a decreased latency to remain on the accelerating rotarod, indicating a global impairment of movement coordination. Long term (11 day) oral administration of nicotine improved this deficit and induced a mild but significant increase in bidirectional rotations. These results suggest a mild enhancement of DA-mediated activity through nicotinic receptor activation.
Nicotine influences DA transmission by activation of nicotinic acetylcholine receptors on DA neurons in the striatum [89-93] as well as on a variety of other neurotransmitter systems that are known to mediate nigrostriatal DA release, including glutamtergic, GABAergic, noradrenalinergic and cholinergic systems [52,53,94-98]. In advanced PD patients, improvements in motor and cognitive symptoms were observed with administration of nicotine [55,56,99] however some studies showed no effect [100-102]. In animal models, the effects of nicotine on rotation behavior after unilateral 6-OHDA administration have also been inconsistent [57,60,103-105]. For example it was determined that nicotine administered orally before but not after unilateral lesions improved amphetamine-induced turning impairments in rats [105], suggesting only a neuroprotective role. In contrast, nicotine decreased apmorphine-induced contralateral rotations and improved glutamatergic transmission when administered 2 weeks after 6-OHDA lesions in rats [57].
With the many differences between studies in humans and animal PD models in the duration, dose and mode of nicotine administration, it has been difficult to gauge the therapeutic value of nicotine in late stages of PD. One notable difference between this and previous studies was that spontaneous (drug-naïve) turning were not assessed in previous studies. In unilateral rodent models, amphetamine or apomorphine are typically used to induce rotational movement which anti-PD therapies are then administered to reverse. Evidence suggests that drug-induced rotational behavior may not utilize the same sensory motor integration pathways that are impaired in PD [65] and their administration may preclude evaluation of novel anti-PD therapies. Thus, we believe it was advantageous to test for the effects of anti-PD compounds using spontaneous motor measures.
Interestingly, it was shown that the nicotinic acetylcholine system interacts with the FGF-2 gene [106] and this association mediates neuroprotective and trophic effects by FGF [107]. Nicotine-induced increases in the proliferation of neuronal precursor cells is accompanied by upregulation of FGF-2 mRNA and the effects of nicotine are mediated by activation of FGFR1 via FGF-2 [108].
In contrast to the balanced effects of nicotine on rotational movement, acute L-DOPA drastically reversed the directionality of rotations. This was also observed in other unilateral rodent models [72,109]. L-DOPA exerts its therapeutic effects by increasing tonic extracellular DA and evoking postsynaptic response by hypersensitive DA-recipient cells [110,111]. Thus, the reversal of turning likely reflects synaptic DA hypersensitivity on the lesioned side, a phenomenon typically revealed with an approximately 80 + % loss of DA terminals [65,68]. L-DOPA also had a negative impact on rotarod performance. In 6-OHDA-lesioned rats, L-DOPA induced prolonged twisting postures caused by exaggerated torsional movements of axial and neck musculature [112]. Mice in this study also appeared to make similar dyskinetic movements, which likely negatively affected their ability to remain on the rotarod. The activation of hypersensitive postsynatptic DA receptors in the lesioned hemisphere by L-DOPA may reflect dyskinesias observed in bilateral models and human patients.
5. CONCLUSION
The th-fgfr1(tk-) mouse model, featuring a developmental disruption of FGFR1 signaling in midbrain DA neurons, demonstrated robust unilateral dopaminergic nigrostriatal damage and PD-related movement impairments. These results, consistent with findings in FGF-2 knockout mice, suggest a role of FGF signaling and related growth factors in the pathophysiology of PD. These models may be advantageous for modeling damaged nigrostriatal DA systems and motor impairments in PD and for the evaluation of novel therapeutic approaches. Also, this report adds to the growing body of research suggesting that nicotinic compounds may facilitate coordinated movement in PD and provide an alternative to L-DOPA therapy.
![]()
![]()
REFERENCES
- Rodriguez-Oroz, M.C., et al. (2009) Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. The Lancet Neurology, 8, 1128- 1139. doi:10.1016/S1474-4422(09)70293-5
- Adams, J.D., Jr., Chang, M.L. and Klaidman, L. (2001) Parkinson’s disease—Redox mechanisms. Current Medicinal Chemistry, 8, 809-814. doi:10.2174/0929867013372995
- Jenner, P. (2003) Oxidative stress in Parkinson’s disease. Annals of Neurology, 53, S26-36; discussion S36-38.
- Harman, D. (2006) Free radical theory of aging: An update: Increasing the functional life span. Annals of the New York Academy of Sciences, 1067, 10-21. doi:10.1196/annals.1354.003
- Connor, B. and Dragunow, M. (1998) The role of neuronal growth factors in neurodegenerative disorders of the human brain. Brain Research Reviews, 27, 1-39. doi:10.1016/S0165-0173(98)00004-6
- Grothe, C. and Timmer, M. (2007) The physiological and pharmacological role of basic fibroblast growth factor in the dopaminergic nigrostriatal system. Brain Research Reviews, 54, 80-91. doi:10.1016/j.brainresrev.2006.12.001
- Lin, L.F., et al. (1993) GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science, 260, 1130-1132. doi:10.1126/science.8493557
- Andereggen, L., et al. (2009) Effects of GDNF pretreatment on function and survival of transplanted fetal ventral mesencephalic cells in the 6-OHDA rat model of Parkinson’s disease. Brain Research, 1276, 39-49. doi:10.1016/j.brainres.2009.04.021
- Hadjiconstantinou, M., et al. (1991) Epidermal growth factor enhances striatal dopaminergic parameters in the 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mouse. Journal of Neurochemistry, 57, 479-482. doi:10.1111/j.1471-4159.1991.tb03776.x
- Hyman, C., et al. (1991) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature, 350, 230-232. doi:10.1038/350230a0
- Lindholm, P., et al. (2007) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature, 448, 73-77. doi:10.1038/nature05957
- Petrova, P., et al. (2003) MANF: A new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. Journal of Molecular Neuroscience, 20, 173-188. doi:10.1385/JMN:20:2:173
- Mogi, M., et al. (1999) Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neuroscience Letters, 270, 45-48. doi:10.1016/S0304-3940(99)00463-2
- Howells, D.W., et al. (2000) Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Experimental Neurology, 166, 127-135. doi:10.1006/exnr.2000.7483
- Nagatsu, T., et al. (2000) Changes in cytokines and neurotrophins in Parkinson’s disease. Journal of Neural Transmission Supplementa, 60, 277-290.
- Parain, K., et al. (1999) Reduced expression of brainderived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport, 10, 557-561. doi:10.1097/00001756-199902250-00021
- Tooyama, I., et al. (1994) Retention of basic fibroblast growth factor immunoreactivity in dopaminergic neurons of the substantia nigra during normal aging in humans contrasts with loss in Parkinson's disease. Brain Research, 656, 165-168. doi:10.1016/0006-8993(94)91378-1
- Choi, J.M., et al. (2011) Analysis of mutations and the association between polymorphisms in the cerebral dopamine neurotrophic factor (CDNF) gene and Parkinson disease. Neuroscience Letters, 493, 97-101. doi:10.1016/j.neulet.2011.02.013
- Chen, L., et al. (2011) The 712A/G polymorphism of Brain-derived neurotrophic factor is associated with Parkinson’s disease but not major depressive disorder in a Chinese han population. Biochemical and Biophysical Research Communications, 408, 318-321. doi:10.1016/j.bbrc.2011.04.030
- Wang, G., et al. (2008) Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. The American Journal of Human Genetics, 82, 283-289. doi:10.1016/j.ajhg.2007.09.021
- Mizuta, I., et al. (2008) Calbindin 1, fibroblast growth factor 20, and alpha-synuclein in sporadic Parkinson’s disease. Human Genetics, 124, 89-94. doi:10.1007/s00439-008-0525-5
- Krejci, P., et al. (2009) Molecular pathology of the fibroblast growth factor family. Human Mutation, 30, 1245-1255. doi:10.1002/humu.21067
- Mason, I. (2007) Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nature Reviews Neuroscience, 8, 583-596. doi:10.1038/nrn2189
- Mayer, E., et al. (1993) Basic fibroblast growth factor promotes the survival of embryonic ventral mesencephalic dopaminergic neurons—I. Effects in vitro. Neuroscience, 56, 379-388. doi:10.1016/0306-4522(93)90339-H
- Li, A., et al. (2006) Apomorphine-induced activation of dopamine receptors modulates FGF-2 expression in astrocytic cultures and promotes survival of dopaminergic neurons. The FASEB Journal, 20, 1263-1265. doi:10.1096/fj.05-5510fje
- Giacobini, M.M., et al. (1993) Fibroblast growth factors enhance dopamine fiber formation from nigral grafts. Brain Research Developmental Brain Research, 75, 65- 73. doi:10.1016/0165-3806(93)90066-J
- Date, I., et al. (1993) Enhanced recovery of the nigrostriatal dopaminergic system in MPTP-treated mice following intrastriatal injection of basic fibroblast growth factor in relation to aging. Brain Research, 621, 150-154. doi:10.1016/0006-8993(93)90312-B
- Otto, D. and Unsicker, K. (1993) FGF-2-mediated protection of cultured mesencephalic dopaminergic neurons against MPTP and MPP+: Specificity and impact of culture conditions, non-dopaminergic neurons, and astroglial cells. Journal of Neuroscience Research, 34, 382-393. doi:10.1002/jnr.490340403
- Zawada, W.M., et al. (1996) Growth factors rescue embryonic dopamine neurons from programmed cell death. Experimental Neurology, 140, 60-67. doi:10.1006/exnr.1996.0115
- Peng, J., et al. (2008) Fibroblast growth factor 2 enhances striatal and nigral neurogenesis in the acute 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neuroscience, 153, 664-670. doi:10.1016/j.neuroscience.2008.02.063
- Casper, D. and Blum, M. (1995) Epidermal growth factor and basic fibroblast growth factor protect dopaminergic neurons from glutamate toxicity in culture. Journal of Neurochemistry, 65, 1016-1026. doi:10.1046/j.1471-4159.1995.65031016.x
- Hsuan, S.L., Klintworth, H.M. and Xia, Z. (2006) Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. Journal of Neuroscience, 26, 4481- 4491. doi:10.1523/JNEUROSCI.4922-05.2006
- Mayer, E., Fawcett, J.W. and Dunnett, S.B. (1993) Basic fibroblast growth factor promotes the survival of embryonic ventral mesencephalic dopaminergic neurons—II. Effects on nigral transplants in vivo. Neuroscience, 56, 389-398. doi:10.1016/0306-4522(93)90340-L
- Timmer, M., et al. (2004) Enhanced survival, reinnervation, and functional recovery of intrastriatal dopamine grafts co-transplanted with Schwann cells overexpressing high molecular weight FGF-2 isoforms. Experimental Neurology, 187, 118-136. doi:10.1016/j.expneurol.2004.01.013
- Murase, S. and McKay, R.D. (2006) A specific survival response in dopamine neurons at most risk in Parkinson’s disease. Journal of Neuroscience, 26, 9750-9760. doi:10.1523/JNEUROSCI.2745-06.2006
- Timmer, M., et al. (2007) Fibroblast growth factor (FGF)-2 and FGF receptor 3 are required for the development of the substantia nigra, and FGF-2 plays a crucial role for the rescue of dopaminergic neurons after 6-hydroxydopamine lesion. Journal of Neuroscience, 27, 459-471. doi:10.1523/JNEUROSCI.4493-06.2007
- Klejbor, I., et al. (2006) Fibroblast growth factor receptor signaling affects development and function of dopamine neurons—inhibition results in a schizophrenia-like syndrome in transgenic mice. Journal of Neurochemistry, 97, 1243-1258. doi:10.1111/j.1471-4159.2006.03754.x
- Moller, A., (1992) Mean volume of pigmented neurons in the substantia nigra in Parkinson’s disease. Acta Neurolpgica Scandinavica Supplementum, 137, 37-39.
- Ma, S.Y., et al. (1996) A quantitative morphometrical study of neuron degeneration in the substantia nigra in Parkinson’s disease. Journal of the Neurological Sciemces, 140, 40-45. doi:10.1016/0022-510X(96)00069-X
- Klejbor, I., et al. (2009) Serotonergic hyperinnervation and effective serotonin blockade in an FGF receptor developmental model of psychosis. Schizophrenia Research, 113, 308-321. doi:10.1016/j.schres.2009.06.006
- Kucinski, A., et al. (2012) alpha7 neuronal nicotinic receptor agonist (TC-7020) reverses increased striatal dopamine release during acoustic PPI testing in a transgenic mouse model of schizophrenia. Schizophrenia Research, 136, 82-87. doi:10.1016/j.schres.2012.01.005
- Jenner, P. (2008) Molecular mechanisms of L-DOPAinduced dyskinesia. Nature Reviews Neuroscience, 9, 665-677. doi:10.1038/nrn2471
- Linazasoro, G. (2005) New ideas on the origin of Ldopa-induced dyskinesias: Age, genes and neural plasticity. Trends in Pharmacological Sciences, 26, 391-397. doi:10.1016/j.tips.2005.06.007
- Grace, A.A. (2008) Physiology of the normal and dopaminedepleted basal ganglia: Insights into levodopa pharmacotherapy. Movement Disorders, 23, S560-S569. doi:10.1002/mds.22020
- Quik, M., et al. (2010) Chronic nicotine treatment increases nAChRs and microglial expression in monkey substantia nigra after nigrostriatal damage. Journal of Molecular Neuroscience, 40, 105-113. doi:10.1007/s12031-009-9265-9
- Gotti, C., et al. (2010) Nicotinic acetylcholine receptors in the mesolimbic pathway: Primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. The Journal of Neurosciemce, 30, 5311- 5325. doi:10.1523/JNEUROSCI.5095-09.2010
- Le Novere, N., et al. (1999) Involvement of alpha 6 nicotinic receptor subunit in nicotine-elicited locomotion, demonstrated by in vivo antisense oligonucleotide infusion. Neuroreport, 10, 2497-2501. doi:10.1097/00001756-199908200-00012
- Zhou, F.M., Liang, Y. and Dani, J.A. (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nature Neuroscience, 4, 1224-1229. doi:10.1038/nn769
- Dani, J.A. and Bertrand, D. (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annual Review of Pharmacology and Toxicology, 47, 699-729. doi:10.1146/annurev.pharmtox.47.120505.105214
- Quik, M., et al. (2004) Loss of alpha-conotoxinMIIand A85380-sensitive nicotinic receptors in Parkinson’s disease striatum. Journal of Neurochemistry, 88, 668-679. doi:10.1111/j.1471-4159.2004.02177.x
- Wonnacott, S. (1997) Presynaptic nicotinic ACh receptors. Trends in Neurosciences, 20, 92-98. doi:10.1016/S0166-2236(96)10073-4
- Exley, R. and Cragg, S.J. (2008) Presynaptic nicotinic receptors: A dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. British Journal of Pharmacology, 153, S283-S297. doi:10.1038/sj.bjp.0707510
- Zhou, F.M., Wilson, C.J. and Dani, J.A. (2002) Cholinergic interneuron characteristics and nicotinic properties in the striatum. Journal of Neurobiology, 53, 590-605. doi:10.1002/neu.10150
- Maskos, U. (2008) The cholinergic mesopontine tegmentum is a relatively neglected nicotinic master modulator of the dopaminergic system: Relevance to drugs of abuse and pathology. British Journal of Pharmacology, 153, S438- S445. doi:10.1038/bjp.2008.5
- Itti, E., et al. (2009) Dopamine transporter imaging under high-dose transdermal nicotine therapy in Parkinson’s disease: An observational study. Nuclear Medicine Communication, 30, 513-518.
- Villafane, G., et al. (2007) Chronic high dose transdermal nicotine in Parkinson’s disease: An open trial. European Journal of Neurology, 14, 1313-1316. doi:10.1111/j.1468-1331.2007.01949.x
- Meshul, C.K., et al. (2002) Nicotine alters striatal glutamate function and decreases the apomorphineinduced contralateral rotations in 6-OHDA-lesioned rats. Experimental Neurology, 175, 257-274. doi:10.1006/exnr.2002.7900
- Huang, L.Z., et al. (2011) Nicotinic receptor agonists decrease L-dopa-induced dyskinesias most effectively in partially lesioned parkinsonian rats. Neuropharmacology, 60, 861-868. doi:10.1016/j.neuropharm.2010.12.032
- Quik, M., et al. (2007) Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Annals of Neurology, 62, 588-596. doi:10.1002/ana.21203
- [61] Quik, M., et al. (2009) Multiple roles for nicotine in Parkinson’s disease. Biochemical Pharmacology, 78, 677- 685. doi:10.1016/j.bcp.2009.05.003
- [62] Bialowas, J., et al. (1979) The relationship between catecholamine levels in the hypothalamus and amygdala under influence of glucose overloading in hungry and sated rats. Polish Journal of Pharmacology & Pharmacy, 31, 325-335.
- [63] Stachowiak, M.K., et al. (1984) Apparent sprouting of striatal serotonergic terminals after dopamine-depleting brain lesions in neonatal rats. Brain Research, 291, 164- 167. doi:10.1016/0006-8993(84)90665-6
- [64] Ungerstedt, U. and Arbuthnott, G.W. (1970) Quantitative recording of rotational behavior in rats after 6-hydroxydopamine lesions of the nigrostriatal dopamine system. Brain Research, 24, 485-493. doi:10.1016/0006-8993(70)90187-3
- [65] Deumens, R., Blokland, A. and Prickaerts, J. (2002) Modeling Parkinson’s disease in rats: An evaluation of 6-OHDA lesions of the nigrostriatal pathway. Experimental Neurology, 175, 303-317. doi:10.1006/exnr.2002.7891
- [66] Castaneda, E., et al. (2005) Assessment of recovery in the hemiparkinson rat: Drug-induced rotation is inadequate. Physiology & Behavior, 84, 525-535. doi:10.1016/j.physbeh.2005.01.019
- [67] Iancu, R., et al. (2005) Behavioral characterization of a unilateral 6-OHDA-lesion model of Parkinson’s disease in mice. Behaviournal Brain Research, 162, 1-10. doi:10.1016/j.bbr.2005.02.023
- [68] Creese, I., Burt, D.R. and Snyder, S.H. (1977) Dopamine receptor binding enhancement accompanies lesion-induced behavioral supersensitivity. Science, 197, 596-598. doi:10.1126/science.877576
- [69] Schwarting, R.K. and Huston, J. (1996) Unilateral 6- hydroxydopamine lesions of meso-striatal dopamine neurons and their physiological sequelae. Progress in Neurobiology, 49, 215-266. doi:10.1016/S0301-0082(96)00015-9
- [70] Keller, R., et al. (1976) Liquid chromatographic analysis of catecholamines routine assay for regional brain mapping. Life Sciences, 19, 995-1003. doi:10.1016/0024-3205(76)90290-3
- [71] Zhang, W.Q., et al. (1988) Increased dopamine release from striata of rats after unilateral nigrostriatal bundle damage. Brain Research, 461, 335-342. doi:10.1016/0006-8993(88)90264-8
- [72] Rozas, G., Guerra, M.J. and Labandeira-Garcia, J.L. (1997) An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Research Protocols, 2, 75-84. doi:10.1016/S1385-299X(97)00034-2
- [73] Lundblad, M., et al. (2004) A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: Relation to motor and cellular parameters of nigrostriatal function. Neurobiology of Disease, 16, 110-123. doi:10.1016/j.nbd.2004.01.007
- [74] Jackson, D., et al. (1988) Inhibition of striatal acetylcholine release by serotonin and dopamine after the intracerebral administration of 6-hydroxydopamine to neonatal rats. Brain Research, 457, 267-273. doi:10.1016/0006-8993(88)90695-6
- [75] Polymeropoulos, M.H., et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276, 2045-2047. doi:10.1126/science.276.5321.2045
- [76] Zimprich, A., et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601-607. doi:10.1016/j.neuron.2004.11.005
- [77] Valente, E.M., et al. (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science, 304, 1158-1160. doi:10.1126/science.1096284
- [78] Bonifati, V., et al. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science, 299, 256-259. doi:10.1126/science.1077209
- [79] Stachowiak, M.K., Maher, P.A. and Stachowiak, E.K. (2007) Integrative nuclear signaling in cell development— A role for FGF receptor-1. DNA and Cell Biology, 26, 811-826. doi:10.1089/dna.2007.0664
- [80] Fang, X., et al. (2005) Control of CREB-binding protein signaling by nuclear fibroblast growth factor receptor-1: A novel mechanism of gene regulation. The Journal of Biological Chemistry, 280, 28451-28462. doi:10.1074/jbc.M504400200
- [81] Maher, P.A. (1996) Nuclear translocation of fibroblast growth factor (FGF) receptors in response to FGF-2. The Journal of Cell Biology, 134, 529-536. doi:10.1083/jcb.134.2.529
- [82] Stachowiak, M.K., et al. (1996) Nuclear accumulation of fibroblast growth factor receptors is regulated by multiple signals in adrenal medullary cells. Molecular Biology of the Cell, 7, 1299-1317.
- [83] Reilly, J.F. and Maher, P.A. (2001) Importin betamediated nuclear import of fibroblast growth factor receptor: Role in cell proliferation. The Journal of Cell Biology, 152, 1307-1312. doi:10.1083/jcb.152.6.1307
- [84] Myers, J.M., et al. (2003) Nuclear trafficking of FGFR1: A role for the transmembrane domain. Journal of Cellular Biochemistry, 88, 1273-1291. doi:10.1002/jcb.10476
- [85] Dunham-Ems, S.M., et al. (2009) Fibroblast growth factor receptor-1 (FGFR1) nuclear dynamics reveal a novel mechanism in transcription control. Molecular Biology of the Cell, 20, 2401-2412. doi:10.1091/mbc.E08-06-0600
- [86] van der Walt, J.M., et al. (2004) Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. The American Journal of Human Genetics, 74, 1121-1127. doi:10.1086/421052
- [87] Ohmachi, S., et al. (2003) Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. Journal of Neuroscience Research, 72, 436-443. doi:10.1002/jnr.10592
- [88] Stachowiak, E.K., et al. (2003) cAMP-induced differentiation of human neuronal progenitor cells is mediated by nuclear fibroblast growth factor receptor-1 (FGFR1). Journal of Neurochemistry, 84, 1296-1312. doi:10.1046/j.1471-4159.2003.01624.x
- [89] Corso, T.D., et al. (2005) Transfection of tyrosine kinase deleted FGF receptor-1 into rat brain substantia nigra reduces the number of tyrosine hydroxylase expressing neurons and decreases concentration levels of striatal dopamine. Molecular Brain Research, 139, 361-366. doi:10.1016/j.molbrainres.2005.05.032
- [90] Champtiaux, N., et al. (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. Journal of Neuroscience, 23, 7820-7829.
- [91] Salminen, O., et al. (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Molecular Pharmacology, 65, 1526-1535. doi:10.1124/mol.65.6.1526
- [92] Grady, S., et al. (1992) Characterization of nicotinic receptor-mediated [3H]dopamine release from synaptosomes prepared from mouse striatum. Journal of Neurochemistry, 59, 848-856. doi:10.1111/j.1471-4159.1992.tb08322.x
- [93] Azam, L., et al. (2005) Alpha-conotoxin BuIA, a novel peptide from Conus bullatus, distinguishes among neuronal nicotinic acetylcholine receptors. The Journal of Biological Chemistry, 280, 80-87.
- [94] Marshall, D.L., Redfern, P.H. and Wonnacott, S. (1997) Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: Comparison of naive and chronic nicotinetreated rats. Journal of Neurochemistry, 68, 1511-1519. doi:10.1046/j.1471-4159.1997.68041511.x
- [95] Pisani, A., et al. (2003) Targeting striatal cholinergic interneurons in Parkinson’s disease: Focus on metabotropic glutamate receptors. Neuropharmacology, 45, 45-56. doi:10.1016/S0028-3908(03)00137-0
- [96] Sharma, G. and Vijayaraghavan, S. (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron, 38, 929-939. doi:10.1016/S0896-6273(03)00322-2
- [97] Lu, Y., et al. (1998) Pharmacological characterization of nicotinic receptor-stimulated GABA release from mouse brain synaptosomes. Journal of Pharmacology and Experimental Therapeutics, 287, 648-657.
- [98] Grady, S.R., et al. (2001) Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: A function mediated by a different nAChR than dopamine release from striatum. Journal of Neurochemistry, 76, 258-268. doi:10.1046/j.1471-4159.2001.00019.x
- [99] Clarke, P.B. and Reuben, M. (1996) Release of [3H]- noradrenaline from rat hippocampal synaptosomes by nicotine: Mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. British Journal of Pharmacology, 117, 595-606. doi:10.1111/j.1476-5381.1996.tb15232.x
- [100] Kelton, M.C., et al. (2000) The effects of nicotine on Parkinson's disease. Brain and Cognition, 43, 274-282.
- [101] Lemay, S., et al. (2004) Lack of efficacy of a nicotine transdermal treatment on motor and cognitive deficits in Parkinson’s disease. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 28, 31-39. doi:10.1016/S0278-5846(03)00172-6
- [102] Vieregge, A., et al. (2001) Transdermal nicotine in PD: A randomized, double-blind, placebo-controlled study. Neurology, 57, 1032-1035. doi:10.1212/WNL.57.6.1032
- [103] Clemens, P., et al. (1995) The short-term effect of nicotine chewing gum in patients with Parkinson’s disease. Psychopharmacology (Berl), 117, 253-256. doi:10.1007/BF02245195
- [104] Gregorio, M.L., et al. (2009) Nicotine induces sensitization of turning behavior in 6-hydroxydopamine lesioned rats. Neurotoxicity Research, 15, 359-366. doi:10.1007/s12640-009-9041-1
- [105] Janhunen, S., Tuominen, R.K. and Ahtee, L. (2005) Comparison of the effects of nicotine and epibatidine given in combination with nomifensine on rotational behaviour in rats. Neuroscience Letters, 381, 314-319. doi:10.1016/j.neulet.2005.02.038
- [106] Huang, L.Z., et al. (2009) Nicotine is neuroprotective when administered before but not after nigrostriatal damage in rats and monkeys. Journal of Neurochemistry, 109, 826-837. doi:10.1111/j.1471-4159.2009.06011.x
- [107] Moffett, J., Kratz, E. and Stachowiak, M.K. (1998) Increased tyrosine phosphorylation and novel cis-acting element mediate activation of the fibroblast growth factor-2 (FGF-2) gene by nicotinic acetylcholine receptor. New mechanism for trans-synaptic regulation of cellular development and plasticity. Molecular Brain Research, 55, 293-305. doi:10.1016/S0169-328X(98)00010-2
- [108] Belluardo, N., et al. (2000) Neurotrophic effects of central nicotinic receptor activation. Journal of Neural Transmission Supplementa, 60, 227-245.
- [109] Mudo, G., et al. (2007) Acute intermittent nicotine treatment induces fibroblast growth factor-2 in the subventricular zone of the adult rat brain and enhances neuronal precursor cell proliferation. Neuroscience, 145, 470-483. doi:10.1016/j.neuroscience.2006.12.012
- [110] Morelli, M., et al. (1993) L-dopa stimulates c-fos expression in dopamine denervated striatum by combined activation of D-1 and D-2 receptors. Brain Research, 623, 334-336. doi:10.1016/0006-8993(93)91449-3
- [111] Bunney, B.S., Aghajanian, G.K. and Roth, R.H. (1973) Comparison of effects of L-dopa, amphetamine and apomorphine on firing rate of rat dopaminergic neurones. Nature New Biology, 245, 123-125.
- [112] Grace, A.A. and Bunney, B.S. (1984) The control of firing pattern in nigral dopamine neurons: Single spike firing. Journal of Neuroscience, 4, 2866-2876.
- [113] Winkler, C., et al. (2002) L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: Relation to motor and cellular parameters of nigrostriatal function. Neurobiology of Disease, 10, 165- 186. doi:10.1006/nbdi.2002.0499