Health
Vol. 4 No. 4 (2012) , Article ID: 18849 , 5 pages DOI:10.4236/health.2012.44031
Desmoplastic small round cell tumor of the abdomen: A case report and literature review of therapeutic options
![]()
1Department of Medical Oncology, Hassan II University Hospital, Fez, Morocco;
*Corresponding Author: hafida.benhammane@gmail.com
2Department of Pathology, Hassan II University Hospital, Fez, Morocco
3Department of General Surgery, Hassan II University Hospital, Fez, Morocco
4Department of Radiology, Hassan II University Hospital, Fez, Morocco
Received 22 December 2011; revised 18 January 2012; accepted 6 February 2012
Keywords: Desmoplastic Small Round Cell Tumor; Abdomen; Histology; Chemotherapy; Surgery; Outcome
ABSTRACT
Desmoplastic small round cell tumor (DSRCT) is a rare and highly aggressive variety of sarcoma arising typically from abdominal or pelvic peritoneum. Diagnosis and treatment approaches of this entity are complex and require a skilled, experienced, multidisciplinary team. Authors report their experience with a case of an intra-abdominal DSRCT arising in a 37-year-old young man in order to discuss the clinico-pathological and radiological behavior of this tumor and underline diagnostic and therapeutic difficulties.
1. INTRODUCTION
Desmoplastic small round cell tumor (DSRCT) is a rare and highly aggressive sarcoma occurring mainly in children and young adults [1]. The first case was described in 1989 by Gerald and Rosai [2]. This tumor has predilection for involvement of the peritoneum. A specific chromosomal translocation t (11; 22) (p13; q12) involving two genes: EWS which is characteristic of Ewing’s sarcoma and WT1 which is a Wilms tumor gene, has been documented in DSRCT [3].
This malignancy puts diagnostic and therapeutic problems; indeed, diagnosis can be suspected by radiological and histological features but it is asserted only by immuno-histochimic and cytogenetic study because of the big number of differential diagnoses and the anatomopathologic poly-morphism [2,3].
No curative treatment has been yet documented; current therapeutic options include multiagent chemotherapy and aggressive surgical debulking and radiotherapy [4,5]. The addition of hyperthermic intra-peritoneal chemotherapy in the multimodal approach has been reported in very few cases but no effect on survival has been clearly demonstrated [6].
The prognosis of this disease is poor with a Median survival of 17 months approximately [7].
We report a case of an intra-abdominal DSRCT in a 37 -year-old young man who was treated with combination chemotherapy and surgery.
2. CASE PRESENTATION
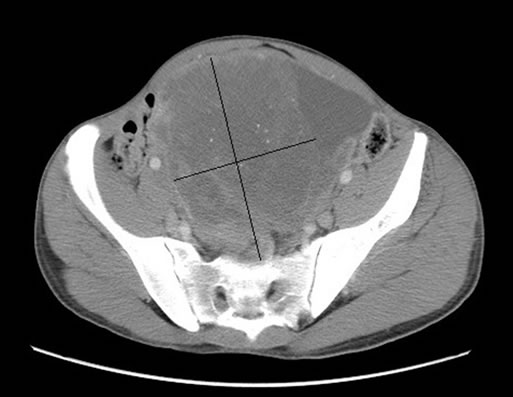
A 37-year-old man presented with a 3 months history of abdominal distention with discomfort, constipation and 20 Kg weight loss. His past medical history included pleural tuberculosis treated 20 years ago. Physical examination revealed an 18 cm sized large abdominal mass arising from the pelvis. Computed tomography (CT) of the abdomen objectified two intraperitoneal masses measuring 195 mm and 44 mm long axis respectively in intimate contact with the intraperitoneal organs with pelvic ascites and retroperitoneal lymph nodes; These masses were not confined to any organ-based primary site (Figure 1).
CT-guided biopsy of the mass has been performed illustrating morphological features of DSRCT, and the diagnosis was confirmed by immunohistochemistry which revealed positivity for cytokeratin and desmin antigen (Figures 2 and 3).
Induction chemotherapy regimen based on anthracyclin (60 mg/m2/cycle) and ifosfamide (9 g/m2/cycle) was received. The evaluation after 3 cycles of chemotherapy was considered to be in favor of a stable disease by the RECIST criteria, we stopped chemotherapy due to poor
 (a)
(a)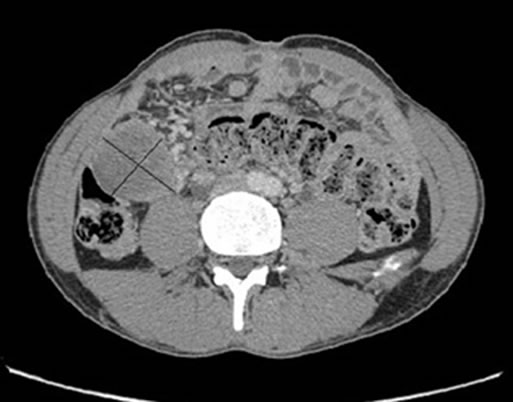 (b)
(b)
Figure 1. abdomino-pelvic CT showing heterogeneous masses arising from the pelvis measuring 195 mm (a) and 44 mm (b) long axis, with multiples lymph nodes and in intimate contact with intraperitoneal organs.
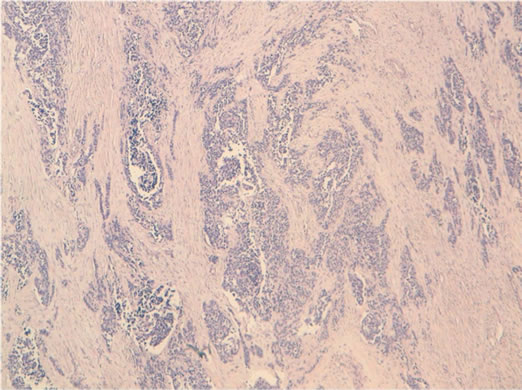
Figure 2. Cellular proliferation of uniform small round cells surrounded by a prominent desmoplastic stroma (HES × 100).
tolerance and we planned a palliative surgical debulking which included the excision of both masses with subtotal omentectomy, rectosigmoidectomy and terminal colostomy. Histological examination confirmed diagnosis of DSRCT with no therapeutic effect. CT evaluation after
 (a)
(a)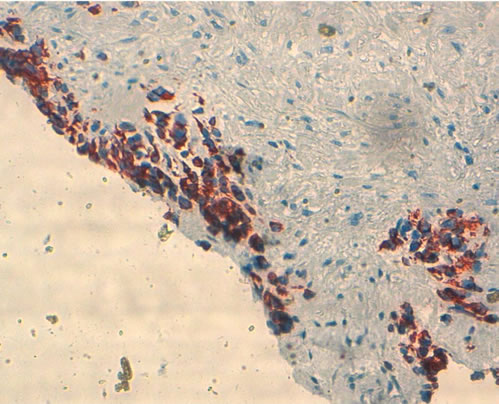 (b)
(b)
Figure 3. Immunohistochemical analysis showing strong immunoreactivity for epithelial marker -cytokeratin antigen- (a) and positivity for muscular marker -desmin antigen- (b).
surgery demonstrated: Disappearance of two large intra peritoneal masses with significant reduction in size and number of lymph nodes. Currently, the patient is still alive with a follow up of 18 months from initial diagnosis.
3. DISCUSSION
Desmoplastic round cell tumor (DRCT) is a rare variety of sarcoma and a distinct clinico-pathological entity that has been described recently in 1989 [2]. The incidence of DRSCT is difficult to estimate because of its rarity; no more than 300 cases have been reported in the literature. This aggressive malignancy is found predominantly in pediatric age group and young adults with a sex ratio (male to female) of 3:1. The age ranged from 10 to 16 years in 73% cases [8].
The cell of origin is not yet known. However, it is postulated to originate from the serosal lining because it is frequently found in the mesothelial-lined surface [9]. But an epithelial, neurogenic and blastematic origin is also discussed [10].
DSRCT typically arises from abdominal or pelvic peritoneum. Very rarely, DSRCT can arise from head and neck, base of skull, paratesticular region, ovary, brain and thoracic viscera [1].
Diagnosis of intra-abdominal DSRCT is difficult and it is usually obtained at an advanced stage. It may be first suggested by the imaging findings which are useful also for staging and for a guided biopsy. However, only histological analysis with an immunohistochemic and cytogenetic study led to the confirmation of diagnosis [11, 12]. In fact, the clinical manifestations are usually nonspecific; typically, the tumor produces few symptoms until it is large enough to compress or invade surrounding structures. Symptoms include abdominal discomfort/ distension, abdominal pain, weight loss, or change in bowel habits. Radiologically, the most characteristic imaging feature of DRSCT of the abdomen is single or multiple, lobulated peritoneal soft tissue masses without an apparent organ of origin. These abnormalities may be associated with ascites, lymph nodes, diffuse nodular peritoneal thickening or with distant metastasis. The lier, lung and bone marrow are common sites of metastases [1, 11].
In some cases Imaging features are not pathognomonic and can be difficult to interpret due to several differential diagnosis that overlap with the appearance of DSRCT such as Peritoneal carcinomatosis from various primary malignancies, malignant peritoneal mesothelioma, germ cell neoplasm, malignant lymphoma, malignant melanoma, and other soft-tissue sarcomas such as malignant fibrous histiocytoma. In addition, DSRCT can simulate the appearance of a benign disease such as desmoid fibromatosis, and tumefactive inflammatory processes [11].
The individualization of DSRCT as an antomo-clinical entity is based on morphological and especially immunohistochemical criteria: DSRCT are characterized by a multidirectional and phenotypic expression [12]. Macroscopically, the tumor presents bosselated grey surfaces with areas of necrosis. Microscopically, tumor manifests as islands of small round cells with hyperchromatic nuclei and scanty cytoplasm surrounded by desmoplastic stroma [13]. Similar morphologic appearances are observed in other small round-cell tumors such as Ewing sarcoma, rhabdomyosarcoma, primitive neuroectodermal tumor, neuroblastoma and Wilms tumor. On the immunohistochemistry, DSRCT express, simultaneously markers of epithelial (keratin and EMA), mesenchymal (vimentin), myogenic (desmin) and neural (neurospecific enolase and CD56) tissues. CD99 antigen, which is highly sensitive to Ewing sarcoma/PNET, can be positive in DSRCT, but with cytoplasm staining pattern [14]. Genetically, the reciprocal translocation t (11; 22) (p13; q12) resulting in EWS-WT1 gene fusion has more recently been discovered to be specific to DSRCT [12].
Therapeutic options of DSRCT of the abdomen are not well documented due to its rarity. Regarding the aggressiveness of the disease, treatment is based on multimodal therapy including an extensive surgery when it is feasible, systemic chemotherapy with or without abdominal radiotherapy [12]. It has been reported that the combination of three modalities has shown best results leading to an overall response rate of 39% and a 3-year survival rate of approximately 50% [1,12] as compared to each modality used separately. However, the impact of cytoreductive surgery on survival remains unclear. Some authors suggest an improvement in survival with surgery with a median survival of 34 months compared to 14 months for operable and inoperable tumors, respectively [1]. Similarly, Lal in its cohort has reported a significant impact of surgery on overall survival with 3 year survival of 58% as compared to 0% in no resectable disease [15], the benefit is greater after complete resection but frequently this act is not feasible due to the extensive involvement as the case of our patient. In contrast, other authors have concluded that surgery has no significant benefit on survival [16,17].
There are only a few reports on the role of chemotherapy in DSRCT. Multiagent chemotherapy leads to approximately 40% tumor response [1,12]. The cytotoxic agents that seem to be effective in DSRCT are cyclophosphamide, ifosfamide, adriamycin, vincristine, etoposide and topotecan [12]. Kushner et al. reported some complete remission with The P6 protocol which has seven courses of chemotherapy (Courses 1, 2, 3 and 6 consisted of HD-CAV, high-dose cyclophosphamide 2100 mg/m2/day on days 1 and 2, doxorubicin 75 mg/m2/day and vincristine 2.0 mg/m2/day on days 1, 2 and 3. Courses 4, 5 and 7 consisted of ifosfamide 1.8 g/m2/day and etoposide 100 mg/m2/day for 5 days) and the progression free survival could be prolonged [4]. Other authors had highlighted particularly the role of anthracycline based chemotherapy in some metastatic cases [18]. In fact, chemotherapy may be beneficial in patients with DSRCT; However, disease recurrence is the rule, often within 6 months [6] except two longest survivors reported in the literature [12].
There are few data on the role of Radiation therapy. This approach is considered essentially to consolidate residual disease or to palliate symptoms. Its use for curative purposes is limited by peritoneal dissemination and radiosensitivity of inra-abdominal organs [8,12].
The current novel therapy is the use of multimodal therapy including hyperthermic intraperitoneal chemotherapy (HIPEC). It has been reported in very few cases with contradictory data. In a series of 7 adult treated by cytoreductive surgery and HIPEC, mean overall survival did not improve significantly [17]. Nevertheless, Hayes Jordan in a retrospective review found that patients with HIPEC had almost double relapse-free survival time compared with patients who received surgery without HIPEC (8.85 versus 5.46 months), but the difference was not statistically significant because of few patients, better outcome in pediatric patients was observed [19]. Hence, more patients are needed to determine the effectiveness of HIPEC in DSRCT. Recently, a possible role of imatinib (tyrosine kinase inhibitor) has been proposed in DSRCT expressing c-kit antigen suggesting a possible clinical benefit [20].
4. CONCLUSION
DSRCT is a rare aggressive neoplasm characterized by a distinct histological appearances and a unique cytogenetic profile. The peritoneal cavity is the commonest site. The treatment guidelines have been difficult to define because of the small number of patients. Current multimodality approach may prolong survival but rarely achieves cure. Further prospective data are needed and molecular characterization of DSRCT should be performed to determine a specific targeted therapy.
5. ACKNOWLEDGEMENTS
All authors have made significant contributions by making diagnosis, treatment and intellectual input in the case and writing the manuscript.
The authors declare that they have no competing interests.
REFERENCES
- Biswas, G., Laskar, S., Banavali, S.D., Gujral, S., Kurkure, P.A., Muckaden, M., Parikh, P.M. and Nair, C.N. (2005) Desmoplastic small round cell tumor: Extra abdominal and abdominal presentations and the results of treatment. Indian Journal Cancer, 42, 78-84. doi:10.4103/0019-509X.16696
- Gerald, W.L. and Rosai, J. (1989) Case 2 desmoplastic small round cell with divergent differentiation. Pediatric Pathology, 9, 177-183. doi:10.3109/15513818909022347
- Gerald, W.L., Rosai, J. and Ladanyi, M. (1995) Characterization of genomic breakpoint and chemeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proceedings of the National Academy of Sciences, 92, 1028-1032. doi:10.1073/pnas.92.4.1028
- Kushner, B.H., LaQuaglia, M.P., Wollner, N., Meyers, P.A., Lindsley, K.L. and Ghavimi, F. (1996) Desmoplastic small round cell tumor prolonged progression free survival with aggressive multimodality therapy. Journal of Clinical Oncology, 14, 1526-1531.
- Marina, N.M., Pappo, A.S., Parham, D.M., Cain, A.M., Rao, B.N. and Poquette, C.A. (1999) Chemotherapy dose intensification for pediatric patients with Ewing’s family of tumors and desmoplastic small round blue cell tumors: A feasibility study at St. Jude children’s research hospital. Journal of Clinical Oncology, 17, 180-190.
- Lauridant, G., Philippin, N., Ledem, F., Lemoine, S., Gouerant, C., Guillemet, P., et al. (2010) Optimal treatment with systemic chemotherapy, complete surgical excision and hyperthermic intraperitoneal chemotherapy for a desmplastic small round cell tumor in an adulte male patient. Gastroentérologie Clinique et Biologique, 34, 321-324.
- Kinra, P. and Pujahari, A.K. (2008) Desmoplastic small round cell tumor abdomen. Indian Journal of Pathology and Microbiology, 51, 449-450. doi:10.4103/0377-4929.42526
- Jellouli, M., Mekki, M., Krichene, I., Zakhama, A., Belghith, M., Jouini, R., Sahnoun, L., Rammeh, S., Harbi, A. and Nouri, A. (2003) Intra-abdominal desmoplastic small round cell tumor in childhood: Case report and review of the literature. Bulletin de cancer, 12, 1063-1066.
- Kim, J.-W., Park, J.H., Cho, H.J. and Kwon, J.-H. (2009) A case of desmoplastic small round cell tumor diagnosed in a young female patient. Cancer Research and Treatment, 4, 233-236. doi:10.4143/crt.2009.41.4.233
- Farhat, F., Culine, S., L’homme, C., Duvillard, P., Terrier Lacombe, M.J., Michel, G., Soulie, P., Theodore, C., Lotz, J.P. and Droz, J.P. (1995) Tumeurs desmoplastiques à petites cellules rondes de l’adulte, une nouvelle entité. Bulletin de Cancer, 82, 665-673.
- Pickhardt, P.J., Fisher, A.J., Balfe, D.M., Dehner, L.P. and Huettner, P.C. (1999) Desmoplastic small round cell tumor of the abdomen: Radiologichistopathologic correlation. Radiology, 210, 633-638.
- Stuart-Buttlea, C.E., Smarta, C.J., Pritchardb, S., Martinc, D. and Welch, I.M. (2008) Desmoplastic small round cell tumour: A review of literature and treatment options. Surgical Oncology, 17, 107-112 doi:10.1016/j.suronc.2007.11.005
- Chang, F. (2005) Desmoplastic small round cell tumours: Cytologic, histologic, and immunochemical features. Archives of Pathology and Laboratory Medicine, 5, 728-732.
- Ordonez, N.G. (1998) Desmoplastic small round cell tumor: A histopathologic study of 39 cases with emphasis on unusual histological patterns. American Journal of Surgical Pathology, 22, 1303-1313. doi:10.1097/00000478-199811000-00001
- Lal, D.R., Su, W.T., Loh, K.C. and La Quaglia, M.P. (2005) Results of multimodal treatment for desmoplastic small round cell tumour. Journal of Paediatric Surgery, 40, 251-255. doi:10.1016/j.jpedsurg.2004.09.046
- Livaditi, E., Mavridis, G. and Soutis, M., (2006) Diffuse intraabdominal desmoplastic small round cell tumor: A tenyear experience. European Journal of Paediatric Surgery, 16, 423-437. doi:10.1055/s-2006-924736
- Gil, A., Paortilla, A., Brun, E., et al. (2004) Clinical perspective on desmoplastic small round cell tumour. Oncology, 67, 231-242. doi:10.1159/000081323
- Mrabti, H., Kaikani, W., Ahbeddou, N., Abahssain, H., El Khannoussi, B., Amrani, M. and Errihani, H. (2011) Metastatic desmoplastic small round cell tumor controlled by an anthracycline-based regimen: Review of the role of chemotherapy. Journal of Gastrointestinal Cancer, 43, 103-109. doi:10.1007/s12029-011-9260-6
- Hayes-Jordan, A., Green, H., Fitzgerald, N., Xiao, L.C. and Anderson, P. (2010) Novel treatment for desmoplastic small round cell tumor: Hyperthermic intraperitoneal perfusion. Journal of Pediatric Surgery, 45, 1000-1006. doi:10.1016/j.jpedsurg.2010.02.034
- Chao, J., Budd, G.T., Chu, P., Frankel, P., Garcia, D., Junqueira, M., Loera, S., Somlo, G., Sato, J. and Chow, W.A. (2010) Phase II clinical trial of imatinib mesylate in therapy of KIT and/or PDGFRalpha-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Anticancer Research, 30, 547-552.