Journal of Biophysical Chemistry
Vol.3 No.2(2012), Article ID:19431,18 pages DOI:10.4236/jbpc.2012.32018
Cancer chemoprevention through the induction of apoptosis by natural compounds*
![]()
1Department of Tumor Pathology, Graduate School of Medicine, Gifu University, Gifu, Japan; #Corresponding Author: takutt@toukaisaibou.co.jp
2Department of Pathology, Fujita Health University School of Medicine, Dengakugakubo, Kutsukake-cho, Toyoake, Japan
3The Tohkai Institute of Cytopathology: Cancer Research and Prevention (TCI-CaRP), Minami-uzura, Gifu, Japan
Received 26 January 2012; revised 28 February 2012; accepted 15 March 2012
Keywords: Cancer Chemoprevention; Apoptosis; Natural Compounds
ABSTRACT
As cell and tissue homeostasis are mediated by the balance between proliferation and apoptosis, controlling this balance is important for cancer chemoprevention. Cancer chemoprevention can be achieved by the use of natural, synthetic or biologic compounds that reverse, suppress or prevent the development of epithelial malignancies. Natural compounds including flavonoids are able to reduce oxidative stress, which is the most likely mechanism mediating the protective effects against cancer development. In addition, in vitro and in vivo studies have suggested that flavonoids, such as (-)-epigallocatechin-3-gallete (EGCG), quercetin, and curcumin, act by induction of apoptosis. Several natural compounds inhibit cell proliferation and angiogenesis. Certain natural products have been shown to inhibit the activation of nuclear factor kappa B (NF-κB) and Akt signaling pathways, both of which are known to maintain a homeostatic balance between cell survival and apoptosis. Understanding the mechanism of these natural products will contribute to the development of more specific preventive strategies against cancer development. Here we focus on the ability of natural cancer chemopreventive agents to induce apoptosis, and attempt to provide evidence for the preventive and therapeutic effects of natural compounds, EGCG, quercetin, and curcumin, in a succinct manner highlightingκand Akt signaling pathways in vivo.
1. INTRODUCTION
Epidemiological studies have shown that diet consisting of a high rich in fruit and vegetable reduces the risk of several types of cancer [1]. Intake of fruit and vegetables has been successfully used in the prevention of chronic diseases associated with oxidative stress conditions, including cancer [2,3]. The cancer preventing properties of fruit and vegetables have been ascribed, at least in part, to their high content of polyphenols [4]. The majority of polyphenols present in food are flavonoids and phenolic acids that are an integral part of the human diet. Laboratory rodent studies have shown that polyphenols have cancer-preventing properties and considered to be potential chemopreventive agents [5-7]. They can influence important cellular and molecular mechanisms associated with multiple carcinogenic steps, such as expression of key proteins in signal transduction pathways (e.g., mitogen activated protein kinases (MAPKs) or activator protein (AP)-1), the transcription factor nuclear factor-kappa B (NF-κB) and its its downstream gene products, modulation of cell-cycle regulation and induction of apoptosis [7], which affect cell differentiation, proliferation and apoptosis, immune responses and metabolism of carcinogens [4].
Apoptosis is conceivably the most potent defense against cancer development since it is the mechanism used by metazoans to eliminate deleterious cells. Many chemopreventive agents have been shown to induce apoptosis in transformed cells both in vitro and in vivo. Induction of apoptosis appears to be associated with their effectiveness in modulating carcinogenic processes [8]. Since apoptosis provides a physiologic mechanism for eliminating initiated or abnormal cells, dietary factors affecting apoptosis can influence carcinogenesis. In fact, activation of apoptosis in pre-cancerous cells is one of the most important mechanisms of cancer chemoprevention by dietary factors [9].
In this review, we focus on the apoptosis-inducing properties of several natural compounds present in the human diet and describe their beneficial effects against cancer development. Understanding the mechanism of these natural products will contribute to the development of more specific preventive strategies against cancer.
2. CHEMOPREVENTIVE COMPOUNDS IN FOOD
An effective chemopreventive agent should preferably intervene early in the process of carcinogenesis to eliminate pre-malignant cells before they acquire malignant character. Many chemopreventive agents are able to block or delay the promotion and/or progression of pre-malignant or malignant cells by modulating cell proliferation and/or differentiation [8,10] and, therefore, should be chronically administered to individuals with a higher risk of cancer development. However, using this approach, even minor adverse effects would be unacceptable: obstacles to the use of chemoprevention for many cancers include long-term toxicity and the development of chemoresistance [8]. These issues can limit the feasibility and success of conventional forms of chemoprevention for many cancers. Alternative approaches involve the use of agents that eliminate cells expeditiously. Targeted delivery of apoptosis-inducing agents to tumor cells would also prevent the need for chronic exposure, limiting the risk of long term toxicity and/or the development of chemoresistance [8].
An ideal chemopreventive agent should be selective for damaged or transformed cells, display a significant bioavailability in the target lesion and have more than one mechanism of action. Moreover, it should be highly effective, easy to administer, and inexpensive. Dietary compounds are particularly attractive because of longstanding exposure to them by humans, their relative lack of toxicity, and encouraging indications from epidemiological studies [11]. Indeed, numerous dietary compounds and micronutrients are emerging that have considerable potential for hindering in vivo deleterious oxidative processes and inducing apoptosis in cancer cells. One potentially important drawback of dietary compounds is their possible low bioavailability after ingestion [12,13].
Food contains several promising chemopreventive compounds [14-16]. Plant polyphenols, ubiquitous in a diet rich in fruit and vegetables, are thought to be responsible for the cancer protective effects ascribed to this type of diet [4,5,13]. In fact, numerous phenolic compounds have been shown to display anti-proliferative and cytotoxic effects towards several tumor cells, presenting toxic effects that specifically target cancer cells rather than normal cells [17-20]. Dietary polyphenols are predominantly consumed through fruit and beverages (juice, wine, tea, coffee, chocolate and beer), with the exception of vegetables, cereals and olive derivatives that are mainly associated with the Mediterranean diet [21-23]. Their average daily intake has been reported to be approximately 1 g [13,24], which is much greater than the intake of all other classes of dietary anti-oxidants. For example, this value is approximately 10-times higher than vitamin C intake and 100-times greater than the intake of both vitamin E and carotenoids [24].
In addition to the anti-oxidant properties, polyphenols exert interesting biological abilities in animal experiments and in vitro systems. The compounds are able to trap and scavenge free radicals, decrease leukocyte immobilization, induce apoptosis, inhibit cell proliferation and angiogenesis, and exhibit phytoestrogenic activity [25-27]. Dietary polyphenols interfere with signal transduction pathways related to the carcinogenesis process, thereby acting as chemopreventive agents. They include the suppression of NF-κB and activating protein (AP-1) activation, inhibition of the mitogen-activated proteins (MAPKs)-, protein kinasesand growth-factor receptormediated pathways, cell cycle arrest, induction of apoptosis, anti-oxidant and anti-inflammatory effects, and suppression of angiogenesis [7].
The chemopreventive properties of a variety of compounds can be directly related to their pro-apoptotic properties, most probably exerted via the intrinsic (mitochondrial) apoptotic pathway [8,10] (Table 1).
Pro-apoptotic diet-derived compounds can conceivably protect against cancer by enhancing elimination of initiated, precancerous cells. Polyphenols have been shown to induce pro-apoptotic responses in malignant or premalignant cells. Many studies have described the proapoptotic properties of dietary polyphenols in a varietry of human cancer cell lines derived from colon, prostate, lung, breast cancers, and leukemia [28]. A major catechin, EGCG, among green tea catechins is the most potent polyphenolic compound with respect to inducing apoptosis and inhibiting proliferation in cancer cells: it induces apoptosis in and suppresses growth of human cancer cell lines, such as breast adenocarcinoma (MCF-7 and MDA-MB-231 cells) [29], oral squamous carcinoma [30], leukemia [31], breast [32], lung [33], prostate [34] and colon [35] cancers.
Several studies have indicated that phenolic concentrations leading to apoptosis are within the micromolar range. For example, quercetin reportedly induces apoptosis at concentrations ranging from 29 M to 150 M [36,37]. In general, the effective concentrations required for induction of apoptosis are higher than those leading to growth inhibition. However, some studies have reported the induction of apoptosis without inhibition of cell proliferation (e.g. EGCG effect on H661 lung cancer cells) [38]. Further increases in polyphenol concentration may cause necrosis of all cell lines tested [39].
Recent studies have proposed that different dietary phenolic compounds may act synergistically, together with established anti-cancer agents and act as enhancers of anti-carcer drugs [40,41] (Table 2). For example, the synergistic effects of EGCG and curcumin in pre-malignant and malignant human oral epithelial cells [42], resveratrol and quercetin in human pancreatic cancer cells [36], quercetin and cisplatin in human laryngeal Hep2 cells [43] and HeLa cells [44], and EGCG and sulindac or tamoxifen against human PC-9 lung cancer cells [45] have been described. This recognized synergy among dietary phenolics and conventional synthetic drugs provides an interesting approach to combination therapy as well as for the pre-treatment of neoplastic cells with polyphenolic agents. This strategy has been able, in some cases, to even overcome chemoresistance [46].
The anti-carcinogenic and cytotoxic activities of polyphenols are largely determined by structural parameters, as much as their anti-oxidant potencies [20,47-50]. Despite their close resemblance, their bioactivity varies considerably upon minor structural modifications, since such modifications often induce significant conformational changes [7,51,52]. This implies an important drawback in the understanding of the effects of polyphenols in human health, when considering the huge number of different compounds (>8000) [12].
3. APOPTOSIS
Although the term apoptosis was first used in 1972 [53] to describe a morphologically distinct form of cell death, several components of this concept have only recently been described in detail. The process of programmed cell death is characterized by distinct morphological characteristics and energy-dependent biochemical mechanisms. Apoptotic cells show morphological changes such as cell shrinkage, pyknosis and extensive plasma membrane blebbing leading to the formation of apoptotic bodies, which are subsequently phagocytosed by macrophages
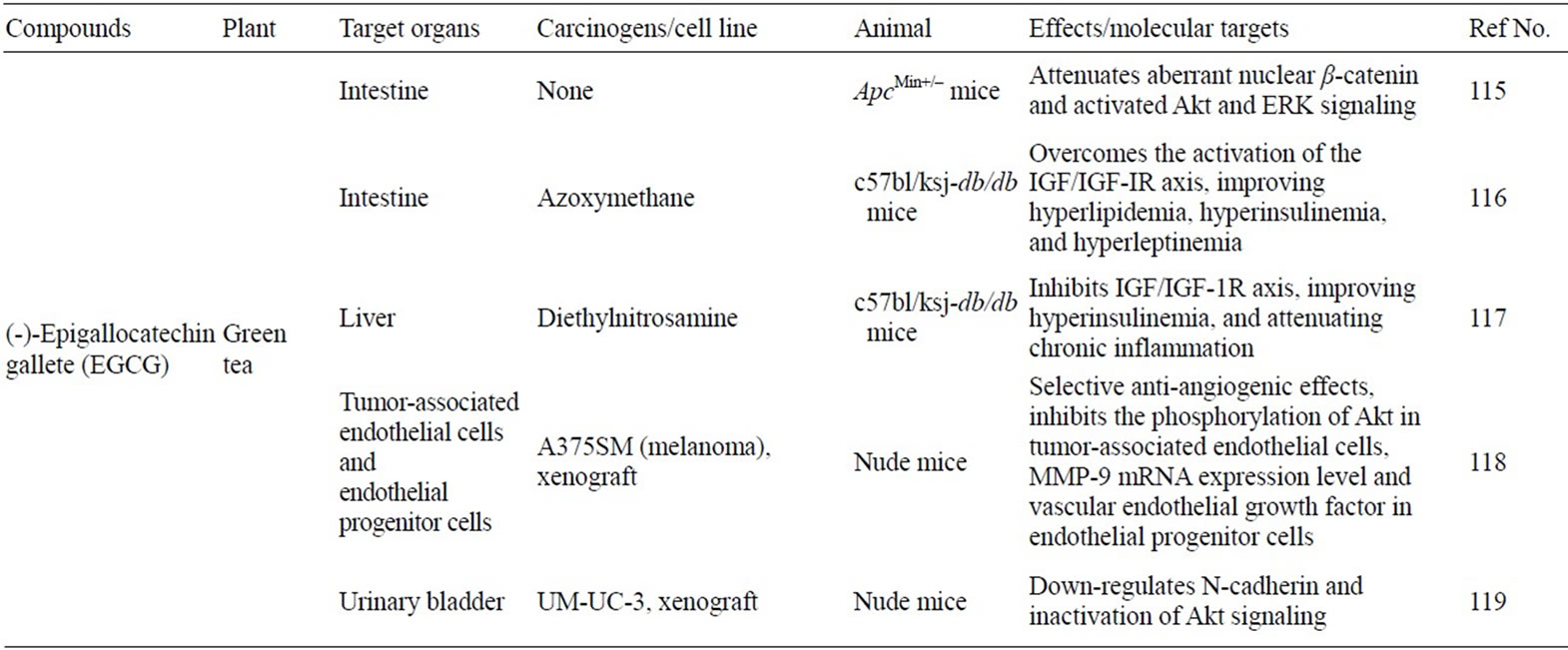
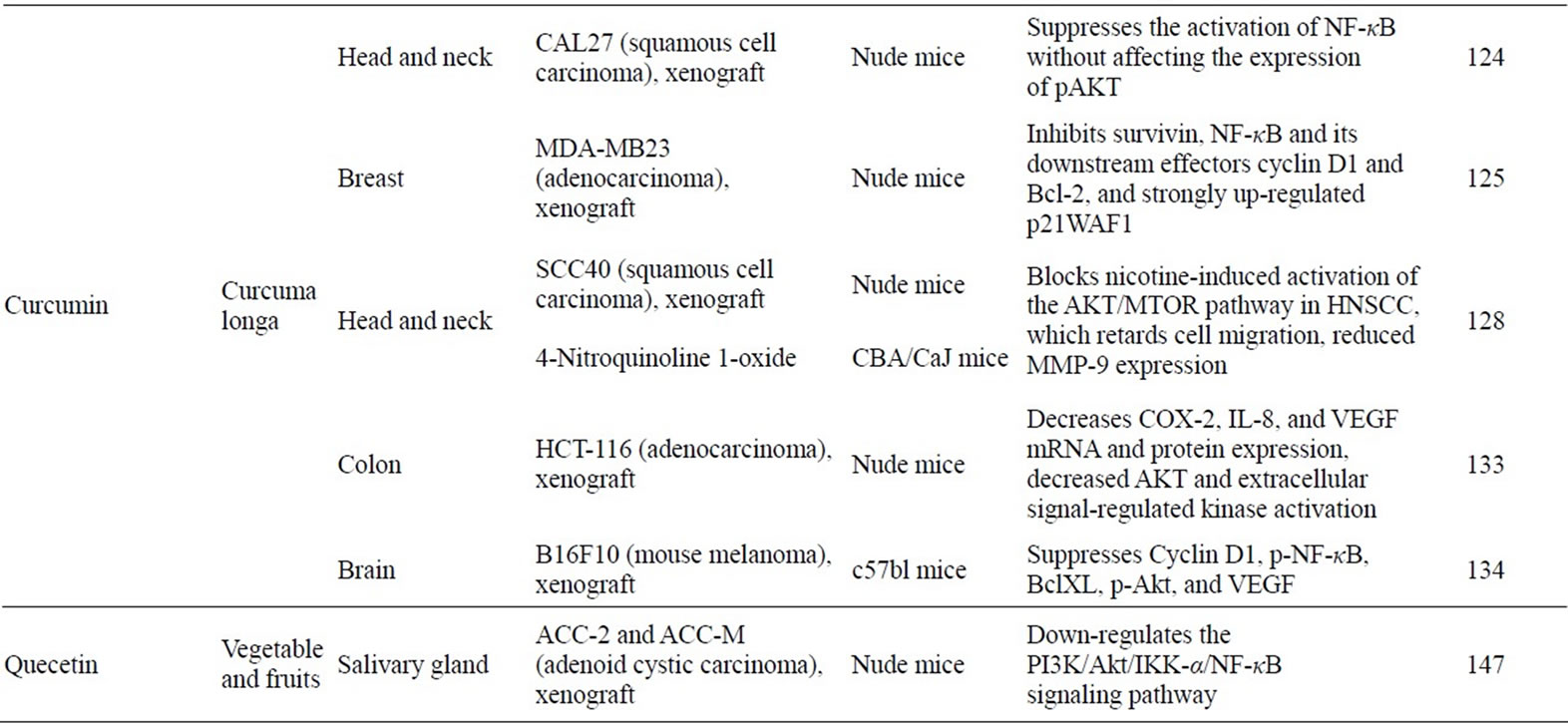
Table 1. Anticancer effect and mechanisms of natural compounds through NF-κB or PI3K/Akt pathway in vivo.
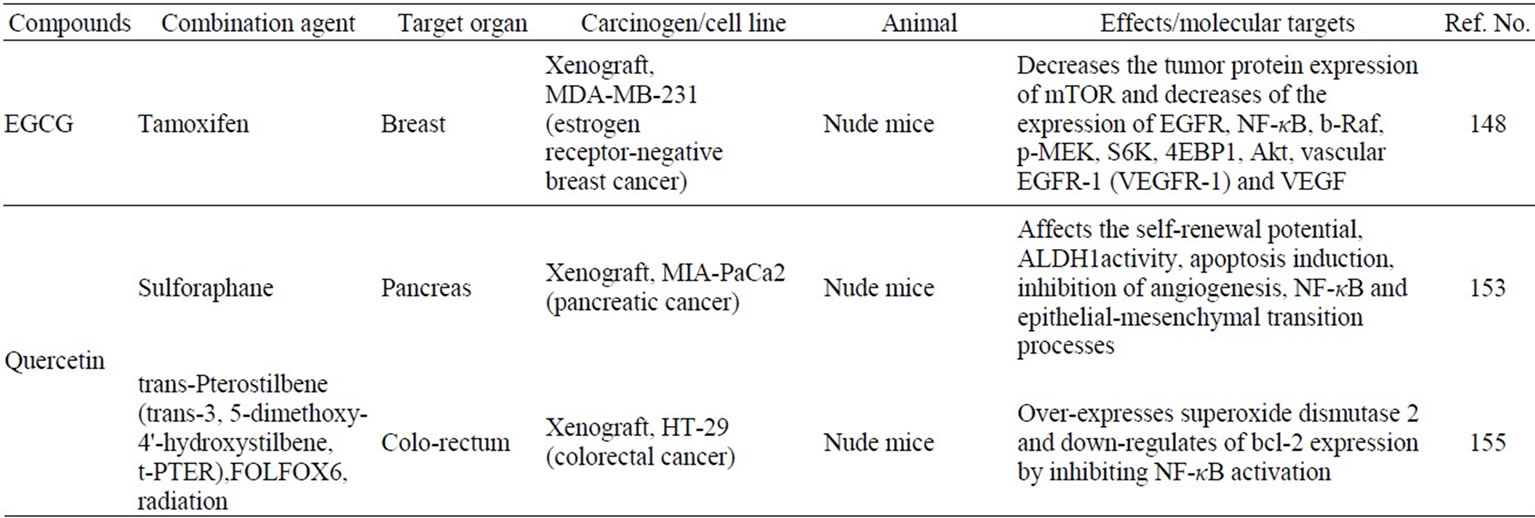
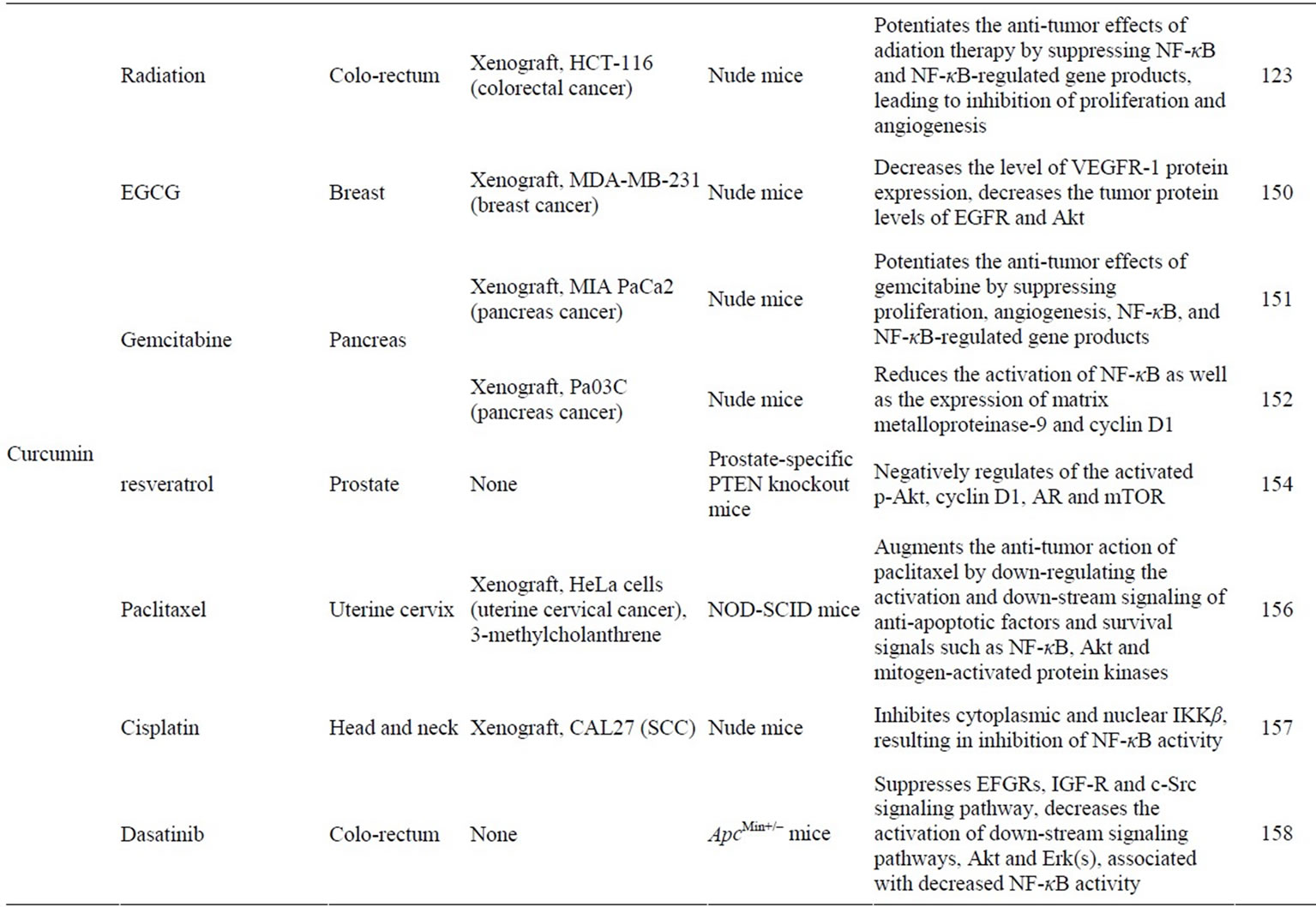
Table 2. Synergistic induction of apoptosis by combining natural compounds and anticancer drugs/radiation through NF-κB or PI3K/ Akt pathway in vivo.
parenchymal or neoplastic cells. Apoptosis rarely causes an inflammatory response, since 1) apoptotic cells do not release their constituents into the surrounding interstitial tissue, 2) apoptotic cells are quickly phagocytosed by cells in the surrounding tissue, preventing secondary necrosis, and 3) the engulfing cells do not produce inflammatory cytokines [54].
Apoptosis can be initiated by receiving extracellular or intracellular signals including growth factor withdrawalUVor gamma-irradiation, chemotherapeutic agents, heat shock, nutrient deprivation, and by a family of transmembrane proteins called death receptors. These signals are transduced to adapter proteins and transmitted to specific cysteine proteases called “initiator caspases”. At this point the cell is committed to undergo apoptosis, followed by “execution of cells” (mediated by sequential activetion of the so-called “executioner caspases”), systematic disintegration of cell structure and phagocytosis of the cell corpses [55].
Caspases (cysteine-dependent aspartate-specific proteases) are typically activated iduring the early stages of apoptosis. This family of proteins is synthesized as inactive zymogens but, once activated, can begin a proteolytic cascade, which results in the cleavage of key cellular components required for normal cellular function, including structural proteins in the cytoskeleton and nuclear proteins such as DNA repair enzymes. Caspases can also activate other degradative enzymes such as DNAases, which begin to cleave the DNA in the nucleus. To date, 14 different members of the caspase-family (Table 3) have been described in mammals [55-57]. The ten major pro-apoptotic caspases can be classified as initiators (caspase-2, -8, -9, -10), effectors or executioners (caspase-3, -6, -7) and inflammatory caspases (caspase-1, -4, -5) [55,58]. Other caspases that have been identified to date (caspase-11, -12, -13 and -14) are involved in specific apoptotic processes or expressed solely in specific types of tissue [55].
It is currently accepted that apoptosis can occur via two main pathways: the extrinsic or death receptor (Figure 1) and intrinsic or mitochondrial (Figure 2) pathways [59]. The extrinsic pathway initiated extracellularly via activation of cell surface receptors by specific molecules known as pro-apoptotic ligands including CD95L/ FasL (receptor CD95/FasR), and Apo2L/TRAIL (receptors DR4, DR5) [53]. The Apo2 ligand has sparked growing interest within the oncology field due to its reported ability to selectively trigger cancer cell death [60,61]. Once activated, the death domains of these receptors, bind to the adaptor protein Fas-associated death domain (FADD), resulting in the assembly of the death-inducing signaling complex (DISC), and recruitment and assembly of the initiator caspase-8 and -10 [62]. Once activated, caspase-8 directly activates caspase-3 to initiate degradation of the cell. Active caspase-8 can also cleave Bid (pro-apoptotic) to tBid, which binds to the mitochondrial membrane to facilitate the release of cytochrome c and initiate the intrinsic pathways. This allows “cross-talk” between the two main pathways and amplifies the apoptotic signaling from death receptors.
The extrinsic and intrinsic pathways both end at the point of the execution phase. Execution caspases activate a cytoplasmic endonuclease, which degrades nuclear material, and proteases that degrade nuclear and cytoskeletal proteins. Such executioners including caspase-3 (considered to be the most important executioner caspase) can be activated by any of the initiator caspases (caspase-8, -9 or -10).
In addition to p53, the extrinsic and intrinsic pathways are also regulated by NF-κB, the ubiquitin proteosome system and the phosphatidylinositol-3-kinase (PI3K) pathway [63]. NF-κB is one of the most studied transcription factors in mammalian cells. Its function has been implicated in inflammation, cell proliferation, differentiation, apoptosis, cell survival and tumorigenesis [64]. NF-κB describes a ubiquitously expressed family of five proteins: p65 (RelA), p50, p52, c-Rel and RelB. Many stimuli produce survival responses in cells that are mediated by NF-κB. Indeed, overall reduction in NF-κB activity has been associated with an increased apoptotic index in many cell types [65]. Inactive NF-κB is bound to IKB inhibitory proteins. Once activated, NF-κB exhibits both
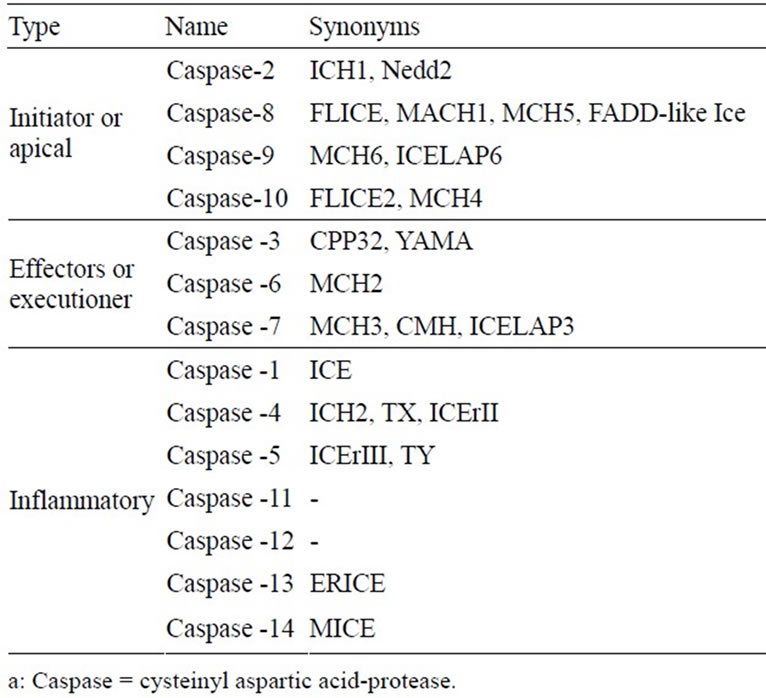
Table 3. Caspasesa involved in apoptosis or programmed cell death.
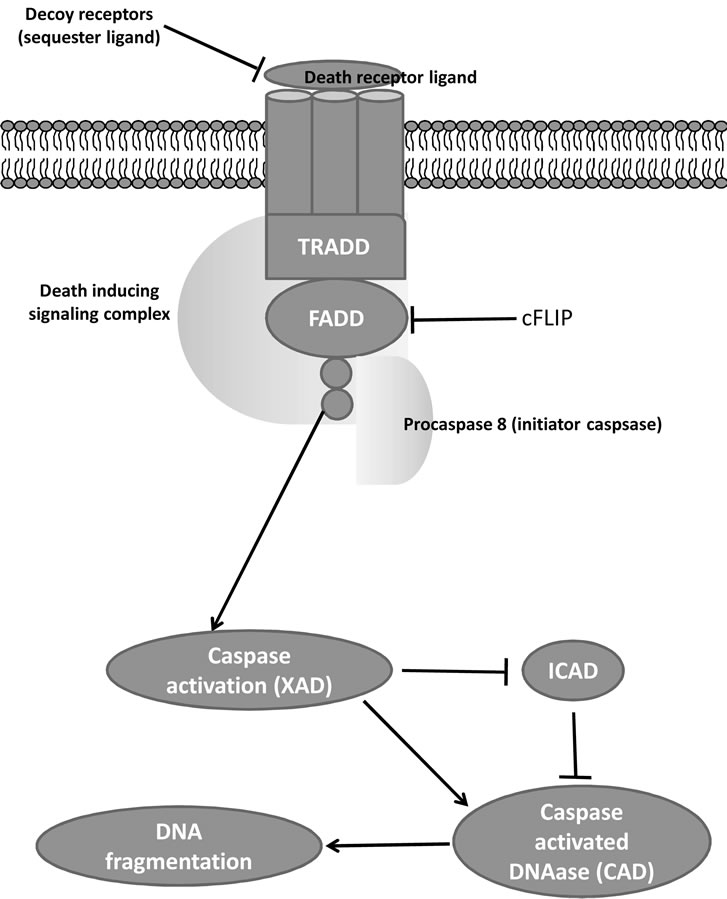
Figure 1. Extrinsic pathway.
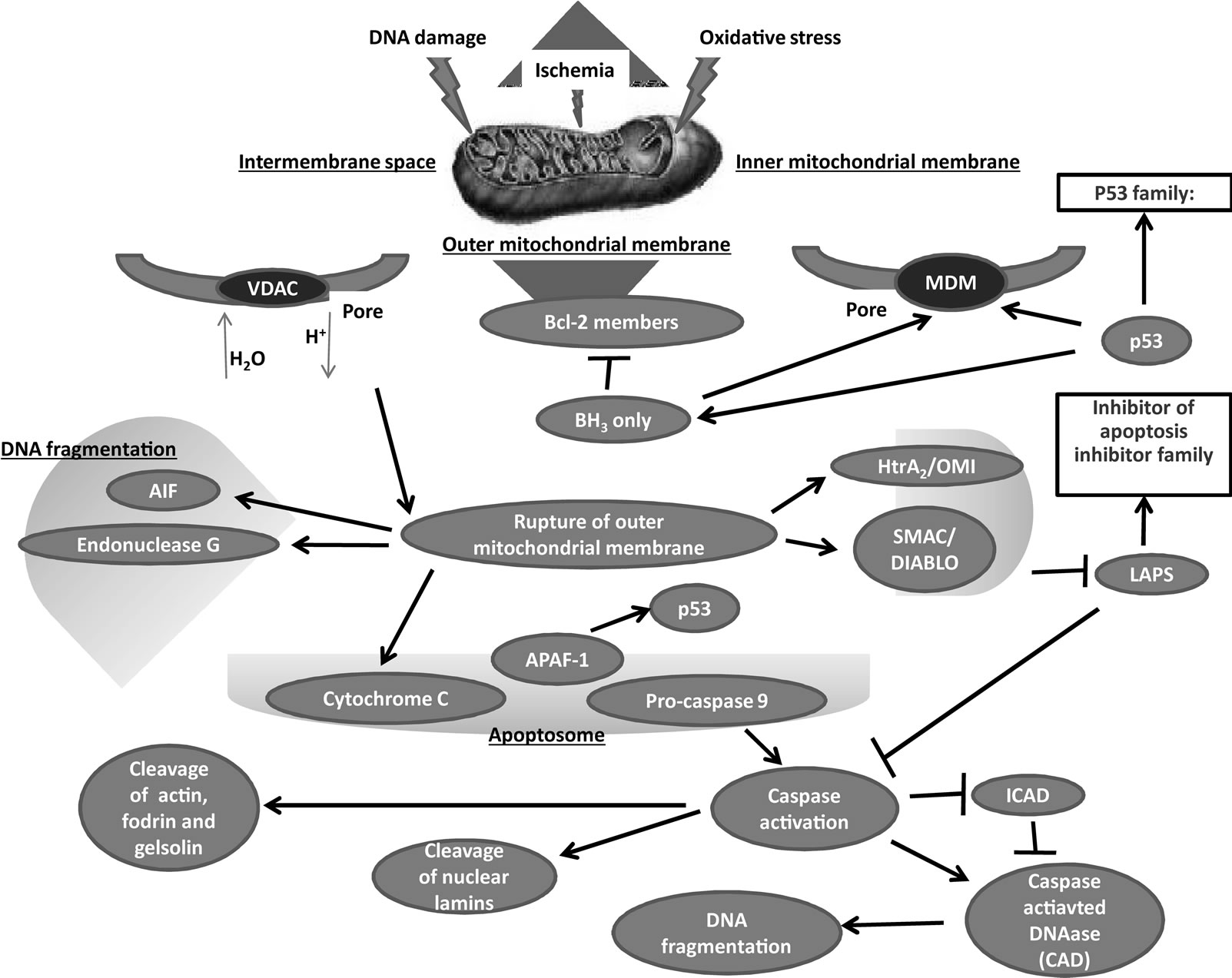
Figure 2. Intrinsic pathway.
anti-apoptotic and pro-apoptotic functions. Physiologically, NF-κB induces resistance to apoptosis through activation of inhibitor of apoptosis (IAP) and X-linked IAP. In addition, NF-κB activation has been shown to inhibit p53-dependent apoptosis following expression of the oncogene AP12/MALT1 [66]. This NF-κB directed survival response is associated with increased expression of anti-apoptotic proteins. Thus, it is not surprising that NF-κB expression is dysregulated in various disease states including chronic inflammation and cancer. In contrast, some stimuli that lead to activation of NF-κB may induce apoptosis, probably via the activation of proapoptotic proteins such as c-myc, p53 and caspase-1 [67].
The PI3K signaling pathway is also crucial for many aspects of cell growth, survival and tissue neo-vascularization and is frequently up-regulated in many cancers [68]. The PI3Ks are a family of related enzymes that are capable of phosphorylating the 3 position hydroxyl group of the inositol ring of PI(4,5)P2 to generate PI(3,4,5)P3
[69]. Upon activation of the PI3K pathway by many growth factors such as epidermal growth factor (EGF), PI(3,4,5) P3 is produced on the inner side of the plasma membrane and binds to Akt. Akt inactivates pro-apoptotic factors such as Bad, which controls the release of cytochrome c [70,71], procaspase-9 and Forkhead transcription factors (such as FOXO). Akt also activates anti-apoptotic genes including the cyclic-AMP response element-binding protein and IΚB kinase leading to NF-κB nuclear localization and the subsequent transcription of pro-survival genes such as Bcl-xL, caspase inhibitors and c-Myb [72,73]. Overexpression of Akt has anti-apoptotic effects in various cell types resulting in resistance to cell death [74].
4. CANCER CHEMOPREVENTION AND APOPTOSIS
Cancer is a pathologic condition where the normal mechanisms of cell cycle regulation are dysfunctional either due to excessive cell proliferation, insufficient apoptosis or both [75-77]. Suppression/inhibition of apoptosis during carcinogenesis is known to play a role in the development and progression of cancers [77-79]. At present, it is accepted that cell populations are tightly regulated by their rates of proliferation, differentiation and death. When the homeostatic balance is disrupted in such a way that clonal outgrowth of mutated cell populations occurs, the development of a tumor will followed [77-79].
In simple terms, one can define carcinogenesis as a multistage process where a normal cell becomes transformed into one with a malignant phenotype. Cells become initiated by the acquisition of an activating mutation in an oncogene or an inactivating mutation in a tumor suppressor gene (initiation). Several additional factors confer these cells a growth advantage (promotion), which allow the cell to survive while accumulating abnormal characteristics and ultimately progressing to a metastatic tumor (progression) [80]. Carcinogenesis is a complex process driven by tight interactions between oncogene activation, tumor suppressor inactivation and cell death machinery. Early in transformation, activated oncogenes that drive the cell to uncontrolled proliferation simultaneously trigger apoptosis, probably as a safety mechanism to remove cells carrying oncogenic mutations [81]. Later in tumorigenesis, the supply of nutrients and oxygen becomes limited, with the tumor cells undergoing hypoxia-induced apoptosis [82]. In order to survive, tumor cells acquire apoptotic-inhibiting mutations (reduced apoptosis) [82]. Failures in normal apoptotic pathways contribute to carcinogenesis by creating a permissive environment for genetic instability and accumulation of mutations, promoting resistance to immune-based destruction, overriding cell-cycle checkpoints (that would normally induce apoptosis), facilitating growth factor/hormone-independent cell survival, supporting anchorage-independent survival during metastasis, reducing dependence of oxygen and nutrients, and conferring resistance to cytotoxic anticancer drugs and radiation [77]. Thus, inhibition of apoptosis can lead to tumor development.
In animal models, most chemical initiators are unable to initiate tumor growth unless a tumor promoter is subsequently applied. Many tumor promoters inhibit apoptosis in vitro [83]. Activation of apoptosis is thus being considered to be one of the most promising therapeutic approaches in cancer therapy [63,77,84]. Tumor cells can acquire resistance to apoptosis, for instance, by overexpressing anti-apoptotic proteins such as Bcl-2 or down-regulatinf/mutating pro-apoptotic proteins such as Bax, the expression of both being regulated by the p53 tumor suppressor gene [85,86]. p53 is a transcription factor essential for the prevention of cancer formation, which can be damaged by radiation, several chemicals and viruses such as human papillomavirus (HPV). The p53 pathway is ubiquitously lost in human cancer either by p53 gene mutation or by loss of cell signaling upstream and downstream of p53 in cancers that express the WT p53 gene [87]. Therefore, despite the enthusiasm towards apoptosis based-drugs, possible difficulties are also being anticipated such as selection of apoptosisresistant tumor cells and systemic toxicity [84].
Several epidemiological studies, later evaluated by meta-analysis, have identified associations between certain dietary factors and cancer that either increase or decrease cancer risk [15,88,89]. It is currently accepted that diet can affect the overall process of carcinogenesis by different mechanisms: its constituents may contain cancer-causing substances as well as many cancer preventive agents. These dietary agents can retard or prevent the process of carcinogenesis by multiple mechanisms, namely 1) enhanced detoxification of the carcinogenic intermediates through induction of phase 2 drug metabolizing enzymes, 2) reduced carcinogenic activation due to suppression of cytochrome P450-dependent monooxygenases, 3) perturbations in cell cycle progression, 4) selective promotion of apoptosis in cancerous or precancerous cells, and 5) inhibition of angiogenesis and metastasis formation [16]. Since apoptosis provides a physiologic mechanism for eliminating abnormal cells, dietary factors affecting apoptosis can have an important effect on carcinogenesis. Conceivably, dietary factors that activate apoptosis in pre-cancerous cells offer a cancer preventive mechanism. In fact, most initiated cells are destroyed by apoptosis before they become malignant and develop into a tumor [79]. Increased understanding in the field of cancer has led to the conviction that most human malignancies should be fought on multiple fronts: in addition to cancer therapy, cancer prevention has become an important means of controlling cancer [8]. Common prevention strategies include avoiding exposure to known cancercausing agents, enhancement of host-defense mechanisms against cancer, life style modifications and chemoprevention [8].
The term chemoprevention refers to the use of agents to slow the progression of, reverse or inhibit carcinogenesis, and was first introduced by Sporn and co-workers in the mid-1970’s [90]. Animal studies, clinical trials and in vitro studies have examined the anticancer activity of numerous putative chemopreventive agents. These studies strongly suggest that the anti-cancer activities of many of these compounds involve the induction of apoptosis, and support the notion that apoptosis is a novel target for cancer chemoprevention [8,10]. Moreover, the pro-apoptotic properties of a variety of chemopreventive agents, like those of many conventional and experimental cancer chemotherapeutic agents, appear to be related to mitochondrial alterations in tumor cells [10,91]. In fact, several classes of chemopreventive agents contain members that trigger mitochondrial disruption and/or mitochondrialmediated apoptosis (intrinsic pathway) in tumor cells in vitro, although other agents may induce apoptosis via the death receptor pathway [9].
Chemotherapy aims to kill cancer cells, in the hope of preventing further cancer progression. Chemoprevention, on the other hand, involves administering non-toxic agents to individuals who may be at an increased risk for cancer. Moreover, surgical and traditional therapeutic approaches (chemotherapy and radiation) are, at present, unable to control most cancer types. Thus, the development of new chemopreventive strategies is required [10,11,92,93]. Chemopreventive compounds can be classified into two major groups: 1) blocking agents, which prevent carcinogens from reaching or reacting with critical target sites, and 2) suppressing agents, which stop the evolution of the pre-neoplastic process. Given that the initiation and progression phases are relatively transient and irreversible events, it seems logical that chemopreventive agents should intervene at the prodromal promotion phase. Three decades of research suggest that chemoprevention is a promising strategy to reduce the incidence of cancer, both in well-defined high-risk groups and in the general population [10-12,92,93].
Of great importance, aberrant NF-κB regulation and Akt activation has been observed in many cancers. To prevent the development and progression of cancers, the strategy should target the cell signaling pathways that are deregulated in malignant tumors. Aberrant regulation of NF-κB and the signaling pathways that control its activity are involved in cancer development and progression, as well as in drug resistance, especially during chemotherapy and radiotherapy [94]. Blocking NF-κB can cause tumor cells to cease proliferation or become more sensitive to the action of antitumor agents [95]. Changes in Akt activator expression observed in human precancerous tissues that might be targeted for chemoprevention [96]. Inhibition of Akt signaling has been associated with the biological actions of numerous chemopreventive compounds. Thus far, several chemopreventive agents including, green tea polyphenols, curcumin, and quercetin have shown their various activities in the inhibition of carcinogenesis through the regulation of major cell signaling pathways such as Akt and NF-κB. Therefore, those are the subject of intense study. Agents capable of suppressing Akt and/or NF-κB activation have therapeutic promise and potential to inhibit carcinogenesis. We currently review dietary cancer chemopreventive compounds include EGCG, curcumin, and quercetin, capable of functioning in this capacity.
Representative dietary cancer chemopreventive compounds include EGCG, curcumin, and quercetin.
4.1. EGCG
Cancer prevention by green tea and its constituents been studied in different animal models of carcinogenesis [97]. The major catechins (a group of polyphenols) in green tea are EGCG, (-)-epigallocatechin (EGC), (-)- epicatechin-3-gallete (ECG) and (-)-epicatechin. EGCG, the most abundant and most studied catechin, exhibits significant growth inhibitory effects in cancer cells. More importantly, after treatment of a primary cell line such as normal epithelial cells with EGCG, there is no observable toxicity at doses that are used for cancer inhibition studies [98,99]. A collection of reviews have commented on the possible mechanistic effects of EGCG in multiple cell lines and have noted similarities in regard to growth inhibition and cell cycle arrest [100-102].
EGCG reportedly affects the transcription factors p53 and NF-κB leading to a change in the ratio of Bax/Bcl-2 in a manner that favors apoptosis [103]. The induction of apoptosis by other green tea catechins has been evaluated in a dose dependent manner (i.e. ECG > EGCG > EGC > EC) [104].
EGCG treatment may lead to a significant doseand time-dependent inhibition of activation and translocation of NF-κB to the nucleus by suppressing the degradation of IΚBα in the cytoplasm [105,106]. EGCG may also inhibit the ATPand IL-1β-induced activation of NF-κB [107].
EGCG has been found to inhibit PI3K/Akt activation, resulting in the modulation of Bcl-2 family proteins and leading to the enhanced apoptosis of bladder cancer cells [108]. EGCG has also been shown to inhibit vascular endothelial growth factor (VEGF)-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt, suggesting that EGCG has an inhibitory effect on the Akt signaling pathway [109, 110]. Further studies have also demonstrated that constitutive activation of Akt, EGFR and Stat3 was inhibited in both YCU-H891 head and neck squamous cell carcinoma and MDA-MB-231 breast carcinoma cell lines treated with EGCG [111].
A dose-dependent increase in p53 was observed after EGCG treatment of LNCaP cells, which carry WT p53, but not in DU145 cells carrying mutant p53 [112]. EGCG was also shown to stabilize p53 and cause up-regulation of its transcriptional activity, resulting in the activation of its downstream targets such as p21WAF1 and Bax, and the induction of apoptosis [113]. In human liver cancer cells, a significant increase in the expression of p53 and p21WAF1 protein that lead to cell cycle arrest was reported after EGCG treatment [114]. Table 1 summarizes the action/mechanisms of three selected compounds that affect NF-κB and Akt-phosphatidylinositol 3-kinase (PI3K) activity in vivo system.
Administration of EGCG to Apcmin/+ mice, an animal model of human intestinal carcinogenesis, via their drinking fluid was found to significantly decrease small intestinal tumor formation [115]. Shimizu et al. have recently reported that EGCG prevents obesity-related colonic and liver tumorigenesis by inhibiting the phosphorylation of the insulin like growth factor-1 receptor (IGF-1R), extracellular signal-regulated kinase (ERK), Akt, glycogen synthase kinase-3beta (GSK-3β), signal transducers and activators of transcripton 3 (Stat3), and c-Jun NH2-terminal kinase (JNK) proteins, and improving hyperinsulinemia in carcinogen-induced mouse model [116,117]. EGCG suppressed the growth of melanoma cells in nude mice with impaired angiogenesis by inhibiting PI3K/Akt signaling specifically in tumor-associated endothelial cells and peripheral blood-derived endothelial cells [118]. Continuous feeding with EGCG to mice prior to and during establishment of bladder carcinoma xenografts in vivo revealed >50% reduction in mean tumor volume without detactable toxicity [119].
4.2. Curcumin
It is widely accepted that curcumin, a yellow pigment found in the rhizome of the spice turmeric, has potent cancer chemopreventive activity in various animal carcinogenesis models [120]. Curcumin is known to be a strong inhibitor of NF-κB. Curcumin has been shown to inhibit IKK, suppress both constitutive and inducible NF-κB activation, and potentiate tumor necrosis factor (TNF)-induced apoptosis [121]. Recent studies have shown that curcumin suppresses the constitutive activation of NF-κB [122] and sensitizes human colorectal cancer xenografts in nude mice to gamma-radiation by targeting NF-κB regulated gene products [123]. Treatment with a liposomal formulation of curcumin resulted in a dose-dependent growth suppression of cancer cells and decreased activation of NF-κB [124]. The findings that expression of NF-κB target genes, including cyclin D1, cyclooxygenase (COX)-2, matrix metalloproteinase-9, Bcl-2, Bcl-xL, Mcl-1L and Mcl-1S, was reduced by the treatment with a liposomal formulation of curcumin indicate that curcumin acts the NF-κB pathway which is involved in carcinogenesis. Al-Hujaily et al. have demonstrated that a novel curcumin analogue, PAC, significantly reduced tumour size, and triggered apoptosis in breast cancer tumor xenografts by inhibiting expression of survivin, NF-κB and its downstream effectors, and strongly up-regulated p21 (WAF1) [125]. Moreover, clinical trials have shown that curcumin down-regulates the expression of NF-κB and COX-2 in peripheral blood mononuclear cells from patients with pancreatic cancer [126]. These results clearly indicate that curcumin inhibits tumor growth by affecting the NF-κB signaling in vitro and in vivo.
Curcumin also exhibits an inhibitory effect on Akt signaling. Recent studies have shown that curcumin doseand time-dependently inhibits the phosphorylation of Akt and mammarian trget of rapamycin (mTOR), and their downstream targets in prostate cancer cells [127]. Inhibition of the Akt/mTOR pathway by curcumin results in suppression of the growth of SCC40 xenografts, and curcumin at 15 mg significantly increases in survival in the 4-nitroquinoline 1-oxide--induced head and neck squamous cell carcinoma survival study [128].
Curcumin has also been shown to inhibit the proliferation of cisplatin-resistant ovarian cancer cells via the inhibition of Akt activation [129]. It has also been reported that an analog of curcumin, 4-hydroxy-3-methoxybenzoic acid methyl ester (HMBME), targets the Akt signaling pathway, inhibits the proliferation of cancer cells and induces apoptosis [130]. Likewise, HMBME was shown to decrease the level of phosphorylated Akt, inhibit Akt kinase activity, and reduce the DNA-binding activity of NF-κB [130]. Several reports also suggest that curcumin has molecular targets within the Akt signaling pathways and that the inhibition of Akt activity may facilitate inhibition of proliferation and induction of apoptosis in cancer cells [131,132]. A curcumin derivative, diphenyl difluoroketone, significantly inhibited the colon cancer xenograft Akt and ERK phospholyration in mice [133]. In other study, solubilized curcumin effectively blocked brain tumor formation in the mice that had already received an intracerebral bolus of mouse melanoma cells (B16F10) by suppressing p-Akt, Cyclin D1, p-NF-κB, Bcl-xL and VEGF [134].
4.3. Quercetin
Quercetin, a powerful anti-oxidant, is consumed by humans as part of their diet [135]. Although the quercetin content of foods has not been systematically analyzed, it is found in many fruits, vegetables and beverages. The chemopreventive effects of quercetin against DNA damage and precancerous changes in cells were recently demonstrated both in vitro and in vivo. Quercetin was found to arrest the progression of cervical neoplasia in Swiss albino mice [136].
Quercetin may offer a defense against the detrimental effects of carcinogenic chemicals and can induce apoptosis via the mitochondrial pathway [137]. Although the anti-carcinogenic mechanisms of quercetin are not well known, quercetin specifically inhibits p21-Ras expression in human colon cancer cell lines and in primary colorectal cancers [138]. Reports suggest that quercetin has DNA-damaging and pro-oxidant property in cells [139]. One proposed mechanism involves the inhibition of Akt/ protein kinase B (PKB) phosphorylation, an upstream kinase of the pro-survival protein kinase cascade involveing PI3K. Significant down-regulation of Bcl-2 and Bcl-xL along with Cu-Zn SOD, which could lead to an increase in ROS, has been reported after quercetin treatment in certain human cancer cell lines [43,140].
Survivin, which binds directly to and inactivates caspases was inhibited by quercetin, resulting in the activation of caspases [43]. One promising therapeutical approach for the induction of apoptosis in cancer cells is using TNF-related apoptosis-including ligand (TRAIL) [141]. Quercetin has been shown to potentially arrest human prostate cancer cells and mediate activation of caspase and poly(ADP-ribose)polymerase (PARP) cleavage [142,143]. A combined treatment of TRAIL and quercetin was found to enhance TRAIL-induced cytotoxicity by activating caspases and inhibiting phosphorylation of Akt [144].
A principal approach for treatment of tumors is based on the fact that cancer cells are resistant to CD95-mediated apoptosis. Alterations in the CD95 system result in the escape of tumor cells from this defense system. Sensitivity to CD95 can be restored by treating the cells with quercetin, thus making the cancer cells susceptible to apoptosis [145]. One study has demonstrated the inhibitory effects of quercetin on H2O2-induced apoptosis via mediation of the AP-1-mediated apoptotic pathway. The mechanistic action of quercetin as an anti-apoptotic compound is attributed to its ability to inhibit MAPK pathways and reduce expression of genes participating in the JNK-c-JUN/AP-1 and ERK-cFOS/AP-1 pathways [146].
In an azoxymethane (AOM)-induced rat colon cancer model, dietary administration wirh quercetin and curcumin decreased the number of aberrant crypt foci (ACF), putataive precursor lesions for colonic adenocarcinoma, by 4- and 2-fold, respectively, compared with the controls [137]. Western blot analyses of caspase-9, Bax (proapoptotic) and Bcl-2 (anti-apoptotic) proteins from colon scrapings suggest that quercetin and curcumin induce apoptosis via the mitochondrial pathway. Sun et al. reported that quercetin significantly prevented in vivo growth of human salivary adenoid cystic carcinoma xenografts in nude mice, accompanied by induction of tumor cell apoptosis, suppression of NF-κB nuclear translocation, as well as down-regulation of Akt and IΚB kinase-α activation. Thus, quercetin would be a promising chemotherapeutic agent through its function of down-regulating the PI3K/Akt/IKK-alpha/NF-κB signaling pathway [147].
5. COMBINATION THERAPY
Some naturally occurring chemopreventive compounds are known to act in synergy with other chemopreventive or anti-cancer agnets, as listed in Table 2. The antineoplastic agents have dose-limiting toxicity and drug resistance, thus limiting their clinical application. Development of novel strategies that overcome radioand chemo-resistance and sensitize cancer cells to anti-neoplastic agent can enhance the therapeutic effect of these drugs. Combination treatment with EGCG and tamoxifen was synergistically cytotoxic and enhanced apoptosis in MDA-MB-231 human breast cancer cells and decreased tumor growth in a MCF-7 cell xenograft model [148,149]. The combined treatment with EGCG and curcumin also resulted in synergistic growth inhibition of MDA-MB- 231 [150]. Curcumin and gemsitabin treatment decreased pancreatic tumor volume in vivo model [151,152]. Furthermore, combination of quercetin with sulforaphane, an isothiocyanate enriched in broccoli, exerted synergistic effects by improving apoptosis resistance [153]. Liposomal forms of curcumin plus resveratrol significantly decreased prostatic adeno-carcinoma in prostatespecific PTEN-knockout mice by effectively inhibiting cell growth and induceing apoptosis [154].
Bcl-2 family proteins are regulators of chemoresistance and radioresistance in cancer. In vivo treatment with quercetin and trans-3,5-dimethoxy-4'-hydroxystilbene (t-PTER) altered expression of molecules involved in regulating cancer cell resistance to drugs and radiations [155]. Combined administration with t-PTER+ quercetin, FOLFOX6 (oxaliplatin, leukovorin, and 5-fluorouracil, a first-line chemotherapy regimen), and radiotherapy eliminates colorectal cancer cells growing in vivo leading to long-term survival. Gene expression analysis of a Bcl-2 family of genes revealed that down-regulation of bcl-2 expression via inhibition of NF-κB activation. Curcumin also potentiates the antitumor effects of radiation therapy in colorectal cancer by suppressing NF-κB and NF-κB-regulated gene products, leading to inhibition of proliferation and angiogenesis [123].
In mouse cervical multi-stage squamous cell carcinoma model using 3-methylcholanthrene and a xenograft model of human cervical cancer in mice, the combined treatment with curcumin and paclitaxel induced a synergestic reduction in the tumor incidence as well as tumor volume compared with the individual treatment of paclitaxel or curcumin [156], suggesting that a suboptimal concentration of curcumin augments the antitumor action of paclitaxel by downregulating the activation and downstream signaling of anti-apoptotic factors and survival signals such as NF-κB, Akt and MAPKs that have significant roles in proliferation, survival, angiogenesis and metastasis. By inhibition of NF-κB activity, curcumin augments the anti-tumor action of cisplatin enhancing growth suppression in vivo [157].
Recently, preclinical investigation revealed that combination therapy with curcumin and dasatinib to be highly effective causing an over 95% regression of intestinal adenomas in Apcmin/+ mice, which could be attributed to decrease proliferation and increased apoptosis [158].
6. SUMMARY AND PERSPECTIVES
The pathogenesis of many chronic diseases, including cancer, has been associated with aberrantly regulated apoptosis [14,75,77,78,159]. The synergistic combination of an undesirable proliferative stimulus and an associated defect in the apoptotic pathway seems universal in cancer [76-80]. Epidemiological studies indicate that dietary habits contribute to, at least, one third of all human cancers [160], and suggest that certain dietary components can exacerbate or interfere with carcinogenesis. Apoptosis is likely to be a crucial mechanism in the chemopreventive properties associated with such dietary factors [14].
In addition to the conventional therapeutic agents, numerous dietary components and micronutrients are emerging, which possess considerable potential for hindering in vivo deleterious oxidative processes and inducing apoptosis of cancerous or pre-cancerous cells [160,161], and are therefore being considered as promising chemopreventive agents. A range of dietary compounds can modulate apoptosis and those with pro-apoptotic properties exhibit beneficial effects in animal and in vitro studies by eliminating cancerous cells [9,14,92]. Moreover, some dietary compounds have also shown beneficial effects in clinical trials [93].
A balance of cell proliferation and apoptosis normally maintains cellular homeostasis. However, apoptosis is a very complex process with numerous specific targets within each arm of the apoptotic pathways. Nevertheless, it is very encouraging that single bioactive dietary agents can directly and indirectly influence many of the targets within the apoptotic pathway. In additon, many of these dietary agents appear to exhibit some degree of specificity for neoplastic cells. Furthermore, the protective effects of single agents can be potentiated and/or synergized by other dietary factors suggesting the possibility of combinational approaches for chemoprevention. While dietary interventions seem encouraging for devising new chemopreventive strategies, there are several issues remaining that need to be fully understood. The dose of each agent, duration of exposure, relative bioavailability of each dietary compound and potentially adverse side effects and/or interactions should be considered.
Further research is required to identify the phytochemical-specific molecular mechanisms of the huge number of already recognized bioactive dietary chemopreventive agents. The potential benefits of cancer chemoprevention appear promising given the data obtained from clinical trials and pre-clinical studies.
7. ACKNOWLEDGEMENTS
This review was based on studies supported in part by a Grant-in-Aid for the 3rd Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan, the Grant-in-Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan, the Grants-in-Aid for Scientific Research (Nos. 18592076, 17015016 and 18880030) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the grant (H2010-12) for the Project Research from High-Technology Center of Kanazawa Medical University.
REFERENCES
- Donaldson, M.S. (2004) Nutrition and cancer: A review of the evidence for an anti-cancer diet. Nutrition Journal, 3, 19. doi:10.1186/1475-2891-3-19
- Riboli, E. and Norat, T. (2003) Epidemiologic evidence of the protective effect of fruit and vegetables on cancer risk. American Journal of Clinical Nutrition, 78, 559S-569S.
- Gonzalez, C.A. and Riboli, E. (2006) Diet and cancer prevention: Where we are, where we are going. Nutrition and Cancer, 56, 225-231. doi:10.1207/s15327914nc5602_14
- Nichenametla, S.N., Taruscio, T.G., Barney, D.L. and Exon, J.H. (2006) A review of the effects and mechanisms of polyphenolics in cancer. Critical Reviews in Food Science and Nutrition, 46, 161-183. doi:10.1080/10408390591000541
- Ramos, S. (2008) Cancer chemoprevention and chemotherapy: Dietary polyphenols and signalling pathways. Molecular Nutrition and Food Research, 52, 507-526. doi:10.1002/mnfr.200700326
- Lambert, J.D. Hong, J., Yang, G.Y., Liao, J. and Yang, C.S. (2005) Inhibition of carcinogenesis by polyphenols: Evidence from laboratory investigations. American Journal of Clinical Nutrition, 81, 284S-291S.
- Fresco, P., Borges, F., Diniz, C. and Marques, M.P. (2006) New insights on the anticancer properties of dietary polyphenols. Medical Research Reviews, 26, 747-766. doi:10.1002/med.20060
- Sun, S.Y., Hail, N. Jr. and Lotan, R. (2004) Apoptosis as a novel target for cancer chemoprevention. Journal of National Cancer Institute, 96, 662-672. doi:10.1093/jnci/djh123
- Martin, K.R. (2006) Targeting apoptosis with dietary bioactive agents. Experimental Biolology and Medicine (Maywood), 231, 117-129.
- Hail, N. Jr. (2005) Mitochondria: A novel target for the chemoprevention of cancer. Apoptosis: An International Journal on Programmed Cell Death, 10, 687-705. doi:10.1007/s10495-005-0792-8
- Hail, N. Jr., Cortes, M., Drake, E.N. and Spallholz, J.E. (2008) Cancer chemoprevention: A radical perspective. Free Radical Biology and Medicine, 45, 97-110. doi:10.1016/j.freeradbiomed.2008.04.004
- Manach, C., Williamson, G., Morand, C., Scalbert, A. and Remesy, C. (2005) Bioavailability and bioefficacy of polyphenols in humans I. Review of 97 bioavailability studies. American Journal of Clinical Nutrition, 81, 230S-242S.
- Scalbert, A., Manach, C., Morand, C., Remesy, C. and Jimenez, L. (2005) Dietary polyphenols and the prevention of diseases. Critical Reviews in Food Science and Nutrition, 45, 287-306. doi:10.1080/1040869059096
- Watson, W.H., Cai, J. and Jones, D.P. (2000) Diet and apoptosis. Annual Review of Nutrition, 20, 485-505. doi:10.1146/annurev.nutr.20.1.485
- Riboli, E., Norat, T. (2001) Cancer prevention and diet: Opportunities in Europe. Public Health Nutrition, 4, 475- 484. doi:10.1079/PHN2001158
- Stan, S.D., Kar, S., Stoner, G.D. and Singh, S.V. (2008) Bioactive food components and cancer risk reduction. Jpournal of Cellular Biochemistry, 104, 339-356. doi:10.1002/jcb.21623
- Lepley, D.M., Li, B., Birt, D.F. and Pelling, J.C. (1996) The chemopreventive flavonoid apigenin induces G2/M arrest in keratinocytes. Carcinogenesis, 17, 2367-2375. doi:10.1093/carcin/17.11.2367
- Skaper, S.D., Fabris, M., Ferrari, V., Dalle Carbonare, M. and Leon, A. (1997) Quercetin protects cutaneous tissueassociated cell types including sensory neurons from oxidative stress induced by glutathione depletion: Cooperative effects of ascorbic acid. Free Radical Biology and Medicine, 22, 669-678. doi:10.1016/S0891-5849(96)00383-8
- Agullo, G., Gamet-Payrastre, L., Manenti, S., Viala, C., Remesy, C., Chap, H. and Payrastre, B. (1997) Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: A comparison with tyrosine kinase and protein kinase C inhibition. Biochemical Pharmacology, 53, 1649-1657. doi:10.1016/S0006-2952(97)82453-7
- Sergediene, E., Jonsson, K., Szymusiak, H., Tyrakowska, B., Rietjens, I.M. and Cenas, N. (1999) Prooxidant toxicity of polyphenolic antioxidants to HL-60 cells: Description of quantitative structure-activity relationships. FEBS Letters, 462, 392-396. doi:10.1016/S0014-5793(99)01561-6
- Bianco, A.D., Muzzalupo, I., Piperno, A., Romeo, G. and Uccella, N. (1999) Bioactive derivatives of oleuropein from olive fruits. Journal of Agricultural and Food Chemistry, 47, 3531-3534. doi:10.1021/jf981240p
- Hollman, P.C. and Katan, M.B. (1999) Dietary flavonoids: Intake, health effects and bioavailability. Food and Chemical Toxicology, 37, 937-942. doi:10.1016/S0278-6915(99)00079-4
- Visioli, F. and Galli, C. (2002) Biological properties of olive oil phytochemicals. Critical Reviews in Food Science and Nutrition, 42, 209-221. doi:10.1080/10408690290825529
- Scalbert, A. and Williamson, G. (2000) Dietary intake and bioavailability of polyphenols. Journal of Nutrition, 130, 2073S-2085S.
- Higdon, J.V. and Frei, B. (2003) Tea catechins and polyphenols: Health effects, metabolism, and antioxidant functions. Critical Reviews in Food Science and Nutrition, 43, 89-143. doi:10.1080/10408690390826464
- Nijveldt, R.J., Van Nood, E., Van Hoorn, D.E., Boelens, P.G., Van Norren, K. and Van Leeuwen, P.A. (2001) Flavonoids: A review of probable mechanisms of action and potential applications. American Journal of Clinical Nutrition, 74, 418-425.
- Fotsis, T., Pepper, M.S., Aktas, E., Breit, S., Rasku, S., Adlercreutz, H., Wahala, K., Montesano, R. and Schweigerer, L. (1997) Flavonoids, dietary-derived inhibittors of cell proliferation and in vitro angiogenesis. Cancer Research, 57, 2916-2921.
- Ramos, S. (2007) Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. Journal of Nutritional Biochem, 18, 427-442. doi:10.1016/j.jnutbio.2006.11.004
- Meeran, S.M., Patel, S.N., Chan, T.H. and Tollefsbol, T.O. (2011) A novel prodrug of epigallocatechin-3-gallate: Differential epigenetic hTERT repression in human breast cancer cells. Cancer Prevention Research (Phila), 4, 1243-1254. doi:10.1158/1940-6207.CAPR-11-0009
- Haghiac, M. and Walle, T. (2005) Quercetin induces necrosis and apoptosis in SCC-9 oral cancer cells. Nutrition and Cancer, 53, 220-231. doi:10.1207/s15327914nc5302_11
- Cheng, S., Gao, N., Zhang, Z., Chen, G., Budhraja, A., Ke, Z., Son, Y.O., Wang, X., Luo, J. and Shi, X. (2010) Quercetin induces tumor-selective apoptosis through downregulation of Mcl-1 and activation of Bax. Clinical Cancer Research, 16, 5679-5691. doi:10.1158/1078-0432.CCR-10-1565
- Chou, C.C., Yang, J.S., Lu, H.F., Ip, S.W., Lo, C., Wu, C.C., Lin, J.P., Tang, N.Y., Chung, J.G., Chou, M.J., Teng, Y.H. and Chen, D.R. (2010) Quercetin-mediated cell cycle arrest and apoptosis involving activation of a caspase cascade through the mitochondrial pathway in human breast cancer MCF-7 cells. Archives of Pharmacal Research, 33, 1181-1191. doi:10.1007/s12272-010-0808-y
- Nguyen, T.T., Tran, E., Nguyen, T.H., Do, P.T., Huynh, T.H. and Huynh, H. (2004) The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis, 25, 647- 659. doi:10.1093/carcin/bgh052
- Senthilkumar, K., Elumalai, P., Arunkumar, R., Banudevi, S., Gunadharini, N.D., Sharmila, G., Selvakumar, K. and Arunakaran, J. (2010) Quercetin regulates insulin like growth factor signaling and induces intrinsic and extrinsic pathway mediated apoptosis in androgen independent prostate cancer cells (PC-3). Molecular and Cellular Biochemistry, 344, 173-184. doi:10.1007/s11010-010-0540-4
- Van Erk, M.J., Roepman, P., Van der Lende, T.R., Stierum, R.H., Aarts, J.M., Van Bladeren, P.J. and Van Ommen, B. (2005) Integrated assessment by multiple gene expression analysis of quercetin bioactivity on anticancer-related mechanisms in colon cancer cells in vitro. European Journal of Nutrition, 44, 143-156. doi:10.1007/s00394-004-0503-1
- Mouria, M., Gukovskaya, A.S., Jung, Y., Buechler, P., Hines, O.J., Reber, H.A. and Pandol, S.J. (2002) Food-derived polyphenols inhibit pancreatic cancer growth through mitochondrial cytochrome C release and apoptosis. International Journal of Cancer, 98, 761-769. doi:10.1002/ijc.10202
- Ramos, S., Alia, M., Bravo, L. and Goya, L. (2005) Comparative effects of food-derived polyphenols on the viability and apoptosis of a human hepatoma cell line (HepG2). Journal of Agricultural and Food Chemistry, 53, 1271-1280. doi:10.1021/jf0490798
- Galati, G., Teng, S., Moridani, M.Y., Chan, T.S. and O’Brien, P.J. (2000) Cancer chemoprevention and apoptosis mechanisms induced by dietary polyphenolics. Drug Metabolism and Drug Interactions, 17, 311-349. doi:10.1515/DMDI.2000.17.1-4.311
- Giovannini, C., Scazzocchio, B., Vari, R., Santangelo, C., D’Archivio, M. and Masella, R. (2007) Apoptosis in cancer and atherosclerosis: Polyphenol activities. Annali dell’Istituto Superiore di Sanita, 43, 406-416.
- Sarkar, F.H. and Li, Y. (2006) Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Research, 66, 3347-3350. doi:10.1158/0008-5472.CAN-05-4526
- Chan, M.M., Fong, D., Soprano, K.J., Holmes, W.F. and Heverling, H. (2003) Inhibition of growth and sensitization to cisplatin-mediated killing of ovarian cancer cells by polyphenolic chemopreventive agents. Journal of Cellular Physiology, 194, 63-70. doi:10.1002/jcp.10186
- Khafif, A., Schantz, S.P., Chou, T.C., Edelstein, D. and Sacks, P.G. (1998) Quantitation of chemopreventive synergism between (-)-epigallocatechin-3-gallate and curcumin in normal, premalignant and malignant human oral epithetlial cells. Carcinogenesis, 19, 419-424. doi:10.1093/carcin/19.3.419
- Sharma, H., Sen, S. and Singh, N. (2005) Molecular pathways in the chemosensitization of cisplatin by quercetin in human head and neck cancer. Cancer Biology and Therapy, 4, 949-955. doi:10.4161/cbt.4.9.1908
- Jakubowicz-Gil, J., Paduch, R., Piersiak, T., Glowniak, K., Gawron, A. and Kandefer-Szerszen, M. (2005) The effect of quercetin on pro-apoptotic activity of cisplatin in HeLa cells. Biochemical Pharmacology, 69, 1343-1350. doi:10.1016/j.bcp.2005.01.022
- Suganuma, M., Okabe, S., Kai, Y., Sueoka, N., Sueoka, E., Fujiki, H. (1999) Synergistic effects of (--)-epigallocatechin gallate with (--)-epicatechin, sulindac, or tamoxifen on cancer-preventive activity in the human lung cancer cell line PC-9. Cancer Research, 59, 44-47.
- Hwang, J.T., Ha, J. and Park, O.J. (2005) Combination of 5-fluorouracil and genistein induces apoptosis synergistically in chemo-resistant cancer cells through the modulation of AMPK and COX-2 signaling pathways. Biochemical and Biophysical Research Communications, 332, 433- 440. doi:10.1016/j.bbrc.2005.04.143
- Passi, S., Picardo, M. and Nazzaro-Porro, M. (1987) Comparative cytotoxicity of phenols in vitro. Biochemical Journal, 245, 537-542.
- Moridani, M.Y., Galati, G. and O’Brien, P.J. (2002) Comparative quantitative structure toxicity relationships for flavonoids evaluated in isolated rat hepatocytes and HeLa tumor cells. Chemico-Biological Interactions, 139, 251- 264. doi:10.1016/S0009-2797(02)00005-4
- Siraki, A.G., Chan, T.S. and O’Brien, P.J. (2004) Application of quantitative structure-toxicity relationships for the comparison of the cytotoxicity of 14 p-benzoquinone congeners in primary cultured rat hepatocytes versus PC12 cells. Toxicological Sciences, 81, 148-159. doi:10.1093/toxsci/kfh182
- Depeint, F., Gee, J.M., Williamson, G. and Johnson, I.T. (2002) Evidence for consistent patterns between flavonoid structures and cellular activities. Proceedings of the Nutrition Society, 61, 97-103. doi:10.1079/PNS2001133
- Gomes, C.A., Da Cruz, T.G., Andrade, J.L., Milhazes, N., Borges, F. and Marques, M.P. (2003) Anticancer activity of phenolic acids of natural or synthetic origin: A structureactivity study. Journal of Medical Chemistry, 46, 5395-5401. doi:10.1021/jm030956v
- Teixeira, S., Siquet, C., Alves, C., Boal, I., Marques, M.P., Borges, F., Lima, J.L. and Reis, S. (2005) Structureproperty studies on the antioxidant activity of flavonoids present in diet. Free Radical Biology and Medicine, 39, 1099-1108. doi:10.1016/j.freeradbiomed.2005.05.028
- Kerr, J.F., Wyllie, A.H. and Currie, A.R. (1972) Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer, 26, 239-257. doi:10.1038/bjc.1972.33
- Kurosaka, K., Takahashi, M., Watanabe, N. and Kobayashi, Y. (2003) Silent cleanup of very early apoptotic cells by macrophages. Journal of Immunology, 171, 4672-4679.
- Elmore, S. (2007) Apoptosis: A review of programmed cell death. Toxicologic Pathology, 35, 495-516. doi:10.1080/01926230701320337
- Denault, J.B. and Salvesen, G.S. (2002) Caspases: Keys in the ignition of cell death. Chemical Reviews, 102, 4489- 4500. doi:10.1021/cr010183n
- Wang, Z.B., Liu, Y.Q. and Cui, Y.F. (2005) Pathways to caspase activation. Cell Biology International, 29, 489-496. doi:10.1016/j.cellbi.2005.04.001
- Rai, N.K., Tripathi, K., Sharma, D. and Shukla, V.K. (2005) Apoptosis: A basic physiologic process in wound healing. International Journal of Lower Extremity Wounds, 4, 138- 144. doi:10.1177/1534734605280018
- Ziegler, D.S. and Kung, A.L. (2008) Therapeutic targeting of apoptosis pathways in cancer. Current Opinion in Oncology, 20, 97-103. doi:10.1097/CCO.0b013e3282f310f6
- [61] Bucur, O., Ray, S., Bucur, M.C. and Almasan, A. (2006) APO2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in prostate cancer therapy. Frontiers in Bioscience, 11, 1549-1568. doi:10.2741/1903
- [62] Xiang, H., Nguyen, C.B., Kelley, S.K., Dybdal, N. and Escandon, E. (2004) Tissue distribution, stability, and pharmacokinetics of Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in human colon carcinoma COLO205 tumor-bearing nude mice. Drug Metabolism and Disposition, 32, 1230-1238. doi:10.1124/dmd.104.000323
- [63] Wang, J., Chun, H.J., Wong, W., Spencer, D.M. and Lenardo, M.J. (2001) Caspase-10 is an initiator caspase in death receptor signaling. Proceedings of the National Academy of Science of the Uited States of America, 98, 13884- 13888. doi:10.1073/pnas.241358198
- [64] Ghobrial, I.M., Witzig, T.E. and Adjei, A.A. (2005) Targeting apoptosis pathways in cancer therapy. CA—A Cancer Journal for Clinicians, 55, 178-194. doi:10.3322/canjclin.55.3.178
- [65] Maldonado, V., Melendez-Zajgla, J. and Ortega, A. (1997) Modulation of NF-kappa B, and Bcl-2 in apoptosis induced by cisplatin in HeLa cells. Mutation Research, 381, 67-75. doi:10.1016/S0027-5107(97)00150-4
- [66] Chen, L.F. and Greene, W.C. (2004) Shaping the nuclear action of NF-kappaB. Nature Reviews Molecular Cell Biology, 5, 392-401. doi:10.1038/nrm1368
- [67] Stoffel, A., Chaurushiya, M., Singh, B. and Levine, A.J. (2004) Activation of NF-kappaB and inhibition of p53-mediated apoptosis by API2/mucosa-associated lymphoid tissue 1 fusions promote oncogenesis. Proceedings of the National Academy of Science of the Uited States of America, 101, 9079-9084. doi:10.1073/pnas.0402415101
- [68] Kuhnel, F., Zender, L., Paul, Y., Tietze, M.K., Trautwein, C., Manns, M. and Kubicka, S. (2000) NFkappaB mediates apoptosis through transcriptional activation of Fas (CD95) in adenoviral hepatitis. Journal of Biological Chemistry, 275, 6421-6427. doi:10.1074/jbc.275.9.6421
- [69] Cantley, L.C. (2002) The phosphoinositide 3-kinase pathway. Science, 296, 1655-1657. doi:10.1126/science.296.5573.1655
- [70] Vanhaesebroeck, B., Leevers, S.J., Ahmadi, K., Timms, J., Katso, R., Driscoll, P.C., Woscholski, R., Parker, P.J. and Waterfield, M.D. (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annual Review of Biochemistry, 70, 535-602. doi:10.1146/annurev.biochem.70.1.535
- [71] Datta, S.R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y. and Greenberg, M.E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell, 91, 231-241. doi:10.1016/S0092-8674(00)80405-5
- [72] del Peso, L., Gonzalez-Garcia, M., Page, C., Herrera, R. and Nunez, G. (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science, 278, 687-689. doi:10.1126/science.278.5338.687
- [73] Barkett, M. and Gilmore, T.D. (1999) Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene, 18, 6910-6924. doi:10.1038/sj.onc.1203238
- [74] Lauder, A., Castellanos, A. and Weston, K. (2001) c-Myb transcription is activated by protein kinase B (PKB) following interleukin 2 stimulation of Tcells and is required for PKB-mediated protection from apoptosis. Molecular and Cellular Biology, 21, 5797-5805. doi:10.1128/MCB.21.17.5797-5805.2001
- [75] Fresno Vara, J.A., Casado, E., de Castro, J., Cejas, P., Belda-Iniesta, C. and Gonzalez-Baron, M. (2004) PI3K/ Akt signalling pathway and cancer. Cancer Treatment Reviews, 30, 193-204. doi:10.1016/j.ctrv.2003.07.007
- [76] Saikumar, P., Dong, Z., Mikhailov, V., Denton, M., Weinberg, J.M. and Venkatachalam, M.A. (1999) Apoptosis: Definition, mechanisms, and relevance to disease. American Journal of Medicine, 107, 489-506. doi:10.1016/S0002-9343(99)00259-4
- [77] Wyllie, A.H., Bellamy, C.O., Bubb, V.J., Clarke, A.R., Corbet, S., Curtis, L., Harrison, D.J., Hooper, M.L., Toft, N., Webb, S. and Bird, C.C. (1999) Apoptosis and carcinogenesis. British Journal of Cancer, 80, 34-37.
- [78] Reed, J.C. (1999) Dysregulation of apoptosis in cancer. Journal of Clinical Oncology, 17, 2941-2953.
- [79] Zhivotovsky, B. and Orrenius, S. (2006) Carcinogenesis and apoptosis: Paradigms and paradoxes. Carcinogenesis, 27, 1939-1945. doi:10.1093/carcin/bgl035
- [80] Thompson, C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456-1462. doi:10.1126/science.7878464
- [81] Hanahan, D. and Weinberg, R.A. (2000) The hallmarks of cancer. Cell, 100, 57-70. doi:10.1016/S0092-8674(00)81683-9
- [82] Hueber, A.O. and Evan, G.I. (1998) Traps to catch unwary oncogenes. Trends in Genetics, 14, 364-367. doi:10.1016/S0168-9525(98)01520-0
- [83] Harris, A.L. (2002) Hypoxia—A key regulatory factor in tumour growth. Nature Reviews Cancer, 2, 38-47. doi:10.1038/nrc704
- [84] Wright, S.C., Zhong, J. and Larrick, J.W. (1994) Inhibition of apoptosis as a mechanism of tumor promotion. FASEB Journal, 8, 654-660.
- [85] Roth, W. and Reed, J.C. (2002) Apoptosis and cancer: When BAX is TRAILing away. Nature Medicine, 8, 216- 218. doi:10.1038/nm0302-216
- [86] Miyashita, T., Krajewski, S., Krajewska, M., Wang, H.G., Lin, H.K., Liebermann, D.A., Hoffman, B. and Reed, J.C. (1994) Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene, 9, 1799-1805.
- [87] Wang, X.W. (1999) Role of p53 and apoptosis in carcinogenesis. Anticancer Research, 19, 4759-4771.
- [88] Bourdon, J.C. (2007) p53 and its isoforms in cancer. British Journal of Cancer, 97, 277-282. doi:10.1038/sj.bjc.6603886
- [89] Key, T.J., Schatzkin, A., Willett, W.C., Allen, N.E., Spencer, E.A. and Travis, R.C. (2004) Diet, nutrition and the prevention of cancer. Public Health Nutrition, 7, 187-200. doi:10.1079/PHN2003588
- [90] Gonzalez, C.A. (2006) Nutrition and cancer: The current epidemiological evidence. British Journal of Nutrition, 96, 42-45. doi:10.1079/BJN20061699
- [91] Sporn, M.B., Dunlop, N.M., Newton, D.L. and Smith, J.M. (1976) Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). Federation Proceedings, 35, 1332-1338.
- [92] Bouchier-Hayes, L., Lartigue, L. and Newmeyer, D.D. (2005) Mitochondria: pharmacological manipulation of cell death. Journal of Clinical Investigation, 115, 2640-2647. doi:10.1172/JCI26274
- [93] Kelloff, G.J., Sigman, C.C. and Greenwald, P. (1999) Cancer chemoprevention: Progress and promise. European Journal of Cancer, 35, 2031-2038. doi:10.1016/S0959-8049(99)00299-3
- [94] Kakizoe, T. (2003) Chemoprevention of cancer—Focusing on clinical trials. Japanese Journal of Clinical Oncology, 33, 421-442. doi:10.1093/jjco/hyg090
- [95] Luqman, S. and Pezzuto, J.M. (2010) NFkappaB: A promising target for natural products in cancer chemo-prevention. Phytotherapy Research: PTR, 24, 949-963.
- [96] Karin, M. (2006) Nuclear factor-kappaB in cancer development and progression. Nature, 441, 431-436. doi:10.1038/nature04870
- [97] Crowell, J.A., Steele, V.E. and Fay, J.R. (2007) Targeting the AKT protein kinase for cancer chemoprevention. Molecular Cancer Therapeutics, 6, 2139-2148. doi:10.1158/1535-7163.MCT-07-0120
- [98] Yang, C.S., Maliakal, P. and Meng, X. (2002) Inhibition of carcinogenesis by tea. Annual Review of Pharmacology and Toxicology, 42, 25-54. doi:10.1146/annurev.pharmtox.42.082101.154309
- [99] Ahmad, N., Feyes, D.K., Nieminen, A.L., Agarwal, R. and Mukhtar, H. (1997) Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. Journl of the National Cancer Institute, 89, 1881-1886. doi:10.1093/jnci/89.24.1881
- [100] Albrecht, D.S., Clubbs, E.A., Ferruzzi, M. and Bomser, J.A. (2008) Epigallocatechin-3-gallate (EGCG) inhibits PC-3 prostate cancer cell proliferation via MEK-independent ERK1/2 activation. Chemico-Biological Interactions, 171, 89-95. doi:10.1016/j.cbi.2007.09.001
- [101] Butt, M.S. and Sultan, M.T. (2009) Green tea: Nature’s defense against malignancies. Critical Reviews in Food Science and Nutrition, 49, 463-473. doi:10.1080/10408390802145310
- [102] Khan, N. and Mukhtar, H. (2008) Multitargeted therapy of cancer by green tea polyphenols. Cancer Letters, 269, 269- 280. doi:10.1016/j.canlet.2008.04.014
- [103] Shimizu, M., Shirakami, Y. and Moriwaki, H. (2008) Targeting receptor tyrosine kinases for chemoprevention by green tea catechin, EGCG. International Journal of Molecular Sciences, 9, 1034-1049. doi:10.3390/ijms9061034
- [104] Hastak, K., Gupta, S., Ahmad, N., Agarwal, M.K., Agarwal, M.L. and Mukhtar, H. (2003) Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene, 22, 4851-4859. doi:10.1038/sj.onc.1206708
- [105] Chung, L.Y., Cheung, T.C., Kong, S.K., Fung, K.P., Choy, Y.M., Chan, Z.Y. and Kwok, T.T. (2001) Induction of apoptosis by green tea catechins in human prostate cancer DU145 cells. Life Sciences, 68, 1207-1214. doi:10.1016/S0024-3205(00)01020-1
- [106] Ahmad, N., Gupta, S. and Mukhtar, H. (2000) Green tea polyphenol epigallocatechin-3-gallate differentially modulates nuclear factor kappaB in cancer cells versus normal cells. Archives of Biochemistry and Biophys, 376, 338-346. doi:10.1006/abbi.2000.1742
- [107] Afaq, F., Adhami, V.M., Ahmad, N. and Mukhtar, H. (2003) Inhibition of ultraviolet B-mediated activation of nuclear factor κB in normal human epidermal keratinocytes by green tea Constituent (-)-epigallocatechin-3-gallate. Oncogene, 22, 1035-1044. doi:10.1038/sj.onc.1206206
- [108] Suganuma, M., Sueoka, E., Sueoka, N., Okabe, S. and Fujiki, H. (2000) Mechanisms of cancer prevention by tea polyphenols based on inhibition of TNF-alpha expression. Biofactors, 13, 67-72. doi:10.1002/biof.5520130112
- [109] Qin, J., Xie, L.P., Zheng, X.Y., Wang, Y.B., Bai, Y., Shen, H.F., Li, L.C. and Dahiya, R. (2007) A component of green tea, (-)-epigallocatechin-3-gallate, promotes apoptosis in T24 human bladder cancer cells via modulation of the PI3K/Akt pathway and Bcl-2 family proteins. Biochemical and Biophysical Research Communications, 354, 852-857. doi:10.1016/j.bbrc.2007.01.003
- [110] Rodriguez, S.K., Guo, W., Liu, L., Band, M.A., Paulson, E.K. and Meydani, M. (2006) Green tea catechin, epigallocatechin-3-gallate, inhibits vascular endothelial growth factor angiogenic signaling by disrupting the formation of a receptor complex. International Journal of Cancer, 118, 1635-1644. doi:10.1002/ijc.21545
- [111] Tang, F.Y., Nguyen, N. and Meydani, M. (2003) Green tea catechins inhibit VEGF-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt molecule. International Journal of Cancer, 106, 871-878. doi:10.1002/ijc.11325
- [112] Masuda, M., Suzui, M., Lim, J.T., Deguchi, A., Soh, J.W. and Weinstein, I.B. (2002) Epigallocatechin-3-gallate decreases VEGF production in head and neck and breast carcinoma cells by inhibiting EGFR-related pathways of signal transduction. Journal of Experimental Therapeutics and Oncology, 2, 350-359. doi:10.1046/j.1359-4117.2002.01062.x
- [113] Gupta, S., Hussain, T. and Mukhtar, H. (2003) Molecular pathway for (-)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Archives of Biochemistry and Biophysics, 410, 177-185. doi:10.1016/S0003-9861(02)00668-9
- [114] Shankar, S., Suthakar, G. and Srivastava, R.K. (2007) Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Frontiers in Bioscience, 12, 5039-5051. doi:10.2741/2446
- [115] Kuo, P.L., Lin, C.C. (2003) Green tea constituent (-)-epigallocatechin-3-gallate inhibits Hep G2 cell proliferation and induces apoptosis through p53-dependent and Fasmediated pathways. Journal of Biomedical Science, 10, 219-227.
- [116] Ju, J., Hong, J., Zhou, J.N., Pan, Z., Bose, M., Liao, J., Yang, G.Y., Liu, Y.Y., Hou, Z., Lin, Y., Ma, J., Shih, W.J., Carothers, A.M. and Yang, C.S. (2005) Inhibition of intestinal tumorigenesis in Apcmin/+ mice by (-)-epigallocatechin-3-gallate, the major catechin in green tea. Cancer Research, 65, 10623-10631. doi:10.1158/0008-5472.CAN-05-1949
- [117] Shimizu, M., Shirakami, Y., Sakai, H., Adachi, S., Hata, K., Hirose, Y., Tsurumi, H., Tanaka, T. and Moriwaki, H. (2008) (-)-Epigallocatechin gallate suppresses azoxymethane-induced colonic premalignant lesions in male C57BL/KsJdb/db mice. Cancer Prevention Research (Phila), 1, 298- 304. doi:10.1158/1940-6207.CAPR-08-0045
- [118] Shimizu, M., Sakai, H., Shirakami, Y., Yasuda, Y., Kubota, M., Terakura, D., Baba, A., Ohno, T., Hara, Y., Tanaka, T. and Moriwaki, H. (2011) Preventive effects of (-)-epigallocatechin gallate on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db Mice. Cancer Prevention Research (Phila), 4, 396-403. doi:10.1158/1940-6207.CAPR-10-0331
- [119] Ohga, N., Hida, K., Hida, Y., Muraki, C., Tsuchiya, K., Matsuda, K., Ohiro, Y., Totsuka, Y. and Shindoh, M. (2009) Inhibitory effects of epigallocatechin-3 gallate, a polyphenol in green tea, on tumor-associated endothelial cells and endothelial progenitor cells. Cancer Science, 100, 1963- 1970. doi:10.1111/j.1349-7006.2009.01255.x
- [120] Rieger-Christ, K.M., Hanley, R., Lodowsky, C., Bernier, T., Vemulapalli, P., Roth, M., Kim, J., Yee, A.S., Le, S.M., Marie, P.J., Libertino, J.A. and Summerhayes, I.C. (2007) The green tea compound, (-)-epigallocatechin-3-gallate downregulates N-cadherin and suppresses migration of bladder carcinoma cells. Journal of Cellular Biochemistry, 102, 377-388. doi:10.1002/jcb.21299
- [121] Aggarwal, B.B., Kumar, A. and Bharti, A.C. (2003) Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Research, 23, 363-398.
- [122] Bharti, A.C., Donato, N., Singh, S. and Aggarwal, B.B. (2003) Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood, 101, 1053-1062. doi:10.1182/blood-2002-05-1320
- [123] Hussain, A.R., Ahmed, M., Al-Jomah, N.A., Khan, A.S., Manogaran, P., Sultana, M., Abubaker, J., Platanias, L.C., Al-Kuraya, K.S. and Uddin, S. (2008) Curcumin suppresses constitutive activation of nuclear factor-kappa B and requires functional Bax to induce apoptosis in Burkitt’s lymphoma cell lines. Molecular Cancer Therapeutics, 7, 3318-3329. doi:10.1158/1535-7163.MCT-08-0541
- [124] Kunnumakkara, A.B., Diagaradjane, P., Guha, S., Deorukhkar, A., Shentu, S., Aggarwal, B.B. and Krishnan, S. (2008) Curcumin sensitizes human colorectal cancer xenografts in nude mice to gamma-radiation by targeting nuclear factor-kappaB-regulated gene products. Clinical Cancer Research, 14, 2128-2136. doi:10.1158/1078-0432.CCR-07-4722
- [125] Wang, D., Veena, M.S., Stevenson, K., Tang, C., Ho, B., Suh, J.D., Duarte, V.M., Faull, K.F., Mehta, K., Srivatsan, E.S. and Wang, M.B. (2008) Liposome-encapsulated curcumin suppresses growth of head and neck squamous cell carcinoma in vitro and in xenografts through the inhibittion of nuclear factor kappaB by an AKT-independent pathway. Clinical Cancer Research, 14, 6228-6236. doi:10.1158/1078-0432.CCR-07-5177
- [126] Al-Hujaily, E.M., Mohamed, A.G., Al-Sharif, I., Youssef, K.M., Manogaran, P.S., Al-Otaibi, B., Al-Haza’a, A., AlJammaz, I., Al-Hussein, K. and Aboussekhra, A. (2011) PAC, a novel curcumin analogue, has anti-breast cancer properties with higher efficiency on ER-negative cells. Breast Cancer Research and Treatment, 128, 97-107. doi:10.1007/s10549-010-1089-3
- [127] Dhillon, N., Aggarwal, B.B., Newman, R.A., Wolff, R.A., Kunnumakkara, A.B., Abbruzzese, J.L., Ng, C.S., Badmaev, V. and Kurzrock, R. (2008) Phase II trial of curcumin in patients with advanced pancreatic cancer. Clinical Cancer Research, 14, 4491-4499. doi:10.1158/1078-0432.CCR-08-0024
- [128] Yu, S., Shen, G., Khor, T.O., Kim, J.H. and Kong, A.N. (2008) Curcumin inhibits Akt/mammalian target of rapamycin signaling through protein phosphatase-dependent mechanism. Molecular Cancer Therapeutics, 7, 2609-2620. doi:10.1158/1535-7163.MCT-07-2400
- [129] Clark, C.A., McEachern, M.D., Shah, S.H., Rong, Y., Rong, X., Smelley, C.L., Caldito, G.C., Abreo, F.W. and Nathan, C.O. (2010) Curcumin inhibits carcinogen and nicotine-induced Mammalian target of rapamycin pathway activation in head and neck squamous cell carcinoma. Cancer Prevention Research (Phila), 3, 1586-1595. doi:10.1158/1940-6207.CAPR-09-0244
- [130] Weir, N.M., Selvendiran, K., Kutala, V.K., Tong, L., Vishwanath, S., Rajaram, M., Tridandapani, S., Anant, S. and Kuppusamy, P. (2007) Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biology and Therapy, 6, 178-184. doi:10.4161/cbt.6.2.3577
- [131] Kumar, A.P., Garcia, G.E., Ghosh, R., Rajnarayanan, R.V., Alworth, W.L. and Slaga, T.J. (2003) 4-Hydroxy-3-methoxy-benzoic acid methyl ester: A curcumin derivative targets Akt/NF kappa B cell survival signaling pathway: Potential for prostate cancer management. Neoplasia, 5, 255- 266.
- [132] Squires, M.S., Hudson, E.A., Howells, L., Sale, S., Houghton, C.E., Jones, J.L., Fox, L.H., Dickens, M., Prigent, S.A. and Manson, M.M. (2003) Relevance of mitogen activated protein kinase (MAPK) and phosphotidy-linositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoptosis by curcumin in breast cells. Biochemical Pharmacology, 65, 361-376. doi:10.1016/S0006-2952(02)01517-4
- [133] Woo, J.H., Kim, Y.H., Choi, Y.J., Kim, D.G., Lee, K.S., Bae, J.H., Min, D.S., Chang, J.S., Jeong, Y.J., Lee, Y.H., Park, J.W. and Kwon, T.K. (2003) Molecular mechanisms of curcumin-induced cytotoxicity: Induction of apoptosis through generation of reactive oxygen species, down-regulation of Bcl-XL and IAP, the release of cytochrome c and inhibition of Akt. Carcinogenesis, 24, 1199-1208. doi:10.1093/carcin/bgg082
- [134] Subramaniam, D., May, R., Sureban, S.M., Lee, K.B., George, R., Kuppusamy, P., Ramanujam, R.P., Hideg, K., Dieckgraefe, B.K., Houchen, C.W. and Anant, S. (2008) Diphenyl difluoroketone: A curcumin derivative with potent in vivo anticancer activity. Cancer Research, 68, 1962- 1969. doi:10.1158/0008-5472.CAN-07-6011
- [135] Purkayastha, S., Berliner, A., Fernando, S.S., Ranasinghe, B., Ray, I., Tariq, H. and Banerjee, P. (2009) Curcumin Blocks Brain Tumor Formation. Brain Research, 1266, 130-138. doi:10.1016/j.brainres.2009.01.066
- [136] Deschner, E.E., Ruperto, J., Wong, G. and Newmark, H.L. (1991) Quercetin and rutin as inhibitors of azoxymethanolinduced colonic neoplasia. Carcinogenesis, 12, 1193-1196. doi:10.1093/carcin/12.7.1193
- [137] De, S., Chakraborty, R.N., Ghosh, S., Sengupta and A., Das, S. (2004) Comparative evaluation of cancer chemopreventive efficacy of alpha-tocopherol and quercetin in a murine model. Journal of Experimental and Clinical Cancer Research, 23, 251-258.
- [138] Volate, S.R., Davenport, D.M., Muga, S.J. and Wargovich, M.J. (2005) Modulation of aberrant crypt foci and apoptosis by dietary herbal supplements (quercetin, curcumin, silymarin, ginseng and rutin). Carcinogenesis, 26, 1450- 1456. doi:10.1093/carcin/bgi089
- [139] Cruz-Correa, M., Shoskes, D.A., Sanchez, P., Zhao, R., Hylind, L.M., Wexner, S.D. and Giardiello, F.M. (2006) Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clinical Gastroenterology and Hepatology, 4, 1035-1038. doi:10.1016/j.cgh.2006.03.020
- [140] Kawanishi, S., Oikawa, S. and Murata, M. (2005) Evaluation for safety of antioxidant chemopreventive agents. Antioxidants and Redox Signaling, 7, 1728-1739. doi:10.1089/ars.2005.7.1728
- [141] Kim, B.M., Choi, Y.J., Han, Y., Yun, Y.S. and Hong, S.H. (2009) N,N-dimethyl phytosphingosine induces caspase-8- dependent cytochrome c release and apoptosis through ROS generation in human leukemia cells. Toxicology and Applied Pharmacology, 239, 87-97. doi:10.1016/j.taap.2009.05.020
- [142] Jung, Y.H., Heo, J., Lee, Y.J., Kwon, T.K. and Kim, Y.H. (2010) Quercetin enhances TRAIL-induced apoptosis in prostate cancer cells via increased protein stability of death receptor 5. Life Sciences, 86, 351-357. doi:10.1016/j.lfs.2010.01.008
- [143] Lee, D.H., Szczepanski, M. and Lee, Y.J. (2008) Role of Bax in quercetin-induced apoptosis in human prostate cancer cells. Biochemical Pharmacology, 75, 2345-2355. doi:10.1016/j.bcp.2008.03.013
- [144] Senthilkumar, K., Elumalai, P., Arunkumar, R., Banudevi, S., Gunadharini, N.D., Sharmila, G., Selvakumar, K. and Arunakaran, J. (2010) Quercetin regulates insulin like growth factor signaling and induces intrinsic and extrinsic pathway mediated apoptosis in androgen independent prostate cancer cells (PC-3). Molecular and Cellular Biochemistry, 344, 173-184. doi:10.1007/s11010-010-0540-4
- [145] Kim, Y.H. and Lee, Y.J. (2007) TRAIL apoptosis is enhanced by quercetin through Akt dephosphorylation. Journal of Cellular Biochemistry, 100, 998-1009. doi:10.1002/jcb.21098
- [146] Russo, M., Palumbo, R., Tedesco, I., Mazzarella, G., Russo, P., Iacomino, G. and Russo, G.L. (1999) Quercetin and anti-CD- 95(Fas/Apo1) enhance apoptosis in HPB-ALL cell line. FEBS Letters, 462, 322-328. doi:10.1016/S0014-5793(99)01544-6
- [147] Ishikawa, Y. and Kitamura, M. (2000) Anti-apoptotic effect of quercetin: Intervention in the JNKand ERK-mediated apoptotic pathways. Kidney International, 58, 1078-1087. doi:10.1046/j.1523-1755.2000.00265.x
- [148] Sun, Z.J., Chen, G., Hu, X., Zhang, W., Liu, Y., Zhu, L.X., Zhou, Q. and Zhao, Y.F. (2010) Activation of PI3K/Akt/ IKK-alpha/NF-kappaB signaling pathway is required for the apoptosis-evasion in human salivary adenoid cystic carcinoma: its inhibition by quercetin. Apoptosis: An International Journal on Programmed Cell Death, 15, 850- 863.
- [149] Scandlyn, M.J., Stuart, E.C., Somers-Edgar, T.J., Menzies, A.R. and Rosengren, R.J. (2008) A new role for tamoxifen in oestrogen receptor-negative breast cancer when it is combined with epigallocatechin gallate. British Journal of Cancer, 99, 1056-1063. doi:10.1038/sj.bjc.6604634
- [150] Sartippour, M.R., Pietras, R., Marquez-Garban, D.C., Chen, H.W., Heber, D., Henning, S.M., Sartippour, G., Zhang, L., Lu, M., Weinberg, O., Rao, J.Y. and Brooks, M.N. (2006) The combination of green tea and tamoxifen is effective against breast cancer. Carcinogenesis, 27, 2424-2433. doi:10.1093/carcin/bgl066
- [151] Somers-Edgar, T.J., Scandlyn, M.J., Stuart, E.C., Le Nedelec, M.J., Valentine, S.P. and Rosengren, R.J. (2008) The combination of epigallocatechin gallate and curcumin suppresses ER alpha-breast cancer cell growth in vitro and in vivo. International Journal of Cancer, 122, 1966-1971. doi:10.1002/ijc.23328
- [152] Kunnumakkara, A.B., Guha, S., Krishnan, S., Diagaradjane, P., Gelovani, J. and Aggarwal, B.B. (2007) Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Research, 67, 3853-3861. doi:10.1158/0008-5472.CAN-06-4257
- [153] Bisht, S., Mizuma, M., Feldmann, G., Ottenhof, N.A., Hong, S.M., Pramanik, D., Chenna, V., Karikari, C., Sharma, R., Goggins, M.G., Rudek, M.A., Ravi and R., Maitra, A. (2010) Systemic administration of polymeric nanoparticle-encapsulated curcumin (NanoCurc) blocks tumor growth and metastases in preclinical models of pancreatic cancer. Molecular Cancer Therapeutics, 9, 2255-2264. doi:10.1158/1535-7163.MCT-10-0172
- [154] Zhou, W., Kallifatidis, G., Baumann, B., Rausch, V., Mattern, J., Gladkich, J., Giese, N., Moldenhauer, G., Wirth, T., Buchler, M.W., Salnikov, A.V. and Herr, I. (2010) Dietary polyphenol quercetin targets pancreatic cancer stem cells. International Journal of Oncology, 37, 551-561.
- [155] Narayanan, N.K., Nargi, D., Randolph, C. and Narayanan, B.A. (2009) Liposome encapsulation of curcumin and resveratrol in combination reduces prostate cancer incidence in PTEN knockout mice. International Journal of Cancer, 125, 1-8. doi:10.1002/ijc.24336
- [156] Priego, S., Feddi, F., Ferrer, P., Mena, S., Benlloch, M., Ortega, A., Carretero, J., Obrador, E., Asensi, M. and Estrela, J.M. (2008) Natural polyphenols facilitate elimination of HT-29 colorectal cancer xenografts by chemoradiotherapy: A Bcl-2- and superoxide dismutase 2-dependent mechanism. Molecular Cancer Therapeutics, 7, 3330-3342. doi:10.1158/1535-7163.MCT-08-0363
- [157] Sreekanth, C.N., Bava, S.V., Sreekumar, E. and Anto, R.J. (2011) Molecular evidences for the chemosensitizing efficacy of liposomal curcumin in paclitaxel chemotherapy in mouse models of cervical cancer. Oncogene, 30, 3139- 3152. doi:10.1038/onc.2011.23
- [158] Duarte, V.M., Han, E., Veena, M.S., Salvado, A., Suh, J.D., Liang, L.J., Faull, K.F., Srivatsan, E.S. and Wang, M.B. (2010) Curcumin enhances the effect of cisplatin in suppression of head and neck squamous cell carcinoma via inhibition of IKKbeta protein of the NFkappaB pathway. Molecular Cancer Therapeutics, 9, 2665-2675. doi:10.1158/1535-7163.MCT-10-0064
- [159] Nautiyal, J., Banerjee, S., Kanwar, S.S., Yu, Y., Patel, B.B., Sarkar, F.H. and Majumdar, A.P. (2011) Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. International Journal of Cancer, 128, 951-961. doi:10.1002/ijc.25410
- [160] Reed, J.C. (2001) Apoptosis-regulating proteins as targets for drug discovery. Trends in Molecular Medicine, 7, 314-319. doi:10.1016/S1471-4914(01)02026-3
- [161] Willett, W.C. (1995) Diet, nutrition, and avoidable cancer. Environmental Health Perspectives, 103, 165-170.
- [162] Howells, L.M. and Manson, M.M. (2005) Prospects for plant-derived chemopreventive agents exhibiting multiple mechanisms of action. Current Medicinal Chemistry—AntiCancer Agents, 5, 201-213. doi:10.2174/1568011053765921
NOTES
#The authors declare no conflict of interest.