Advances in Chemical Engineering and Science
Vol.07 No.01(2017), Article ID:73614,15 pages
10.4236/aces.2017.71007
Hydrothermal Dissolution of Opal in Sodium Hydroxide Lyes for the Synthesis of Water Glass
Thomas Pfeiffer1,2, Dirk Enke3, Robert Roth4, Hans Roggendorf4*
1Institute of Physics, Martin-Luther-University Halle-Wittenberg, Halle, Germany
2OPTERRA Zement GmbH, Karsdorf, Germany
3Institute of Chemical Technology, University of Leipzig, Leipzig, Germany
4Institute of Physics, Martin-Luther-University Halle-Wittenberg, Halle, Germany

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: December 13, 2016; Accepted: January 16, 2017; Published: January 19, 2017
ABSTRACT
Common opal was dissolved in NaOH lyes in rotating autoclaves. The starting material was characterized by X-ray diffraction and adsorption spectroscopy, thermal and chemical analysis, electron and atomic force microscopy. The opal proved to be an Opal-CT with a microstructure consisting of microcrystalline tridymite, traces of low-quartz, and amorphous parts built up by random packings of size distributed amorphous silica colloids. The dissolution conditions have been inspired by the technological process of hydrothermal water glass synthesis by dissolving silica. Temperature and time of the dissolution process as well as initial molar SiO2:Na2O (Rm) ratios of the starting materials were varied systematically. The particle size of the samples was varied, too, but due to the nanoscopic microstructure without greater impact on dissolution kinetics. The process products were analyzed chemically. Additionally, some of them were characterized by X-ray diffraction, viscosimetry and dynamic light scattering. Already after short dissolution times, water glasses with quite high silica concentrations of up to 27 wt.% and SiO2:Na2O ratios of up to 3.7 were obtained. At longer dissolution times low-quartz and analcime precipitated and the SiO2 contents were reduced to about 22 wt.% and Rm to about 2.7. The silica contents in equilibrium with low-quartz were almost independent on temperature.
Keywords:
Water Glass, Opal, Silica, Hydrothermal Dissolution

1. Introduction
The term water glass not only describes alkali-rich silicate glasses but also the liquid phases obtained either when these glasses are dissolved into water or when a silica phase is dissolved in alkaline lyes. Commercially available liquid sodium water glasses have compositions characterised by a molar SiO2:Na2O ratio (Rm) of 1.8 to 4 (ratios of 2.0 and 3.3 are standard compositions) and a SiO2 content of 25 wt.% to 30 wt.% (Weldes and Lange [1] ).
The hydrothermal dissolution of silica materials in sodium lyes is one of the two standard methods for the production of water glasses. The concurring method is dissolving alkaline silicate glasses in H2O at elevated temperatures up to 150˚C. The hydrothermal dissolution of silica is usually restricted to compositions with Rm < 2.7 [1] . Possibly, these concentration limitations are due to chemical saturation. Higher Rm values can be obtained when raw materials with higher reactivities like cristobalite or vitreous silica are used. Hydrothermal dissolution of silica materials requires less energy and thus causes lower CO2 output during production.
The solubility of silica phases is an important topic of silicate chemistry and geochemistry. Solubility of silica at higher pH and temperatures is also important for process technologies concerned with the production of liquid water glasses. The older literature on the solubility of silica was evaluated by Iler [2] . The solubility of silica depends on the solid equilibrium phase coexisting with the aqueous liquid, as well as on temperature, pressure, and pH of the dissolution process and on the particle size of the dissolving silica phase. Iler [2] refers to the work of Stöber [3] that at nearly neutral conditions the solubility of silica decreases in the order of vitreous silica > stishovite > cristobalite > tridymite > quartz > coesite.
The conditions of hydrothermal dissolution in sodium hydroxide comprise elevated temperatures (above 100˚C) and pressures (Dove et al. [4] ) as well as high pH values. Temperature and pressure enhance solubility of silica, whereas the effects of pressure are in most cases not separated from temperature. Fleming et al. [5] calculated the silica solubility at temperatures up to 300˚C and pH values up to 12 in equilibrium with quartz or amorphous silica by thermodynamic means and reported solubilities up to 3 mole SiO2/l. Further results for higher temperatures and pressures are found in publications connected with geochemistry (Gunnarsson et al. [6] ), or in phase diagrams (e.g. Tuttle et al. [7] ).
Opals are minerals with silica as main component. They can be regarded as an impure silica source with a higher reactivity than ordinary quartz or even vitreous silica. According to Iler [2] the term “opal” covers many types of hydrated amorphous silicas. Amorphous means here the absence of sharp X-ray diffraction peaks. But the presence of submicroscopic crystallites like cristobalite or tridymite is possible. Opals were classified by Jones and Segnit [8] according to their structure:
Opal-A: Amorphous materials of either monosized silica colloids forming an ordered microstructure (precious opal) or size distributed statistically packed colloids (sometimes called potch). The ordered microstructures resemble crystal lattices and may serve as Bragg lattices for optical light, making them suitable as gemstones.
Opal-CT: Partly microcrystalline materials which contain predominantly cristobalite in an amorphous matrix.
Opal-C: To a higher degree microcrystalline material with cristobalite and/or tridy- mite. Opal-CT and opal C are also termed common opal.
Flörke et al. [9] reviewed the nomenclature of micro- and non-crystalline silica minerals. According to their data H2O contents of up to 12 wt.% can be incorporated into opals either as hydroxide groups or as molecular water. Opals may contain―besides silica―other hydrated oxides like Al2O3 or Fe2O3.
The dissolution of opal was treated in a paper by Abakov et al. [10] . They dissolved an opal with 88 wt.% silica that possibly contained microcrystalline cristobalite at temperatures up to 175˚C in 1 M NaOH and compared it with results obtained when dissolving other silica sources or alumosilicates under the same conditions. In one test with opal a SiO2 concentration of 10.75 g SiO2/l was reached. The authors explain the high solubility of opal by its nanoscaled microstructure. Ordiales [11] patented the hydrothermal synthesis of sodium water glasses from opals. He achieved a Rm value of 3.3 at a water vapour pressure of 455,000 Pa (»150˚C).
The objectives of the work presented here were to use opal as a model substance for impure but reactive silica sources like diatomite, flint stone, opal or biogenic silica (e.g. rice husk ash) to investigate their dissolution behaviour under the conditions of water glass production. Since Rm values up to 2.7 (e.g. Weldes et al. [1] ) have been reached by hydrothermal quartz dissolution the investigations presented here aimed at Rm > 3. Additionally, it was intended to discuss the results in terms of chemical solubility to close the gap between solubility studies performed in the context of geochemistry (e.g. [5] [6] ) and the high silica concentration observed in sodium water glasses.
2. Experimental Study
A common opal from Minas Gerais, Brazil, was used as silica source. The opal materials were processed and characterized by the following means:
・ The samples were washed with water containing tensides as washing agents, dried at 150˚C, rinsed again with pure water and dried again. Then the samples were crushed and ground in an agate beaker with a planetary ball mill (Retsch S 1000, Retsch, Haan, Germany) and magnetically freed from abraded steel particles.
・ The density of bulk opal was measured by applying Archimedes principle.
・ For sieving a vibratory sieve shaker analysette 3 (Fritzsch, Idar-Oberstein, Germany) was applied. The ground opal was classified into different size fractions. Opal powders with diameters < 90 μm and opal grains with sizes between 250 and 500 μm were used for standard dissolution tests.
・ Chemical analysis was done by X-ray fluorescence analysis (Siemens SRS 3000, Siemens, Munich, Germany, now: Bruker AXS GmbH, Karlsruhe, Germany).
・ The H2O content was determined by thermal analysis (STA 409, Netzsch, Selb, Germany) up to 1400˚C with a heating rate of 10 K/min.
・ The H2O content was characterized also by FT-IR spectrometry (IFS 66, Bruker, Karlsruhe, Germany).
・ The structure of the materials was analysed by X-ray diffraction analysis (URD 63, FPM Freiberg, Freiberg, Germany; CuKα radiation).
・ The surface of the particles was investigated by environmental scanning electron microscopy (ESEM XL 30 FEG, Philips, Eindhoven, Netherlands).
・ Fracture surfaces of samples with a size of 1.2 × 1.5 × 0.1 cm3 before breaking were investigated by atomic force microscopy using an Asylum Research MFP-3D (Oxford Instruments, Santa Barbara, CA, USA) with NSC15-tips (Mikro Masch) in tapping/semicontact mode to get on the one hand surface topography and on the other hand structural surface information, too.
・ The internal porosity was characterized by low-temperature nitrogen adsorption (Sorptomatic 1990, Thermo Scientific, Waltham, MA, USA).
Several dissolution methods like heating the batches in pressure tight containers placed in ovens or heating baths with and without stirring were tried. A few experiments at 30˚C to 100˚C were performed with PFA (perfluoro alkoxy alkanes) vessels with a volume of 125 ml which were magnetically stirred inside a heating bath. The most promising method was the vertical rotation of digestion vessels inside a modified cabinet dryer (WTB ED 53, Binder, Germany), which was used for temperatures above 100˚C. The rotation speed of the vessels was 25 rounds per minute. Two different types of digestion vessels were used, each with a volume of 125 ml: PTFE (polytetrafluoro ethylene) lined steel bombs (type: Parr acid digestion bomb, Bomb No. 4748, company: Parr Instrument GmbH, Frankfurt, Germany) for temperatures ³ 160˚C and PFA vessels (Savillex, Eden Prairie, MN, USA) for lower temperatures.
Dissolution temperatures between 30˚C and 220˚C were applied and the dissolution times ranged from 1 to 20 h. The experiments started at room temperature and ended by rapid cooling by a forced cool air flow inside the drying cabinet. The dissolution time was corrected with respect to a dead time which is due to lower reactivity during heating and cooling the reactors. The dead times ranged between 25 and 90 minutes, depending on temperature, and were determined by evaluation of time dependent dissolution runs.
The following starting materials were used:
・ Common opal (Minas Gerais, Brazil, delivered by Rheinischer Mineralienkontor Krantz, Bonn, Germany).
・ Deionized water.
・ Solid NaOH (p. a. grade; Carl Roth, Karlsruhe, Germany) for the first experiments.
・ Concentrated NaOH lye (caustic soda lye, Akzo Nobel Industrial Chemicals, Bitterfeld, Germany, purity according to [12] ) with a nominal NaOH content of 50 wt.%. The concentration of this NaOH brine was checked by titration with 0.5 M HCl and proved to be 49.5 wt.%.
The following batches have been applied:
・ A: Batch consisting of 5 g opal, 4 g NaOH brine and 11 g H2O. The particle size was <90 μm. Considering the silica content of the applied opal a maximal Rm value = 3 would have been obtained.
・ B: Like A, but with increased opal content of 6.5 g (NaOH and H2O contents remained constant). The particle size was <90 μm. The batch corresponded to Rm = 3.9.
・ C: Like B, but with larger opal particles with a size range from 250 to 500 μm.
・ D: In dissolution tests at 160˚C the Rm value was varied between 2.4 and 6.7. The batch contained 8 g NaOH, 22 g H2O and 8 to 20 g opal with sizes < 90 μm.
・ E: Like D, but with 8 to 20 g opal with sizes between 250 and 500 μm.
The batch size was 20 g, for some investigation larger batches of up to 60 g were used, e. g. to obtain samples for viscosity measurements. The tightness of the containers was checked by weighing before and after thermal treatment. After cooling down to room temperature a liquid phase and a residue were separated by vacuum filtration. Pressures between 30 and 50 kPa were applied for nearly quantitative separation without significant evaporation of H2O. The aim of vacuum filtration was to interrupt ongoing dissolution reactions of finely dispersed silica materials directly after the dissolution tests. A possible concentration change due to evaporation was checked by chemical analysis of a sample before and after vacuum filtration: the silica content of a sample dissolved 20 h at 160˚C (batch C) changed from 25.6 wt.% to 25.9 wt.%, whereas the Na2O content remained constant.
The obtained liquids were characterized by:
・ Titration with 0.5 mole/l hydrochloric acid to analyze the alkaline content (as Na2O).
・ Calcination of the liquid phase by stepwise heating via 100˚C and 400˚C to 900˚C with holding times of 2 h to determine the residual solids content (RSC). The samples were mixed with calcined quartz sand in order to avoid explosive steam evolution. The difference (RSC minus Na2O-content) is equal to the nonalkaline oxide content of the solution if nonoxidic impurities can be neglected.
・ Detailed chemical analysis of two dissolved samples with inductively coupled plasma mass spectrometry (ICP-MS, ELAN 9000, PerkinElmer, Waltham, MA, USA).
・ Since alkali oxides other than Na2O could be neglected and the RSC consisted mainly of silica the obtained data could be applied to calculate SiO2:Na2O ratios of the hydrothermally obtained water glasses. The nonalkaline oxide content was then named SiO2 content for convenience.
・ Density with a pycnometer (Marienfeld, Lauda-Königshofen, Germany).
・ Viscosity (Ubbelohde viscosimeter, Labor-Therm, Germany) and refractive index (Abbe-60-Refractometer, Bellingham + Stanley Ltd., Royal Tunbridge Wells, UK).
・ The size colloidal distributions of two samples were characterized with dynamic light scattering (ALV-5000 EPP, ALV, Langen, Germany) after filtration with a 0.45 μm filter.
The solid residues were washed with H2O to remove alkali species which might react with CO2 and dried gently at 30˚C. Then they were characterized by:
・ X-ray powder diffraction (D8 Advanced X-Ray Diffractometer, Bruker AXS GmbH, Germany).
3. Results
3.1. Characterization of Opal
The investigated opal was supplied in fist sized pieces with translucent yellowish appearance. Brown veins were separating brighter parts. Figure 1 shows a picture of a specimen. The chemical composition obtained by chemical analysis is listed in Table 1. The yellowish colour and the brown veins are probably due to Fe3+. The density was 2.14 ± 0.04 g/cm3 (standard deviation of 3 samples, accuracy of single measurement < 0.001 g/cm3). The diffraction data (Figure 2) showed the presence of microcrystalline tridymite (major phase), possibly low-quartz and amorphous silica. Line width analysis with Scherrers formula (Patterson [13] yielded crystallite sizes of about 10 nm. The water release upon heating was analysed by thermal analysis. The sample lost 2.8 wt.% up to 200˚C (accompanied by an exothermic peak), which can be attributed to capillary water. Further mass losses of 1.1 wt.% between 200˚C and 500˚C and 0.8 wt.% between 500˚C and 1400˚C were observed. The total weight loss summarized to 4.7 wt.%. Iler [2] published data on dehydroxylation of silica surfaces and stated that up to 180˚C absorbed water is set free, whereas at higher temperatures SiOH groups condensate and liberate molecular H2O. Between 600˚C and 700˚C an accelerated water loss of 0.3 wt.% within 100 K was observed, which is connected with the beginning of an exothermic peak. The reason for this accelerated mass loss is seen in structural changes without further investigating it. Low temperature nitrogen adsorption evaluated according to Brunauer, Emmet and Teller [14] yielded a specific surface area of 38 m2/g. The pores had a size distribution with a maximum at about 35 nm and occupied a pore volume of 0.024 cm3/g (=5.1 vol.%). Capillary water in these pores would yield a mass fraction of 2.4 wt.% which is comparable to the mass loss measured up to 200˚C. FT-IR spectroscopy showed bands typical of molecular water as well as hydroxyl groups connected to the silicate network.
The surface of opal grains was investigated by environmental scanning electron microscopy. Micrographs of different phase ensembles on the surface are shown in Figure 3. The platelike crystallites on the inner side of a larger pore (Figure 3(a)) are probably tridymite crystals whereas Figure 3(b) shows a fracture surface at an higher magnification. Another fracture surface was investigated by atomic force microscopy (Figure 4)

Figure 1. Part of opal specimen; size of platelet (right side): 1.5 × 1.2 × 0.1 cm3.

Table 1. Chemical analysis of opal by X-ray fluorescence analysis; H2O content was determined by thermal analysis.
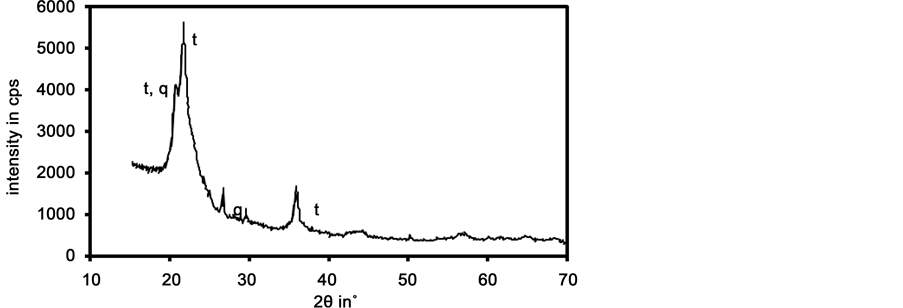
Figure 2. X-ray diffraction pattern of opal; CuKα radiation; t: tridymite (JCPDS 18-1170), q: low-quartz (JCPDS 2-458).

Figure 3. Electron micrographs of common opal; scale bar in the lower right corner; (a) surface inside larger pore, (b) fracture surface.

Figure 4. Atomic force microscope image of opal fracture surface; (a) height image, (b) tapping amplitude image measured for structure analyzation; lateral scale bar in the picture, height or deflection scale on right side.
also at higher magnification. Here a random close packing of colloidal particles with sizes of about 50 to measurement. To summarize, the microstructure of the investigated opal is heterogeneous on a 10 to 100 nm scale. Microcrystalline as well as colloidal microstructures can be identified.
3.2. Dissolution Tests
The dissolution tests with opal batches were performed with rotating autoclaves. After these dissolution tests the obtained materials were separated into a liquid and a solid phase. 20 g batches yielded between 10 and 16 g liquid phase and 4 to 6 g solid phase. A mass loss of 5 wt.% to 20 wt.% of the batch was observed which was mainly due to wall losses caused by the experimental procedure. The vacuum filtration reduced the solid content for about 0.3 wt.% whereas the Na2O content remained constant. This is an indication that filtration has a selective influence on the concentrations. The results are presented in Figures 5-8. As later will be discussed the error range of the silica analysis is between −0.6 wt.% and +0.2 wt.%. The size of the symbols used in Figures 5-8 has approximately that size.
3.2.1. Time Dependence
In Figure 5 the time dependence of the silica contents obtained with batch A at temperatures between 160˚C and 220˚C is shown. Usually, the highest silica contents were obtained after 4 h. At longer dissolution times the silica contents decreased and possibly approximated a constant value.
3.2.2. Dependence on Batch Composition and Particle Size
In Figure 6 dissolution tests at 200˚C with batches A, B, and C are compared. Dissolu-
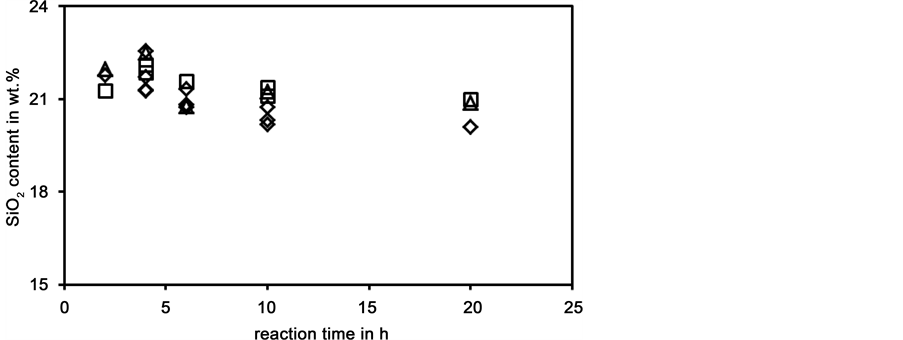
Figure 5. Silica content of liquid phases prepared from batch A as a function of dissolution time; dissolution temperatures: o: 160˚C, ¯: 200˚C, D: 220˚C.
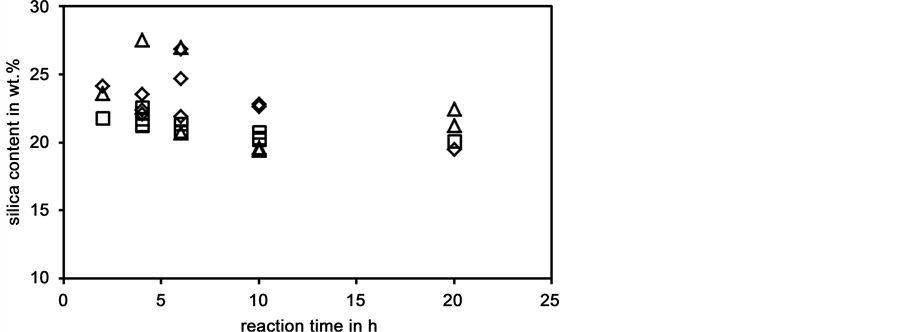
Figure 6. Silica content of liquid phases prepared from batches A (o), B (¯), and C (D) as a function of dissolution time; dissolution temperature 200˚C.
tion tests with batches B and C show a more pronounced maximum of silica contents after 4 or 6 h. On one hand batches B and C contained 30% more opal. Therefore, the higher silica contents in shorter dissolution runs are possibly due to the higher surface area of the dissolving opal grains. On the other hand, the decrease of grain size from batch B to batch C had not the expected effect on kinetics. The decrease of silica content in longer dissolution tests, parallel to observations discussed in connection with Figure 5, leads to the assumption that there is a solubility limitation.
3.2.3. Temperature Dependence
Figure 7 compares the maximal SiO2 contents obtained with batches B and C as a function of temperature. The maximal SiO2 contents obtained with batch C increase with Temperature except at 220˚C B. In the case of batch C the maximal SiO2 contents decrease with temperature, whereas they generally increase with temperature. The final values of the SiO2 contents obtained with batch A are included in the diagram for comparison. It is supposed that these long term silica concentrations are closer to equilibrium values. Then the maximal values (“overshot”) are due to dissolution kinetics and can be governed by the grain size or surface area of the dissolving opal. The experiences with batches B and C indicated that a surplus of silica enhances the achievable silica concentrations.
3.2.4. Dependence on Rm
Therefore, the Rm values of the starting materials were varied over a wide range between 2.4 and 6.7 in batches D and E. The results are shown in Figure 8. With batch D as well as with batch E the silica contents approach constant values of about 25 wt.% and the Rm and approach values of about 3.5, respectively, with increasing initial Rm value of the batch. This shows that compositions close to commercial water glasses with Rm = 3.3 can be reproducibly obtained if reactive starting materials are used. The concentration values reached with batches D and E within 6 h are higher than the long term values reached with batch A (included in Figure 7). Thus, these high values do not present equilibrium values.
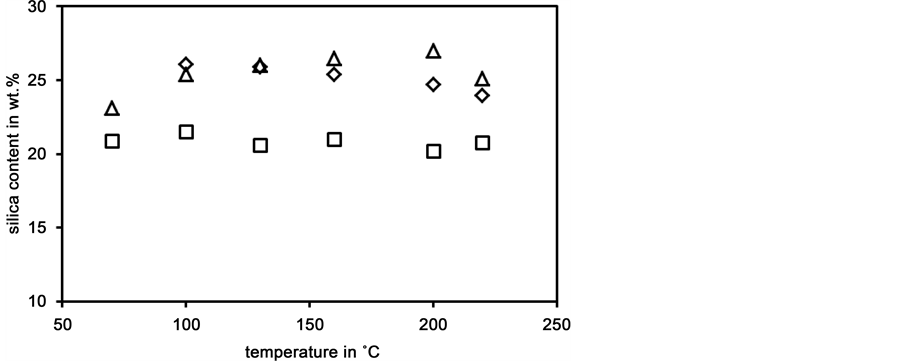
Figure 7. Maxima of silica contents of liquid phases prepared from batches B (¯) and C (D) as a function of dissolution temperature; dissolution times 4 h or 6 h; final value of silica content of liquid phase prepared from batch A (o) at that these temperatures included for comparison.
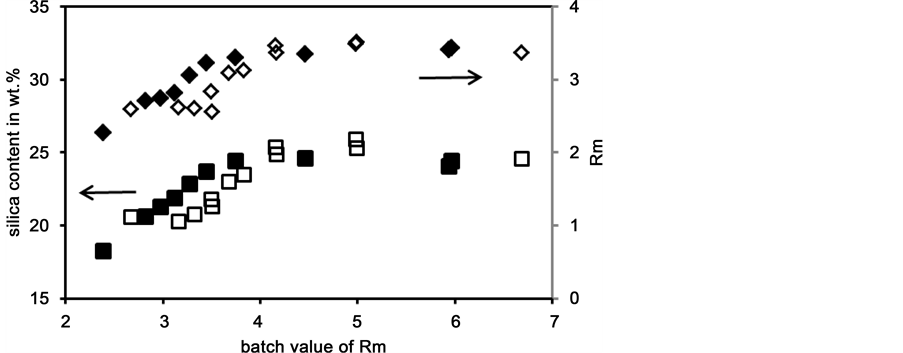
Figure 8. Silica content (n: batch D, o: batch E) and measured values of Rm (¿: batch D, ¯: batch E) of liquid phases as a function of the initial Rm value; dissolution time 6 h; dissolution temperature 160˚C.
3.3. Characterization of the Products
Some of the findings reported above made it necessary to characterize the dissolution products in detail. The chemical composition of two liquid phases with a composition close to commercial sodium water glasses were analysed additionally with inductively coupled plasma spectroscopy. The results are shown in Table 2. The Na2O content was measured by two different methods and allows a control of the analysis: the difference of the results is <0.3 wt.%. The sum of the “impurities” (nonalkaline oxides minus SiO2) is less than 0.3 wt.%. Therefore, the silica content of the “nonalkaline oxides” is larger than 98 wt.%. Together with the observed up concentration of silica by vacuum filtration the measured “silica” contents are about 0.6 wt.% too high. Thus the error range of the silica determination is −0.6 wt.% to +0.2 wt.%. A comparison of the liquid phase composition with the opal composition (Table 1) reveals that the concentration of the impurities is remarkably reduced the Al2O3 content by a factor of 2.5 and the Fe2O3 content by a factor > 10.
Basic properties of the obtained liquids are compared in Table 3 with literature data (Roggendorf et al. [15] ). Density and refractive index agree very well whereas the viscosities deviate to a certain degree. This deviation is due to the lower alkaline contents (Weldes and Lange [1] ) of the water glasses prepared here from opal.
The solid residues contain crystalline phases and probably remnants of the dissolved opal. In a few cases gel-like materials were formed as dissolution remnants, especially at intermediate temperatures and high initial Rm values. The solid residues of two dissolution tests were investigated by X-ray diffraction. One sample was made from batch B, and was treated 20 h at 220˚C. The liquid phase contained 18.5 wt.% SiO2 and 7.8 wt.% Na2O. The diffraction pattern is shown in Figure 9. The second sample was made from batch C and treated under comparable conditions. In this sample the liquid phase contained 19.3 wt.% SiO2 and 8.1 wt.% Na2O. The diffraction patterns of both samples were nearly identical. The major phase was low-quartz which was accompanied by analcime, a zeolite occurring in nature with the chemical formula Na[AlSi2O6]∙H2O. The crystallization of analcime is probably responsible for the reduction of the alumina content of the liquid phase in comparison to the starting materials. A water glass made by dissolving batch C for 20 h at 160˚C was investigated by dynamic light scattering. The sample had a silica content of 25.6 wt.% and Rm = 3.53. Colloidal particles with a
Table 2. Chemical analysis data of two solutions prepared by dissolving opal in NaOH lye; Na2O is determined by titration and the other elements (including Na) by ICP-MS; Main elements are converted to oxide components for comparison; dissolution temperature: 160˚C; dissolution time: 6 h; opal contents 15 g (batch D) and 12.5 g (batch E).
Table 3. Basic properties of obtained water glass prepared with batch E compared with literature data obtained by dissolving a sodium silicate glass; the opal content is related to 20 g batch, two 40 g batches were treated simultaneously and mixed before analysis in order to produce enough liquid phase.

Figure 9. X-ray diffraction pattern of solid residue after hydrothermal treatment of opal in NaOH lye; batch B, dissolution temperature: 220˚C; dissolution time: 20 h; nonalkaline oxide content: 18.5 wt.%, Na2O: 7.8 wt.%, Rm = 2.44; CuKα radiation; a: analcime (Na[AlSi2O6]∙H2O ; JCPDS 2-340), q: low-quartz (JCPDS 2-458).
monomodal size distribution at a radius of 1.6 nm were found. These particles are larger than the “primary” colloids found by Böschel et al. [16] . But the larger sizes interpreted as aggregates in that reference, were not found.
4. Discussion
Opal is an unlikely raw material for water glass production. Nevertheless, it can serve as model for other microcrystalline or amorphous silica sources. The material used in this study is an Opal-CT with a microstructure consisting of microcrystalline tridymite, traces of low-quartz, and amorphous parts build up by random packings of size distributed amorphous silica colloids. Further, it has an internal porosity and an appreciable specific surface area. All these features of the microstructure may contribute to the fast dissolution under hydrothermal conditions. The dissolution conditions were inspired by the hydrothermal synthesis of water glass synthesis by dissolving silica. High concentrations of SiO2 in the liquid phase were reached already after short dissolution times. Therefore, it was not possible to evaluate the kinetics of the early stages of the dissolution process.
In dissolution test with varied initial Rm value it was found that a higher amount of initial silica is needed to reach comparable silica concentrations within the fixed dissolution time, but the deviation is rather small. Therefore, it is concluded that the use of coarser opal grains did not significantly slow down the dissolution process as it was initially hoped. The effects of particle size and specific surface area on dissolution rates was discussed by Diedrich et al. [17] who made aggregation of very small particles, lacking etch pits or different concentrations of reactive surface groups responsible for surface normalized dissolution rates increasing with particle size.
Most of the test runs were evaluated by titration (yielding Na2O contents, other alkalis were only found in traces in chemical analysis) and determination of the solids content via calcination. The “nonalkaline oxides” obtained by subtraction (solid content minus Na2O content) consisted mainly of SiO2, more than 98 wt.% SiO2 were found in chemical analysis. This allows the comparison with commercially produced sodium water glasses. The vacuum filtration which was applied to stop the dissolution reaction after the test run increases the uncertainty level by further 0.3 wt.%.
The starting materials contain appreciable amounts of Al2O3 and Fe2O3 as impurities. The purity of the liquid phase was enhanced by crystallization of analcime. This phase crystallizes under hydrothermal conditions and contains Al in its chemical formula. Additionally, it can incorporate some amounts of iron [18] and serve as adsorbent for cations [19] . The main crystalline phase precipitating during the dissolution tests was low-quartz. It is probable that an equilibrium state saturated with respect to low-quartz is approached. This would also explain the observed concentration development with time which seems to pass a maximum and approaches constant values for the long term. It is therefore supposed that the minimal values obtained especially with batch A are close to the equilibrium solubility of low - quartz under hydrothermal conditions at quite high pH values. These solutions have pH values of about 12 after cooling down to room temperature [15] . The SiO2 concentrations are higher than those calculated from available thermodynamic data (Fleming et al. [5] ), but in these studies only monomeric silicate molecules or anions and not the colloidal silica were taken into account.
The higher limits of silica concentrations of about 25 wt.% to 27 wt.% might have different causes. They might be typical values of critical limiting supersaturation according to La Mer [20] , restricted by viscosity, or caused by solubility limitations due to another phase. Additionally, the colloidal fraction might contribute to the solubility equilibrium.
To answer some of these questions, dissolution tests with quartz and amorphous silica were performed and will be evaluated in future publications.
5. Conclusion
Common opal was used as silica source for hydrothermal water glass production. At temperatures between 100˚C and 220˚C high silica contents of about 25 wt.% and Rm values above 3.0 were reached with 4 to 6 h. The obtained water glasses are comparable to commercials products. At longer dissolution times, the silica concentration of the water glasses decreased and probably approached concentration limitation of silica of about 20 wt.% to 22 wt.% nearly independent of temperature due to a solubility equilibrium. The colloidal fraction might be involved in this equilibrium. The identification of a solubility equilibrium explaining the observed concentration limitations is regarded as new aspect in discussing the chemistry of water glasses. The applied common opal contained appreciable impurity concentrations, especially iron oxide and alumina. Due to crystallization and possibly adsorption at crystallization products (e.g. analcime) the impurity level of the obtained liquid water glasses was remarkably reduced. Low-quartz was identified as major crystalline phase of the solid residue and is supposed to control solubility.
Acknowledgements
The authors gratefully thank S. Brinke and H. Schwalbe (experimental support), S. Sander (dynamic light scattering), B. Niehus (CP-MS analysis), J. Bienias (porosimetry measurements), Dr. N. Kilian (X-ray diffraction), Dr. A. Hauser (atomic force microscopy), F. Syrowatka (scanning electron microscopy) and the work shop of the institute of physics, especially H. Benkwitz, for building the rotation device for steel autoclaves.
Cite this paper
Pfeiffer, T., Enke, D., Roth, R. and Roggendorf, H. (2017) Hydrothermal Dissolution of Opal in Sodium Hydroxide Lyes for the Synthesis of Water Glass. Advances in Chemical Engi- neering and Science, 7, 76-90. http://dx.doi.org/10.4236/aces.2017.71007
References
- 1. Weldes, H.H. and Lange, K.R. (1969) Properties of Soluble Silicates. Journal of Industrial and Engineering Chemistry, 61, 29-44.
https://doi.org/10.1021/ie50712a008 - 2. Iler, R.K. (1979) Chemistry of Silica. Wiley-Interscience, New York.
- 3. Stober, W. (1966) Physical and Chemical Properties of Coesite and Stishovite Compared to Quartz (Translated from German). Beitr. Silikoseforschung, 89, 1-113.
- 4. Dove, P.M. and Rimstidt, J.D. (1994) Silica-Water Interactions. In: Prewitt, P.J., et al., Eds., Reviews in Mineralogy, Vol. 29, Silica-Physical Behavior, Geochemistry and Materials Applications, Mineralogical Society of America, Washington DC, 259-308.
- 5. Fleming, B.A. and Crerar, D.A. (1982) Silicic Acid Ionization and Calculation of Silica Solubility at Elevated Temperature and pH Application to Geothermal Fluid Processing and Reinjection. Geothermics, 11, 15-29.
https://doi.org/10.1016/0375-6505(82)90004-9 - 6. Gunnarsson, I. and Arnorsson, S. (2000) Amorphous Silica Solubility and the Thermodynamic Properties of H4SiO4 in the Range of 0° to 350°C at Psat. Geochimica et Cosmochimica Acta, 64, 2295-2307.
https://doi.org/10.1016/S0016-7037(99)00426-3 - 7. Tuttle, O.F.A. and Friedmann, I.I. (1948) Liquid Immiscibility in the System H2O-Na2O-SiO2. Journal of the American Chemical Society, 70, 919-926.
https://doi.org/10.1021/ja01183a011 - 8. Jones, J.B. and Segnit, E.R. (1971) The Nature of Opal. I. Nomenclature and Constituent Phases. Journal of the Geological Society of Australia, 18, 57-68.
https://doi.org/10.1080/00167617108728743 - 9. Florke, O.W., Graetsch, H., Martin, B. and Wirth, R. (1991) Nomenclature of Micro- and Non-Crystalline Silica Materials—Based on Structure and Microstructure. Neues Jahrbuch für Mineralogie Abhandlungen, 163, 19-42.
- 10. Avakov, V.A. and Vinogradow, B.N. (1974) Comparative Solubility of Silica and Some Alumosilicates (Title Translated from Russian). Izvestiya Vysshikh Uchebnykh Zavedenii, Khimiya i Khimicheskaya Tekhnologiya, 17, 879-882.
- 11. Ordiales, E. (1969) Production of Alkali Silicates from Opal and Similar Minerals (Translated from German). German Patent DE 1969-1900066.
- 12. DIN EN 896 (2012) Chemicals Used for Treatment of Water Intended for Human Consumption-Sodium Hydroxide. Beuth Verlag, Berlin.
- 13. Oelkers, E.H., Helgeson H.C., Shock, E.L., Sverjensky, D.A., Johnson, J.W.A. and Pokrovskii, V.A. (1994) Summary of the Apparent Partial Molal Standard Gibbs Free Energies of Aqueous Species, Minerals, and Gases at Pressures 1 to 5000 Bars and Temperatures 25 to 1000 °C. Journal of Physical and Chemical Reference Data, 24, 1401-1560.
- 14. Patterson, A.L. (1939) The Scherrer Formula for X-Ray Particle Size Determination. Physical Review, 56, 978-982.
https://doi.org/10.1103/PhysRev.56.978 - 15. Brunauer, S., Emmet, P.H. and Teller, E. (1938) Adsorption of Gases in Multimolecular Layers. Journal of the American Chemical Society, 60, 309-319.
https://doi.org/10.1021/ja01269a023 - 16. Roggendorf, H., Grond, W. and Hurbanic, M. (1996) Structural Characterization of Concentrated Alkaline Silicate Solutions by 29Si-NMR Spectroscopy, FT-IR Spectroscopy, Light Scattering, and Electron Microscopy—Molecules, Colloids and Artefacts. Glass Science and Technology, 69, 216-231.
- 17. Boschel, D., Janich, M. and Roggendorf, H. (2003) Size Distribution of Colloidal Silica in Sodium Silicate Solutions Investigated by Dynamic Light Scattering and Viscosity Measurements. Journal of Colloid and Interface Science, 267, 360-368.
https://doi.org/10.1016/j.jcis.2003.07.016 - 18. Diedrich, T., Dybowska, A., Schott, J., Valsami-Jones, E. and Oelkers, E.H. (2012) The Dissolution of SiO2 Nanoparticles as a Function of Particle Size. Environmental Science & Technology, 46, 4909-4915.
https://doi.org/10.1021/es2045053 - 19. Azizi, S.N. and Yousefpour, M. (2011) Isomorphous Substitution of Iron and Nickel into Analcime Zeolite. Zeitschrift für anorganische und allgemeine Chemie, 637, 759-765.
https://doi.org/10.1002/zaac.201100059 - 20. Jannagadha Rao, M. and Gopal Krishna, B. (2013) Naturally Engineered Analcime for Water Treatment Process and Its Calorimetric Properties. International Journal of Science and Research, 160-166.
- 21. La Mer, V.K. (1952) Nucleation in Phase Transitions. Industrial & Engineering Chemistry, 44, 1270-1277.
https://doi.org/10.1021/ie50510a027