Advances in Chemical Engineering and Science
Vol. 2 No. 2 (2012) , Article ID: 18898 , 8 pages DOI:10.4236/aces.2012.22028
Oxidative Consumption of Oral Biomolecules by Therapeutically-Relevant Doses of Ozone
183 Chambers Lane, Willesden Green, London, UK
2Warwick Dentistry, Warwick Medical School, University of Warwick, Warwick, UK
3Institute for Materials Research and Innovation (IMRI), University of Bolton, Bolton, UK
Email: *mg6@bolton.ac.uk
Received October 23, 2011; revised January 4, 2012; accepted February 5, 2012
Keywords: Ozone Therapy; Ozone Scavengers; Oral Biomolecules; Oral Fluids and Tissues; 1H NMR Analysis
ABSTRACT
In view of its potent microbicidal actions, ozone (O3) offers much potential for application as a therapeutic agent in oral health, e.g. in the treatment of dental caries. This oxidant is extremely reactive towards biomolecules present in the oral environment, and in this study we have employed high-resolution proton (1H) nuclear magnetic resonance (NMR) spectroscopy to determine the nature and extent of the oxidation of biomolecules known to be present in carious dentin, plaque and saliva. Phosphate-buffered (pH 7.00) aqueous solutions containing sodium pyruvate, α-D-glucose, L-cys teine and L-methionine (5.00 mM) were treated with gaseous O3 (4.48 mmol.) delivered by a therapeutic O3 generating device. Attack of O3 on methionine and cysteine generated the corresponding primary oxidation products of these substrates, specifically methionine sulphoxide [98% ± 4% (mean ± SEM) yield] and cystine (95% ± 6% yield) respectively, and treatment of pyruvate with this oxidant produced acetate and CO2 via an oxidative decarboxylation process (93% ± 4% yield). Reaction of O3 with α-D-glucose gave rise to formate as a major product (24% ± 2% yield). In conclusion, multicomponent 1H NMR analysis of appropriate chemical model systems provides valuable molecular information regarding the reactivity of O3 towards biomolecules present in the oral environment, information which is of much relevance to its therapeutic mechanisms of action. Moreover, in view of the much higher concentrations of these O3-scavenging biomolecules in oral fluid and/or soft tissue environments than that of O3 applied, they may also serve to offer protection against putative adverse effects inducible by any of this oxidant which escapes from its site of therapeutic application (e.g., at primary root carious lesions).
1. Introduction
Currently, root caries represents a challenging problem to the dental profession in view of a marked increase in the population of elderly patients during the late 20th century. This condition is primarily ascribable to tooth demineralisation processes induced by organic acids (e.g., lactic, pyruvic and further organic acids) generated by bacteria, predominantly Streptococcus mutans [1-3], and recent investigations conducted by Baysan et al. [4-6] have revealed that ozone (O3) exerts powerful bactericidal actions towards this and other pathogens, together with further micro-organisms associated with primary root carious lesions. Indeed, the application of O3 in dental practices may serve as a viable, cost-effective and convenient means of treating dental caries, and it may have the potential to eventually replace conventional drilling and filling procedures which are commonly and frequently employed by dental surgeons [7].
In view of its powerful oxidising actions, O3 has a very extensive redox chemistry in physiological environments, and the oxidation of critical biomolecules is undoubtedly responsible for its broad-spectrum biocidal properties. Indeed, O3 can attack a very wide variety of biomolecules, for example, free or protein-incorporated amino acids such as cysteine, methionine, tryptophan, histidine and tyrosine, carbohydrates such as glucose, amines such as trimethylamine, phenolic adducts, ascorbate and urate [8,9], and, of course, its well-characterised ozonation of carbon-carbon double bonds, e.g. those present in unsaturated or polyunsaturated fatty acids (UFAs and PUFAs respectively) [10-12]. Oxidation of PUFAs by O3 gives rise to the generation of fatty acid ozonides which, on fragmentation, produce aldehydic adducts, the latter putatively serving as ‘biomarkers’ of O3-induced cell and tissue damage and, more specifically, UFA ozonation. Indeed, the reactions of O3 with human skin lipids have been implicated as sources of carbonyl, dicarbonyl and hydroxycarbonyl species detectable in indoor air [13].
Intriguingly, reaction of water-soluble, single electrondonors with O3 primarily generates the ozone radical anion (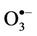 ), a transient adduct which, on protonation, decomposes to hydroxyl radical (
), a transient adduct which, on protonation, decomposes to hydroxyl radical (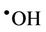 ) and dioxygen. Hence, selected reaction products which putatively arise from the interactions of O3 with biomolecules present in human tissues and biofluids are identical to those which can be generated from the attack of
) and dioxygen. Hence, selected reaction products which putatively arise from the interactions of O3 with biomolecules present in human tissues and biofluids are identical to those which can be generated from the attack of 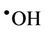 radical on such biomolecular scavengers of this ‘Reactive Oxygen Species’ (ROS).
radical on such biomolecular scavengers of this ‘Reactive Oxygen Species’ (ROS).
In view of the clear indications for the therapeutic application of O3 in the treatment of selected oral diseases, a series of clinical trials involving this agent have recently been completed (e.g. [7]). Moreover, during its therapeutic application, this therapeutic oxidant can gain access to oral fluids such as human saliva, and hence it is of much importance to monitor its reactivity with biomolecules present in this (and other) biofluids (particularly electron-donors), and the products derived from such reactions. Therefore, in this investigation we have performed an evaluation of the oxidising actions of O3 (generated by a therapeutic device for clinical dental treatment) towards a series of biomolecules present in the oral environment. For this purpose, we have employed high resolution proton (1H) nuclear magnetic resonance (NMR) spectroscopy to determine the nature and extent of the oxidation of biomolecules which are known to be present in oral fluids such as human saliva [2] and gingival crevicular fluid (and also in carious dentin and plaque matrices) by O3.
L-methionine and L-cysteine were chosen as model, electron-donating amino acids for these studies since they are present in many salivary proteins and, subsequent to the bacterially-induced proteolysis of such macromolecules, act as precursors to volatile sulphur compounds (VSCs) which predominantly give rise to halitosis (oral malodour). The major VSCs produced in this manner comprise hydrogen sulphide (H2S), dimethyl disulphide (CH3S-SCH3) and methyl mercaptan (CH3SH), the latter accounting for approximately 60% of the total VSCs detectable [14].
The therapeutic and biochemical significance of the results acquired, particularly the ability of salivary biomolecules to protect against the oxidising actions of this agent (i.e. that which may “escape” from its therapeutic application site) are discussed in detail.
2. Materials and Methods
2.1. Materials
Sodium pyruvate, L-methionine, L-cysteine, L-cystine, α-D-glucose, sodium trimethylsilyl-[2,2,3,3-2H4] propionate (TSP) and deuterium oxide (2H2O) were of the highest possible analytical grade and purchased from the Sigma-Aldrich Chemical Co. (Gillingham, Dorset, UK).
2.2. Sample Preparation and Treatment
Aqueous solutions containing sodium pyruvate, alpha-Dglucose, L-cysteine and L-methionine (5.00 mM) were prepared in 40.0 mM phosphate buffer (pH 7.00) which was rigorously deoxygenated with O2-free N2 gas prior to use. 5.00 ml aliquots of these aqueous solutions were divided into two equivalent portions (2.50 ml). The first of these was treated with gaseous O3 synthesised by the HealOzone Unit (CurOzone, USA) for a 10 s period (equivalent to a delivery of 4.84 mmol of this reactive oxygen radical species) and then equilibrated at a temperature of 35˚C for a 60 min. period prior to high-resolution 1H NMR analysis. The second matching group of de-oxygenated (untreated) 2.50 ml volume solutions, which were equilibrated in the same manner, served as essential controls.
Each of the above experiments was repeated three times so that there was a total of n = 4 replicate solutions (each with an initial pre-treatment O3 scavenger concentration of 5.00 mM) for each biomolecule investigated.
2.3. 1H NMR Measurements
One-dimensional (1D) 600 MHz proton NMR spectra of the chemical model systems described above were acquired on a Bruker AMX-600 spectrometer. Typically, 0.60 ml aliquots of untreated or ozonated biomolecule solutions were treated with a 0.10 ml volume of a 1.47 mM solution of TSP in 2H2O (the latter providing a field-frequency lock), the mixtures thoroughly rotamixed and then transferred to 5-mm diameter NMR tubes. Typical pulsing conditions were: 64 FIDs using 32,768 data points, 72˚ pulses and a 3 s pulse repetition rate to allow full spin-lattice relaxation of the hydrogen nuclei in the samples investigated. Chemical shifts were referenced to TSP (internal; final concentration 0.21 mM), and exponential line-broadening functions of 0.30 Hz were employed for purposes of processing.
The intensities of the most prominent 1H NMR resonances of each biomolecule and their corresponding reaction products with O3 were determined by electronic integration via the spectrometer’s proprietary software (XWIN-NMR), and the concentrations of components detectable were determined by comparisons of their resonance areas with that of the added TSP internal standard (final concentration 0.21 mM). Maintenance of the exact integral regions for each spectrum acquired was ensured.
2.4. Spectrophotometric Determination of O3 in Ozonated Phosphate Buffer Solutions
The (soluble) O3 concentration of 40.00 mM phosphate buffer solutions treated with O3 in the same manner as each of the above biomolecule solutions was determined by a modification of the spectrophotometric method of Schechter [15] which involves the oxidation of iodide anion by O3 to form the chromophoric  ion (λmax = 350 nm, e = 2.32 × 104 M−1·cm−1). Absorbance measurements and electronic absorption spectra were recorded on a Unicam UV-2 spectrophotometer.
ion (λmax = 350 nm, e = 2.32 × 104 M−1·cm−1). Absorbance measurements and electronic absorption spectra were recorded on a Unicam UV-2 spectrophotometer.
2.5. Statistical Treatment of Experimental Data
Results were reported as the mean ± between replicates (n = 4) standard error of the mean (SEM) percentage consumption of each biomolecule examined in this study (all pre-treatment biomolecule concentrations were equivalent, i.e. 5.00 mM).
3. Results
3.1. Reaction of Pyruvate with O3
5.00 mM aqueous solutions of the α-keto acid anion pyruvate were treated with O3 as described in the Materials and Methods section in order to investigate the redox reaction occurring between these agents, and proton (1H) NMR analysis of these solutions demonstrated a marked level of oxidative decarboxylation of this alpha-keto acid anion to acetate and CO2 (Figure 1), an observation consistent with the reaction depicted in equation (1). Also consistent with this observation, a singlet resonance located at 1.50 ppm and ascribable to pyruvate hydrate [the enol form of this α-keto acid anion ( )], of much lower intensity than that of the keto form at 2.388 ppm, was also removed from spectra after O3 treatment. The decreases observed in the intensities of the pyruvate and pyruvate hydrate signals were, of course, accompanied by corresponding equivalent increases in that of the acetate-CH3 resonance (δ = 1.92 ppm). The mean ± SEM percentage decrease in the intensities of the combined pyruvateand pyruvate hydrate-CH3 signals was found to be 93% ± 4% (mean ± SEM), although it should be noted that the effective concentration of O3 in the system employed here is limited by its solubility in water, together with its rate and level of consumption by the scavenger employed, and also its catalytically-promoted dissociation to dioxygen during the 5 min. treatment period.
)], of much lower intensity than that of the keto form at 2.388 ppm, was also removed from spectra after O3 treatment. The decreases observed in the intensities of the pyruvate and pyruvate hydrate signals were, of course, accompanied by corresponding equivalent increases in that of the acetate-CH3 resonance (δ = 1.92 ppm). The mean ± SEM percentage decrease in the intensities of the combined pyruvateand pyruvate hydrate-CH3 signals was found to be 93% ± 4% (mean ± SEM), although it should be noted that the effective concentration of O3 in the system employed here is limited by its solubility in water, together with its rate and level of consumption by the scavenger employed, and also its catalytically-promoted dissociation to dioxygen during the 5 min. treatment period.
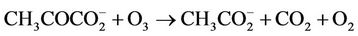 (1)
(1)
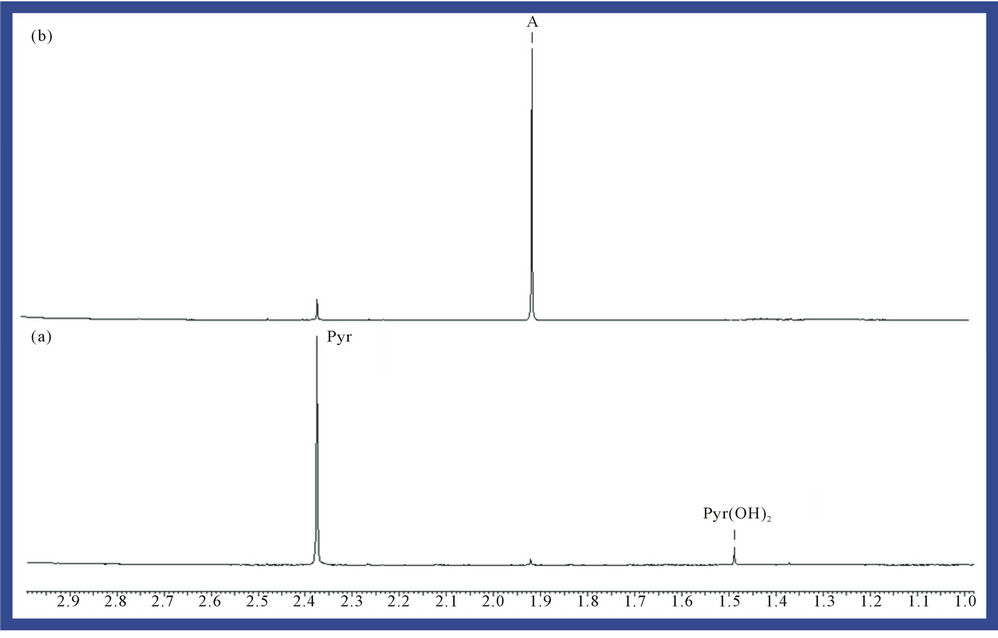
Figure 1. Expanded 1.00 - 3.00 ppm regions of the 600 MHz 1H NMR spectra of a 5.00 mM solution of sodium pyruvate in 40.0 mM phosphate buffer (pH 7.00) (a) prior and (b) subsequent to exposure to gaseous O3 (at a level equivalent to 4.84 mmol. per 5.00 ml of substrate solution) and then equilibrated at 35˚C for a 60 min. period. Abbreviations: Pyr, pyruvate-CH3; Pyr(OH)2, pyruvate hydrate-CH3; A, acetate-CH3.
3.2. Reaction of L-Methionine with O3
1H NMR analysis of reaction products revealed that interaction of O3 with the thiomethyl group (-S-CH3)-containing amino acid L-methionine generated methionine sulphoxide as a major product (Figure 2), a process consistent with equation (2), where R represents H3N+CH (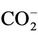 )CH2CH2-. Indeed, the -SO-CH3 protons of this oxidation product has a characteristic singlet resonance located at 2.725 ppm, and it’s concentration is readily monitored by the (relative or normalised) intensity of this signal. Further 1H NMR signals which were consistent with the generation of methionine sulphoxide were multiplets located at 2.30, 3.02 and 3.85 ppm, which correspond to the β-CH2, γ-CH2 and α-CH protons, respecttively, in this oxidation product. No evidence for the production of methionine sulphone (characteristic-SO2- CH3 singlet resonance located at δ = 3.09 ppm), which may arise from the oxidation of methionine sulphoxide by further O3, was provided in these investigations. Under our experimental conditions, the yield of methionine sulphoxide produced was 98% ± 4%, i.e. the reaction was essentially complete and quantitative.
)CH2CH2-. Indeed, the -SO-CH3 protons of this oxidation product has a characteristic singlet resonance located at 2.725 ppm, and it’s concentration is readily monitored by the (relative or normalised) intensity of this signal. Further 1H NMR signals which were consistent with the generation of methionine sulphoxide were multiplets located at 2.30, 3.02 and 3.85 ppm, which correspond to the β-CH2, γ-CH2 and α-CH protons, respecttively, in this oxidation product. No evidence for the production of methionine sulphone (characteristic-SO2- CH3 singlet resonance located at δ = 3.09 ppm), which may arise from the oxidation of methionine sulphoxide by further O3, was provided in these investigations. Under our experimental conditions, the yield of methionine sulphoxide produced was 98% ± 4%, i.e. the reaction was essentially complete and quantitative.
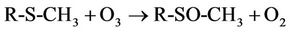 (2)
(2)
3.3. Reaction of L-Cysteine with O3
L-cysteine was also chosen for these chemical model system experiments since we have been unable to detect this thiol in human saliva by 1H NMR analysis in view of its low concentration in the “free” (non-protein-incorporated) state, and also its complex ABX coupling pattern (i.e., no clearly-visible sharp resonances of low multiplicity). 1H NMR analysis demonstrated that exposure of aqueous solutions of this biomolecule [with its characteristic ABX coupling pattern of resonances centred at 3.02 and 3.10 ppm (AB protons) and 3.97 ppm (X proton)] to O3 (Section 2) generated its corresponding disulphide, cystine, as a major product (data not shown). Indeed, a reference spectrum acquired on an authentic sample of L-cystine confirmed its identity (clear 4-line
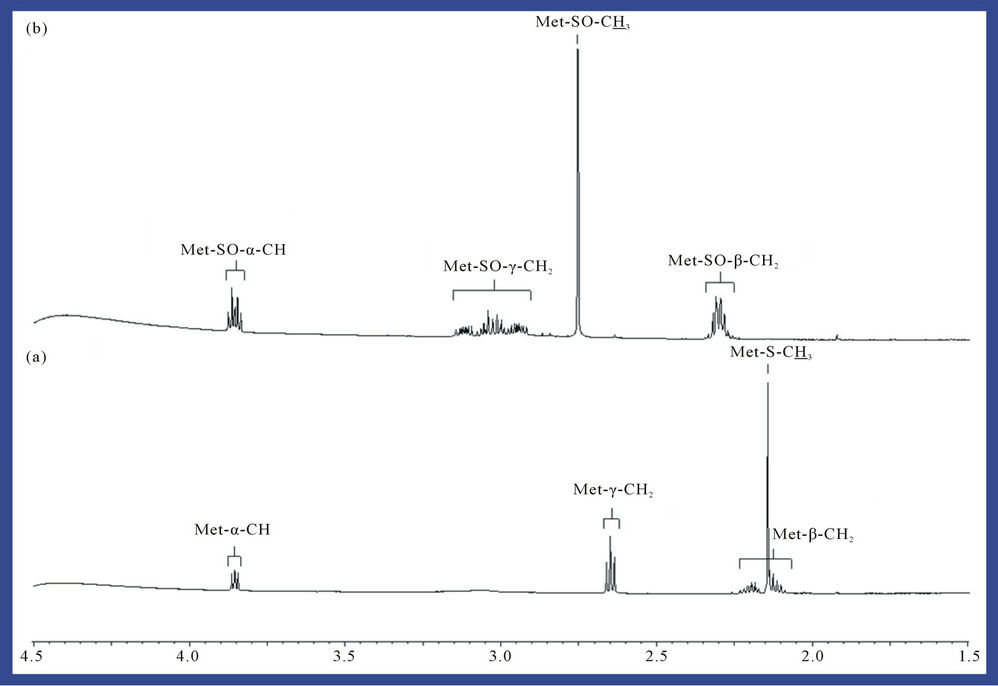
Figure 2. Expanded 1.50 - 4.50 ppm regions of the 600 MHz 1H NMR spectra of a 5.00 mM solution of L-methionine in 40.0 mM phosphate buffer (pH 7.00) (a) before and (b) after exposure to gaseous O3 (at a level equivalent to 4.84 mmol. per 5.00 ml of substrate solution) and then equilibrated at 35˚C for a 60 min. period. Abbreviations: Met-S-CH3, -α-CH, -β-CH2 and -γ-CH2, methionine-S-CH3, α-CH, -β-CH2 and -γ-CH2 resonances respectively; Met-SO-CH3, -α-CH, -β-CH2 and -γ-CH2, methionine sulphoxide-S-CH3, -α-CH, -β-CH2 and -γ-CH2 resonances respectively.
multiplet signals located at 3.20 and 3.41 ppm (AB protons), and a further one at 4.14 ppm (X proton)). Electronic integration of the L-cysteine and cystine resonances demonstrated that 95% ± 6% of the former O3 scavenger was oxidatively transformed to the latter oxidation product. No evidence for the generation of cysteine’s higher oxidation products (such as cysteic acid) was obtained.
These observations are explicable by the stepwise processes described in equations (3)-(5), i.e. deprotonation of the thiol followed by transfer of an electron from the thiolate anion (RS−) to O3, and then combination of two thiyl radicals ( , generated in the reaction depicted in equation (4)) to produce cystine (represented as RSSR, where R = H3N+CH(
, generated in the reaction depicted in equation (4)) to produce cystine (represented as RSSR, where R = H3N+CH( )CH2-).
)CH2-).
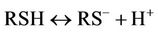 (3)
(3)
 (4)
(4)
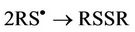 (5)
(5)
However, it should be noted that the aggressivelypowerful oxidant hydroxyl radical (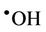 ) can arise from protonation of the
) can arise from protonation of the  species followed by decomposition of the resulting
species followed by decomposition of the resulting  radical adduct (equations (6) and (7)). The
radical adduct (equations (6) and (7)). The  radical generated in this manner can then
radical generated in this manner can then
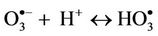 (6)
(6)
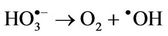 (7)
(7)
also serve to oxidise L-cysteine to cystine via the prior abstraction of an electron from the thiolate anion (equation (8)), a process again forming thiyl radicals which then also combine to yield the corresponding disulphide (equation (5)).
 (8)
(8)
3.4. Reaction of α-D-Glucose with O3
1H NMR analysis also showed that treatment of phosphate-buffered aqueous solutions of α-D-glucose with O3 in the manner described in Section 2 generated formate as a major reaction product (identified as singlet 1H NMR resonance located at δ = 8.46 ppm, Figure 3), an obser-
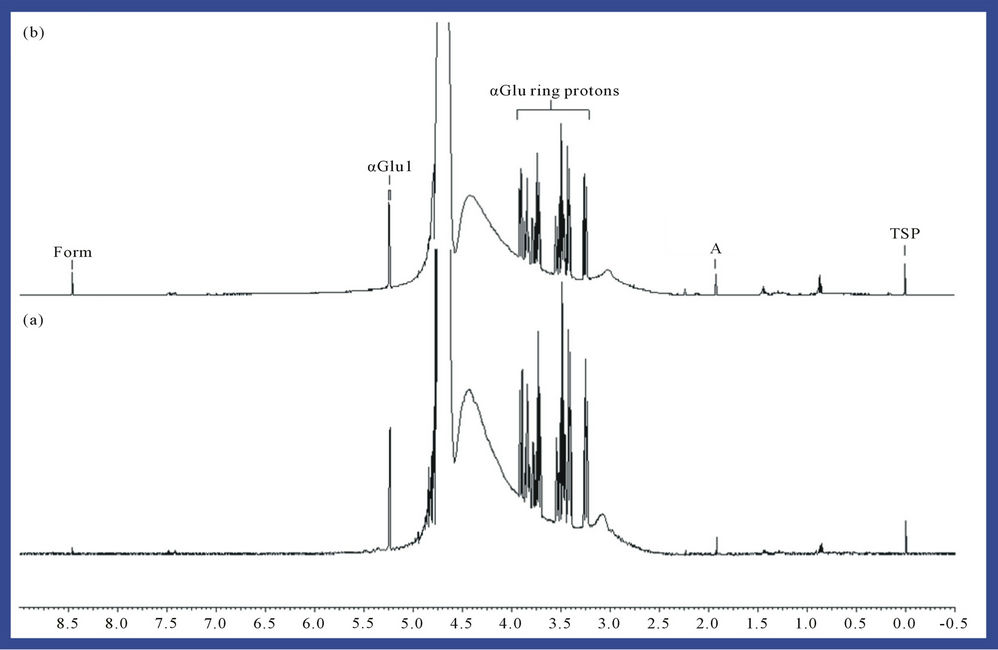
Figure 3. 1H NMR spectrum of a 5.00 Mm solution of α-D-glucose (a) before and (b) following exposure to gaseous O3 (at a level equivalent to 4.84 mmol. per 5.00 ml of substrate solution) and then equilibrated at 35˚C for a 60 min. period. Abbreviations: A, acetate-CH3; Form, formate-H; α-Glu ring protons, i-D-glucose ring protons; α-Glu1, α-D-glucose anomeric ring proton; TSP, trimethylsilyl-[2,2,3,3-2H4] propionate-Si(CH3)3 (internal chemical shift reference and quantitative 1H NMR standard, δ = 0.00 ppm).
vation consistent with previous studies conducted on the interactions of ROS (particularly radiolytically-generated 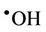 radical) with carbohydrates in general [16]. A mean ± SEM formate concentration of 1.21 ± 0.11 mM (representing a 24% ± 2% yield of this oxidation product) was produced from the 5.00 mM glucose substrate solution.
radical) with carbohydrates in general [16]. A mean ± SEM formate concentration of 1.21 ± 0.11 mM (representing a 24% ± 2% yield of this oxidation product) was produced from the 5.00 mM glucose substrate solution.
4. Discussion
4.1. O3 Scavenging Capacities of the Biomolecules Tested
The differences observed in the oxidising capacity of O3 towards pyruvate, methionine, cysteine and α-glucose can be rationalised in terms of the relative scavenging efficacies of these antioxidants tested (L-methionine ≈ L-cysteine ≈ pyruvate > α-D-glucose). Also of much importance for consideration is their mean salivary (or alternative oral fluid or tissue) concentrations. Indeed, for human saliva, these mean values are in the order pyruvate/pyruvate hydrate (1.67 mM [2]) >> methionine (mean pre-O3 treatment salivary level 274 µM, a value estimated from the data acquired in this investigation) >> cysteine (mean concentration 32 µM [17]): the glucose concentration of this particular oral fluid is highly variable, but little or none of it is detectable by 1H NMR analysis of samples collected following an 8 hr. overnight fasting episode [2].
When expressed relative to tryptophan, the rate constants for the reaction of O3 with methionine, cysteine, and glutathione were found to be 0.77 ± 0.08, 0.88 ± 0.19 and 0.42 ± 0.01 respectively [8] (these relative rate constants correspond to O3 rather than scavenger consumption). Hence, cysteine reacts with O3 at a very similar rate to that of methionine, although it is clear that in human saliva, the latter thioether will serve as a much more effective O3 scavenger than the former thiol in view of its much higher concentration therein (approximately 10-fold greater).
4.2. Therapeutic Relevance of the Results Acquired
The oxidative decarboxylation of pyruvate by O3 provides a mechanistic example of the therapeutic actions exertable by the latter therapeutic agent (e.g. its caries-preventative, cariostatic and further therapeutic properties). Indeed, pryruvic acid is an extremely powerful organic acid (proton donor) since it has a pKa value of 3.20 mM, and therefore is much more powerful than lactic acid (Ka = 0.14 mM) in this context [18]. Indeed, we have previously noted that pyruvic acid may play a significant role in the facilitation of enamel erosion and tooth demineralisation processes [19], and its removal in human saliva, alternative oral fluids, carious dentin and/or plaque by O3 may serve to inhibit the induction or further development of primary root caries lesions, especially since salivary organic acids in their unionised forms readily diffuse into tooth enamel [20].
The oxidative removal of the amino acids methionine and cysteine (both of which are detectable in human saliva, the former by 1H NMR analysis) by O3 represents an observation of some significance in relation to both oral hygiene and clinical periodontology, since hydrogen sulphide (H2S) and methyl mercaptan (CH3SH) are produced from these agents via the metabolic pathways of gram-negative bacteria. Indeed, the enzyme cystine reductase reduces cystine to cysteine, and serine sulphydrase gives rise to the desulphydration of cysteine, a process generating H2S and serine [21,22]. Therefore, data acquired here also provide evidence that O3 may have the ability to clinically suppress oral malodour via the direct oxidative consumption of VSCs and/or their particular amino acid precursors, especially if applied in the form of an O3-containing oral rinse system, i.e. ozonated water. In relation to these investigations, in 1992 Grigor and Roberts [17] found that hydrogen peroxide (H2O2) significantly diminished salivary thiol levels both in vivo and in vitro.
However, it is also conceivable that the bacterial enzyme cystine reductase may reverse the O3-mediated transformation of cysteine to cystine in the oral environment, although it should be noted that O3 may be present in excess over that of total oral fluid or tissue thiol concentrations, especially when the gaseous concentration is considered along with that present in aqueous solution, the latter level being limited to its solubility in water at physiological temperature, estimates of which are influenced and complicated by that present in the local gas phase and also pH values [23]. However, O3 determinations in our laboratory performed by a modification of a previously reported method [15] gave a mean concentration of ca. 20 ppm (0.42 mM) when 40.0 mM phosphate buffer solutions (pH 7.00) were treated with this gaseous oxidant under the same experimental conditions as the biomolecule solutions employed for this study (section 2.1), i.e. at ambient temperature and pressure. Hence, a significant fraction of salivary thiols (or those present in alternative oral environments) are expected to be retained in their oxidised (disulphide) forms following reaction with O3. Furthermore, it is probable that O3 has the capacity to oxidatively inactivate bacterially-derived cystine reductase, together with a range of further enzymes available at its site of therapeutic application.
It is also of much importance to note that methionine is one of only two amino acids encoded by a single codon (AUG) in the standard genetic code (tryptophan, encoded by UGG, is the other) [24]. The codon AUG also represents the “Commencement” message for a ribosome that signals the initiation of protein translation from mRNA. Consequently, methionine is incorporated into the Nterminal position of all proteins in eukaryotes and archaea during translation, although it is usually removed via post-translational modification. This phenomenon is also clearly of much importance to bacteria; however, in this case, the N-formylmethionine derivative is employed as a primary amino acid adduct [25].
Notwithstanding, it is also important to note that singlet oxygen (1O2) is also generated in high yield from the reaction of O3 with a wide range of biomolecules, including the amino acids methionine and cysteine, and the tripeptide glutathione, together with urate, ascorbate, NADH, NADPH and albumin [26] (that involving methionine, which has a thioether group in its side-chain, is depicted in Equation (9)), and therefore this further ROS may also play an important role in perpetuating damage to biomolecules primarily exerted via the actions of O3. Indeed, methionine also readily reacts with 1O2 to again form its corresponding sulphoxide, a process postulated to involve a highly-reactive persulphoxide intermediate [27] [RS+(CH3)-O-O– in the case of this substrate, where R represents H3N+CH( )CH2CH2-].
)CH2CH2-].
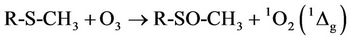 (9)
(9)
Interestingly, for cysteine, the mechanism of cystine generation therefrom has been suggested to involve the reaction of an intermediate sulphoxide species (RSOH) with a second molecule of the cysteine substrate [26] (equation (10)).
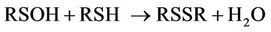 (10)
(10)
4.3. Protective Roles of the Biomolecules Investigated against Adverse Intra-Oral Dissemination of O3
All of the metabolites investigated here also serve to act as biomolecular protectants against any adverse or toxic effects exertable by the “leakage” of therapeutically-applied O3 to soft tissue areas in the oral environment which are or may be accessible by oral fluids containing them (in particular, but not limited to, human saliva). In this manner, these biomolecules have the ability to circumvent any deleterious O3-mediated cell and tissue damage potentially arising from this intra-oral dissemination process.
Indeed, the oxidative modifications of pyruvate, methionine, cysteine and glucose observed here serve as an important examples of the capacity of salivary metabolites to offer protection against any O3 which diffuses away from its site of application, for example, from primary root carious lesions.
4.4. Advantages of 1H NMR Analysis for Evaluations of the Biomolecular Fate of O3 in the Oral Environment
High-resolution, high-field 1H NMR spectroscopy is a technique which offers many advantages over alternative labour-intensive and onerous analytical methodologies for evaluating the biomolecular fate of O3 in oral fluids since (1) it permits the rapid, non-invasive and simultaneous examination of a variety of metabolic species and their O3-induced oxidation status in chemical model systems, and (2) little or no knowledge of sample composition is required prior to analysis. Moreover, chemical shift values, coupling patterns and coupling constants of resonances present in the 1H NMR spectra of such model systems provide much valuable information regarding the molecular nature of oxidation products generated from the interaction of biomolecules with O3.
5. Conclusion
In conclusion, high-resolution 1H NMR spectroscopy provides much valuable molecular information regarding the nature and extent of oral fluid or tissue biomolecule consumption by O3. Such information is clearly of much relevance the potential therapeutic and microbicidal actions of this novel agent in the oral environment, particularly since singlet oxygen (1O2), which is generated as a by-product of O3’s reactions with methionine, cysteine and further biomolecules, can perpetuate oxidative damage to these substrates and hence its microbicidal activity. Furthermore, in view of the large excess of these O3- reactive biomolecules over adventitious O3 in the oral environment, these scavengers are likely to offer remotely-located oral cells and soft tissues protection against any of this oxidant which leaches away from its immediate site of therapeutic application.
REFERENCES
- D. Beighton, E. Lynch and M. R. Heath, “A Microbiological Study of Primary Root Caries Lesions with Different Treatment Needs,” Journal of Dental Research, Vol. 72, No. 3, 1993, pp. 623-629. doi:10.1177/00220345930720031201
- C. J. L. Silwood, M. C. Grootveld, A. W. D. Claxson and E. Lynch, “1H and 13C NMR Spectroscopic Analysis of Human Saliva,” Journal of Dental Research, Vol. 81, No. 6, 2002, pp. 422-427. doi:10.1177/154405910208100613
- E. Lynch, “Kariesbehandlung Mit Ozon,” Die Quintessenz, Vol. 54, 2003, pp. 608-610.
- A. Baysan, R. Whiley and E. Lynch, “Anti-Microbial Effects of a Novel Ozone Generating Device on Micro-Organisms Associated with Primary Root Carious Lesions,” Caries Research, Vol. 34, 2000, pp. 498-501. doi:10.1159/000016630
- A. Baysan, E. Lynch and M. Grootveld, “The Use of Ozone for the Management of Primary Root Caries,” In: T. Albrektsson, D. Bratthall, P. O. Glantz and J. Lindhe, Eds., Tissue Preservation in Caries Treatment, Quintessence Publishing Company Ltd., London, 2001, pp. 49-68.
- A. Baysan and E. Lynch, “Effect of Ozone on the Oral Microbiota and Clinical Severity of Primary Root Caries,” American Journal of Dentistry, Vol. 17, No. 1, 2004, pp. 56-60.
- J. Holmes, “Clinical Reversal of Root Caries Using Ozone, Double-Blind, Randomised, Controlled 18-Month Trial,” Gerodontology, Vol. 20, No. 2, 2003, pp. 106-114. doi:10.1111/j.1741-2358.2003.00106.x
- J. R. Kanovsky and P. D. Sima, “Reactive Absorption of Ozone by Aqueous Biomolecule Solutions: Implications for the Role of Sulfhydryl Compounds as Targets for Ozone,” Archives of Biochemistry and Biophysics, Vol. 316, No. 1, 1995, pp. 52-62. doi:10.1006/abbi.1995.1009
- S. Kermani, A. Ben-Jebria and J. S. Ultman, “Kinetics of Ozone Reaction with Uric Acid, Ascorbic Acid, and Glutathione at Physiologically Relevant Conditions,” Archives of Biochemistry and Biophysics, Vol. 451, No. 1, 2006, pp. 8-16. doi:10.1016/j.abb.2006.04.015
- T. Thornberry and J. P. D. Abbatt, “Heterogeneous Reaction of Ozone with Liquid Unsaturated Fatty Acids: Detailed Kinetics and Gas-Phase Product Studies,” Physical Chemistry and Chemical Physics, Vol. 6, 2004, pp. 84-93. doi:10.1039/b310149e
- M. F. Diaz, F. Hernandez, O. Ledea, J. A. Gavin Sazatornil and J. Moleiro, “1H NMR Study of Methyl Linoleate Ozonation,” Ozone: Science & Engineering: The Journal of the International Ozone Association, Vol. 25, No. 2, 2003, pp. 121-126. doi:10.1080/713610666
- J. J. Thiele, M. G. Traber, T. G. Polefka, C. E. Cross and L. Packer, “Ozone-Exposure Depletes Vitamin E and Induces Lipid Peroxidation in Murine Stratum Corneum,” Journal of Investigative Dermatology, Vol. 108, 1997, pp. 753-757. doi:10.1111/1523-1747.ep12292144
- A. Wisthaler and C. J. Weschler, “Reactions of Ozone with Human Skin Lipids: Sources of Carbonyls, Dicarbonyls, and Hydroxycarbonyls in Indoor Air,” Proceedings of the National Academy of Sciences, Vol. 107, No. 15, 2010, pp. 6568-6575. doi:10.1073/pnas.0904498106
- J. Tonzetich, “Direct Gas Chromatographic Analysis of Sulphur Compounds in Mouth Air in Man,” Archives of Oral Biology, Vol. 16, No. 6, 1971, pp. 587-597. doi:10.1016/0003-9969(71)90062-8
- H. Shechter, “Spectrophotometric Method for Determination of Ozone in Water,” Water Research, Vol. 7, No. 5, 1973, pp. 729-729. doi:10.1016/0043-1354(73)90089-4
- M. Grootveld, E. B. Henderson, A. J. Farrell, D. R. Blake, H. G. Parkes and P. Haycock, “Oxidative Damage to Hyaluronate and Glucose in Synovial Fluid during Exercise of the Inflamed Rheumatoid Joint: Detection of Abnormal Low-Molecular-Mass Metabolities by Proton NMR Spectroscopy,” Biochemical Journal, Vol. 273, No. 2, 1991, pp. 459-467.
- J. Grigor and A. J. Roberts, “Reduction in the Levels of Oral Malodor Precursors by Hydrogen Peroxide: In Vitro and in Vivo Assessments,” Journal of Clinical Dentistry, Vol. 3, No. 4, 1992, pp. 111-115.
- A. E. Martell and R. J. Motekaitis. “The Determination and Use of Stability Constants,” VCH Weinham, Hoboken, 1988.
- C. J. L. Silwood, E. Lynch, S. Seddon, A. Sheerin, A. W. D. Claxson and M. C. Grootveld, “1H-NMR Analysis of Microbial-Derived Organic Acids in Primary Root Carious Lesions and Saliva,” NMR in Biomedicine, Vol. 12, No. 6, 1999, pp. 345-356. doi:10.1002/(SICI)1099-1492(199910)12:6<345::AID-NBM580>3.0.CO;2-C
- J. D. B. Featherstone and B. E. Rodgers, “Effect of Acetic, Lactic and Other Organic Acids on the Formation of Artificial Carious Lesions,” Caries Research, Vol. 15, 1981, pp. 377-385. doi:10.1159/000260541
- J. Tonzetich and P. A. W. Carpenter, “Production of Volatile Sulphur Compounds from Cysteine, Cysteine and Methionine by Human Dental Plaque,” Archives of Oral Biology, Vol. 16, No. 6, 1971, pp. 599-607. doi:10.1016/0003-9969(71)90063-X
- J. Tonzetich, P. W. Johnson and S. K. Ng, “Pathways of Cystine Metabolism in Human Saliva,” Archives of Oral Biology, Vol. 24, No. 1, 1979, pp. 35-39. doi:10.1016/0003-9969(79)90172-9
- R. Battino, T. R. Rettich and T. Tominaga, “The Solubility of Oxygen and Ozone in Liquids,” Journal of Physical Chemistry Reference Data, Vol. 12, No. 2, 1983, pp. 163-178. doi:10.1063/1.555680
- A. Bender, et al., “Adaptive Antioxidant Methionine Accumulation in Respiratory Chain Complexes Explains the Use of a Deviant Genetic Code in Mitochondria,” Proceedings of the National Academy of Sciences, Vol. 105, No. 43, 2008, pp. 16496-16501. doi:10.1073/pnas.0802779105
- R. E Thach, K. F. Dewey, J. C. Brown and P. Doty, “Formylmethionine Codon AUG as an Initiator of Polypeptide Synthesis,” Science, Vol. 153, No. 3734, 1966, pp. 416-418. doi:10.1126/science.153.3734.416
- J. R. Kanovsky and P. Sima, “Singlet Oxygen Production from the Reactions of Ozone with Biological Molecules,” Journal of Biological Chemistry, Vol. 266, No. 14, 1990, pp. 9039-9042.
- R. Bonnett and G. Martinez, “Photobleaching of Sensitisers used in Photodynamic Therapy,” Tetrahedron, Vol. 57, No. 47, 2001, pp. 9513-9547. doi:10.1016/S0040-4020(01)00952-8
NOTES
*Corresponding author.