Food and Nutrition Sciences
Vol.08 No.12(2017), Article ID:81167,13 pages
10.4236/fns.2017.812083
Ascorbic Acid Attenuates Acute Ethanol-Induced Liver Injury in SMP30 Knockout Mice
Eun-Mi Cho, H. M. Arif Ullah, Ahmed K. Elfadl, Myung-Jin Chung, Soong-Koo Kim, Yong Deuk Kim, Eun-Joo Lee, Kyu-Shik Jeong*
College of Veterinary Medicine, Kyungpook National University, Daegu, Republic of Korea
Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 13, 2017; Accepted: December 15, 2017; Published: December 19, 2017
ABSTRACT
Ascorbic acid (AA) is recognized as a free radical scavenger that protects cells from oxidative stress-induced damage. However, no studies have investigated the role of AA in acute alcoholic liver disease using senescence marker protein-30 (SMP30) knockout (KO) mice. SMP30 is a novel 34-kDa protein involved in AA biosynthesis. The present study aimed to elucidate the physiological functions of AA in acute ethanol-induced liver injury using SMP30 KO mice, which cannot synthesize AA in vivo. After a 4-week experimental period, mice were divided into six groups. The following three groups comprised the ethanol treatment groups: WT-E group (wild-type), KV-E group (AA-supplemented), and KT-E group (AA-deficient). Mice were exposed to an acute dose of ethanol (6 g ethanol/kg) administered by gavage once a day for three days. The other three control groups, namely, WT-C, KV-C, and KT-C control groups, received an equal volume of water via oral administration. Analysis of changes in body weight showed that mice in the KT-E group had significant loss of body weight compared to the control, KV-E, and WT-E groups. Behavioral analysis revealed that alcohol exposure significantly increased alcohol sensitivity in the KT-E group, whereas the WT-E, KV-E, and control groups developed ethanol tolerance. Aspartate transaminase (AST) levels in the KT-E group were significantly higher than those in the control, KV-E, and WT-E groups. The number of large and binucleated hepatocytes was significantly higher in the KT-E group than in the KV-E and WT-E groups. In addition, cytochrome P450 2E1 (CYP2E1) was over expressed in the central vein in the KT-E group when compared to the KV-E and WT-E groups. Our current findings indicate that AA supplementation in SMP30 KO mice can alleviate alcohol-induced liver damage by down regulating CYP2E1 expression. These results suggest that reduced CYP2E1 expression is a novel mechanism responsible for AA-induced reduction of ethanol-mediated oxidative stress.
Keywords:
Ascorbic Acid, Alcohol, Liver, SMP30

1. Introduction
Ascorbic acid (AA) is widely known as a water-soluble antioxidant that plays a major role in preventing cellular damage caused by free radicals, highly reactive species leading to cell damage and homeostatic disruption [1] . Various studies have been conducted to investigate the anti-oxidative properties of AA. The antioxidants act at different levels mainly through radical scavenging to detoxify reactive oxygen species (ROS) [2] . A number of reports have suggested that antioxidants, such as vitamin C [3] , vitamin E, and selenium, exert inhibiting effects against free radical-induced damage due to alcohol consumption [4] [5] . Here, we described the role of AA in acute alcohol-induced liver injury in senescence marker protein-30 (SMP30) knockout (KO) mice.
SMP30 is a novel 34-kDa protein whose expression in the liver, kidneys, and lungs decreases with age in an androgen-independent manner [6] [7] . Recently, SMP30 was verified as a gluconolactonase involved in L-ascorbic acid biosynthesis [8] . However, no similar studies have been conducted using SMP30 KO mice as an animal model for AA deficiency.
Alcoholic liver disease (ALD) is caused by excessive alcohol consumption and is characterized by a wide spectrum of hepatic lesions, ranging from steatosis (fatty liver) to severe steatohepatitis, as well as cirrhosis and hepatocellular carcinoma [9] [10] , which represents a major cause of morbidity and mortality in Western countries. Several pathways have been suggested to play key roles in mediating ethanol-induced oxidative stress [11] . Three pathways are primarily involved in the biotransformation process during ethanol oxidation [12] . The primary metabolic pathway is mediated by acetaldehydes produced by alcohol dehydrogenase, which is primarily expressed in the liver and gastrointestinal tract [12] [13] . The second metabolic pathway is mediated by catalase [13] , and the last pathway is mediated by cytochrome P450, particularly cytochrome P450 2E1 (CYP2E1) [14] . Alcohol-induced formation of liver lesions is primarily caused by aldehydes, cytochrome P450, and enzymatic products of catalase, which are toxic compounds produced during ethanol biotransformation [15] .
Oxidative stress can also be induced by elevated fatty acid production due to alcohol uptake [16] . The cytotoxic effects of ethanol biotransformation and ROS generation include enhanced cell injury, apoptosis, and necrosis, which in turn trigger ALD [12] .
In the current study, we focused on the effects of AA on alcohol-induced liver injury in SMP30 knockout (KO) mice, which cannot synthesize AA in vivo.
2. Materials and Methods
2.1. Animal Project
2.1.1. Chemicals
Ascorbic acid (AA; ≥98% purity) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Analytical-grade ethanol was purchased from Duksan Pure Chemicals (Ansan, Korea). All other chemicals were of the highest grade and were purchased from commercial suppliers.
2.1.2. Animals
SMP30 KO mice were generated following a previously described gene targeting technique [16] . SMP30 KO mice and wild-type (C57BL/6) mice were maintained for 4 weeks as follows: 1) WT (wild-type mice, fed with autoclaved mouse chow + tap water), 2) KV (SMP30 KO mice, fed with autoclaved mouse chow + water supplemented with 1.5 g AA/L), and 3) KT (SMP30 KO mice, fed with AA-free diet + tap water). Adult male (7 weeks old) wild-type and SMP30 KO mice weighing 16 - 17 g were obtained from the Pathology Laboratory, College of Veterinary Medicine, Kyungpook National University. Mice were divided into six groups comprising five mice each (5 mice/Group) and housed at 22˚C ± 1˚C with 55% ± 5% relative humidity under a 12 h light/dark cycle. All animal experiments were approved by the Institutional Animal Care and Use Committee at Kyungpook National University in accordance with the National Institutes of Health (NIH) guidelines.
2.1.3. Experimental Protocol
SMP30 KO and wild-type mice were divided into three groups each and maintained for 4 weeks as follows: 1) WT (wild-type mice, fed with autoclaved mouse chow + tap water), 2) KV (SMP30 KO mice, fed with autoclaved mouse chow + water supplemented with 1.5 g AA/L), and 3) KT (SMP30 KO mice, fed with AA-free diet + tap water). After the 4-week experimental period, mice were divided into three ethanol-treated groups (WT-E, KV-E, and KT-E) and three control groups (WT-C, KV-C, and KT-C). Mice in the ethanol-treated groups were exposed to an acute dose of 6 g ethanol/kg body weight administered by gavage once a day for three days (0, 24, and 48 h), whereas control mice received an equal volume of water by oral administration. [17] All animals were sacrificed at 12 h after the third ethanol administration (Table 1).
2.1.4. Genotyping
Mice were maintained in a temperature- and humidity-controlled room under a 12 h light/dark cycle. Genomic DNA was extracted from the tail tip as previously described [18] . SMP30 genotyping was performed via polymerase chain reaction (PCR) using the primers TA4 (5'-CAAGTAACTCTAGGTATGGAC-3'), TS3 (5'-CTAGCCATGGTGGATGAAGAT-3'), and NEO (5'-TCGTGCTTTACG GTATCGCCGCTCCCG ATT-3'). PCR was run according to the following profile: 94˚C for 3 min; 35 cycles of 94˚C 1 min, 55˚C for 1 min, and for 72˚C for 2 min; and 72˚C for 5 min.

Table 1. Animal treatment groups.
a: Acute dose of ethanol (6 g/kg) once a day for 3 days; b: 1.5 g ascorbic acid, (AA/L) supplemented with water.
2.1.5. Measurement of Serum ALT and AST Levels
Blood samples were placed into centrifuge tubes and centrifuged at 800 g at 4˚C for 10 min to separate the serum. Serum alanine transaminase (ALT) and aspartate transaminase (AST) levels were measured on an automated analyzer (Konelab 20, Thermo Clinical Lab systems, Finland). We referred the control values of ALT and AST in normal C57BL/6 mice to baseline hematology and clinical chemistry values as a function of sex and age for Charles River outbred mice.
2.1.6. Histological Examination of Liver Tissue
Previous studies have reported that a single dose of ethanol at 6 g/kg administered by gavage successfully [17] causes liver histopathologies, such as microvesicular steatosis and necrosis in hepatocytes, which are characterized by cell enlargement and nuclear dissolution [19] . In the present study, the same ethanol dose (6 g/kg) was administered to the mice once a day for three days. After sacrificing the mice, a small portion of liver was immediately removed, washed with cold normal saline, and fixed in 10% neutral-buffered formaldehyde solution. Fixed tissue fragments were embedded in paraffin and subjected to histopathological examination. Sections were cut into 6 - 8-μm-thick sections, placed on microscope slides, deparaffinized, and hydrated. Finally, sections were stained with hematoxylin and eosin (H & E) for histological observation of hepatic tissues.
2.1.7. Immunohistochemistry of Liver Tissues
Liver sections (6 - 8 μm) were mounted on glass slides, deparaffinized, and dehydrated in toluene and graded alcohol series. Sections were then blocked with endogenous peroxidase and incubated in 3% hydrogen peroxide (H2O2) solution in methanol at room temperature (RT) for 30 min. The antigen was retrieved by heating the sections in 0.01 mM citrate buffer (at pH 6.0) for 10 min in a microwave. After cooling, tissue sections were washed with phosphate-buffered saline (PBS) and incubated with 10% non-immune goat serum for 1 h at RT to inhibit non-specific reactions. Sections were then incubated with CYP2E1 primary antibody (1:200 dilution; Chemicon International, Inc., U.S.A.). The antigen-antibody complex was visualized via the labeled streptavidin-biotin method using a Histostantin-plus bulk kit (Zymed Laboratories Inc, Southern San Francisco, CA, U.S.A.), followed by incubation with 3,3'-diaminobenzidine tetrahydrochloride (DAB, Zymed Laboratories Inc, San Francisco, CA, U.S.A.) as chromogen. After washing, slides were counterstained with Meyer's hematoxylin and washed with tap water. For the negative control, the primary antibody was replaced with PBS.
2.2. Statistical Analysis
Data were expressed as mean ± standard error of the mean (SEM). Data were analyzed using a one-way analysis of variance (ANOVA), followed by Dunnett’s t-test for multiple comparisons using SPSS software (Release 12.0, SPSS Inc., and U.S.A.). Statistical significance was considered at P < 0.05.
3. Results
3.1. Changes in Body Weight
Mean body weight after ethanol administration was compared with initial body weight for mice in each group. All control groups that were orally administered with water at the same dosage showed higher body weights than the ethanol-treated groups. Mice in the WT-C, KV-C, and KT-C control groups gained an average of 20.3%, 21.7%, and 5.8% of their initial body weights, respectively. In ethanol-treated mice, the KT-E group (AA-deficient) lost an average of 2.2% of their initial body weights, but the WT-E (wild-type) and KV-E groups (AA-supplemented) gained an average of 7.3% and 5.6% of their initial body weights, respectively. Analysis of body weight changes after ethanol administration showed that mice in the KT-E group lost more weight than those in the KV-E and WT-E groups. The above findings confirmed that AA deficiency elevates stress levels on whole body system, which in turn leads to greater body weight loss when administered with an equal dose of ethanol compared to normal groups (Figure 1).
3.2. Changes in Behavior
After the first ethanol administration, mice in all treatment groups exhibited drunk-like behavior and fell down within 15 min. After the second ethanol administration, all mice in the WT-E group fell down, except for two mice. Finally, after the third ethanol administration, only four mice in the KT-E group fell down (Figure 2), indicating that AA supplementation remarkably attenuated ethanol-mediated hypnotic effects. On the other hand, mice in the control, WT-E, and KV-E groups quickly developed tolerance to ethanol, although mice in the KT-E (AA-deficient) group maintained sensitivity to the hypnotic effects of ethanol.
3.3. Changes in Serum Biochemical Parameters
Serum transaminases are sensitive indicators of liver cell injury [20] . Ethanol consumption increases serum levels of Serum alanine transaminase (ALT) and
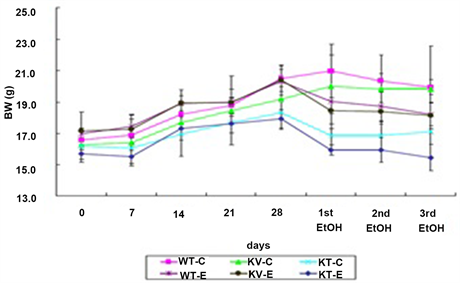
Figure 1. Effect of ascorbic acid (AA) supplementation on body weight throughout the experimental period from day 0 until the third ethanol administration (0 - 31 days). Mice were gavaged with three doses of ethanol (6 g/kg) or water. The graph shows the body weight index of each group. Mice in the KT-E group (AA-deficient) showed greater weight loss than mice in the WT-E and KV-E groups (AA-supplemented). Data are expressed as mean ± SEM (n = 5).

Figure 2. Effect of AA on behavior after repeated ethanol administration (6 g/kg) observed at 10 min after the third ethanol dose. Mice in the WT-E and KV-E groups (AA-supplemented) developed tolerance to ethanol, whereas mice in the KT-E group (AA-deficient) showed sensitivity to ethanol.
aspartate transaminase (AST). As shown in Figure 3, AST expression was significantly upregulated in mice in the KT-E group (P < 0.05), whereas mice in the WT-E and KV-E groups had lower serum levels of ALT and AST. However, mice in the KV-E group, which were supplemented with AA and administered with ethanol, showed highly stable ALT and AST levels that were similar to those in the control groups (Figure 3). These results revealed that AA is a potent suppressor of transaminase production in the serum.
3.4. Histopathological Observation of Liver Tissue
Occurrence of histopathological changes in the liver is a characteristic feature observed during onset and progression of alcoholic liver disease. Here, mice were administered with ethanol at 6 g/kg once a day for 3 days to investigate the
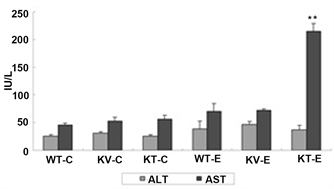
Figure 3. Changes in serum aspartate transaminase (AST) levels at 12 h after the third dose of ethanol (6 g/kg) or water in SMP30 KO and wild-type mice. Data are expressed as mean ± SEM (n = 5). Mice in the KT-E group (AA-deficient) showed significantly higher AST levels (P < 0.01) than those in the WT-E and KV-E groups (present AA). The normal range of AST is 73.6 ± 15.3 IU/L.
corresponding hepatic lesions. H & E staining showed significant histopathological changes in the hepatic tissues of ethanol-exposed mice (Figure 4). To examine the extent of ethanol-induced hepatic damage, we counted the number of enlarged hepatocytes and binucleated hepatocytes. The increase in the number of abnormal and binucleated hepatocytes is a marker of the severity of free radical-induced hepatic damage [21] . We observed a higher number of binucleated hepatocytes in the KT-E group than in the other groups. In particular, binucleated hepatocytes were significantly more pronounced in the KT-E group (AA-deficient) (P < 0.05), but were less evident in the AA-supplemented group (KV-E) and wild-type group (WT-E) (Figure 4). These results demonstrated that AA plays a vital role in suppressing the formation of large and binucleated hepatocytes in the liver.
3.5. AA Downregulated CYP2E1 Expression in Response to Acute Ethanol Exposure
Oxidative metabolism of ethanol by cytochrome P450 2E1 (CYP2E1) is known to contribute to ethanol-induced liver injury by inducing oxidative stress [22] . Thus, CYP2E1 levels in the livers of mice in the ethanol-treated groups and the control groups were evaluated via immunohistochemical staining. Ethanol-inducible CYP2E1 expression was observed in liver sections from all experimental groups. CYP2E1 expression levels in hepatocytes surrounding the central veins of the livers of mice in the control groups (WT-C, KV-C, and KT-C) were considered normal. CYP2E1 expression levels were found to be lower around the areas of pericentral veins in the AA-supplemented (KV-E) and wild-type (WT-E) groups when compared with those in the ethanol-treated group, but were significantly (P < 0.05) higher in the AA-deficient group (KT-E) (Figure 5). Based on these findings, we suggest that AA exerts a protective effect
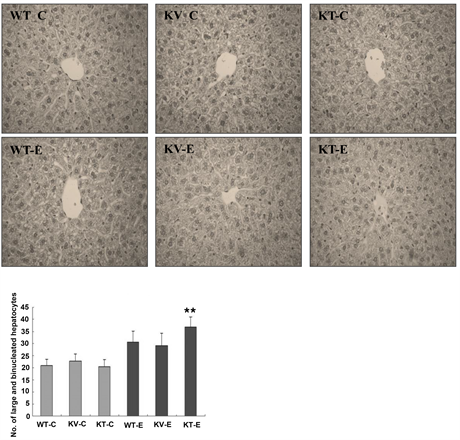
Figure 4. Histopathological changes in the liver tissues of mice after administration of ethanol (6 g/kg) or water. Tissues were subjected to histopathological examination via hematoxylin and eosin (H & E) staining and examined at 400× magnification. No histopathological abnormalities were observed in all the control groups (WT-C, KV-C, and KT-C). Mice in the KT-E group developed binucleated hepatocytes with enlarged nuclei. The number of binucleated hepatocytes with enlarged nuclei was significantly (P < 0.05) higher in the KT-E group (AA-deficient) than in the WT-E (wild-type) and KV-E (AA-supplemented) groups. Data are expressed as mean ± SEM (n = 5).
against ethanol-induced oxidative stress in the liver by inducing CYP2E1 expression.
4. Discussion
Ascorbic acid (AA) is synthesized from D-glucuronate [1] . In the AA synthesis pathway, L-gulonate is converted to L-gulonolactone by a lactonase called SMP30, whose absence leads to AA deficiency in mice [23] . SMP30 KO mice developed symptoms of scurvy when fed with an AA-free diet [8] . AA acts as a free-radical scavenger that is essential in preventing cellular damage caused by the free radicals and free radical-mediated lipid peroxidation [4] . Although multiple studies have evaluated the potential biological role of AA, no similar experimental evaluation has been conducted using SMP30 KO mouse as an animal model. Therefore, we aimed to establish a condition of ethanol exposure to produce liver injury in SMP30 KO mice, to elucidate the in vivo functions of AA.

Figure 5. AA attenuated ethanol-induced oxidative stress by downregulating CYP2E1. Mice were administered with ethanol (6 g/kg) or water. Results of immunohistochemical (IHC) staining revealed that CYP2E1 expression was significantly (P < 0.05) higher around the area of pericentral veins of the liver tissues in KT-E (AA-deficient) mice. On the other hand, CYP2E1 expression was downregulated in the control groups and the KV-E (AA-supplemented) and WT-E groups. Staining intensity was calculated using an image analysis program. Data are expressed as mean ± SEM (n = 5). All sections were observed under 400× magnification.
Oxidative stress plays a vital role in alcoholic hepatic injury [24] [25] . Accumulating evidence has indicated that ethanol metabolism leads to excessive production of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) and superoxide anion radical [26] . ROS-induced cellular stress is exacerbated by bacterial translocation, hypoxia, and release of proinflammatory cytokines [27] . Accumulation of CYP2E1 in mitochondrial membrane leads to local and cellular oxidative stress and plays a vital pathogenic role in the development of alcoholic liver disease (ALD) [28] [29] . Generation of free radicals during ethanol oxidation by cytochrome P4502E1 is an important step in the pathogenesis of ethanol-associated liver injury [30] . CYP2E1 is likely to play a critical role in promoting the generation of ROS, resulting in hepatocyte damage, which is primarily blocked by anti-oxidant enzymes, especially at the pericentral area [31] . Therefore, inhibition of the CYP2E1 pathway is critical for protection against ethanol-induced liver injury [32] . Experimental data have demonstrated that a CYP2E1 induction is correlated with reduced ethanol-mediated hepatotoxicity. The increase in ROS production and lipid peroxidation observed in the microsomes of rats subjected to chronic ethanol exposure was blocked by treatment with chemical inhibitors of CYP2E1 and anti-CYP2E1 immunoglobulin G [33] [34] .
Comparison of body weights and behavioral changes after ethanol administration showed that mice in the KT-E group (AA-deficient) were more sensitive than mice in the KV-E (AA-supplemented) and WT-E (wild-type) groups subjected to an equal ethanol dose. Our experimental study revealed that AA supplementation protects against homeostasis disruption by maintaining regular body weight and developing tolerance to ethanol. On the other hand, mice in the AA-deficient KT-E group showed significant loss of body weight and exhibited sensitivity to the hypotonic effects of ethanol (Figure 1 and Figure 2).
Ethanol oxidation, which can be mediated by the alcohol dehydrogenase (ADH) or the microsomal pathway, results in acetaldehyde production and leads to cell damage and subsequent increase the levels of serum transaminases (AST and ALT) in the blood [35] [36] . In this study, AST levels in the KT-E group were significantly (P < 0.05) higher than those in the KV-E and WT-E groups (Figure 3). However, AA supplementation resulted in marked recovery of AST and ALT to levels similar to normal values, as observed in the KV-E (AA-supplemented) and WT-E (wild-type) groups. These data demonstrate the protective effect of AA against ethanol-induced liver injury.
Under pathological conditions, dead liver cells are replaced through the proliferation of surviving liver cells. Binucleated hepatocytes, which are considered the most reliable markers of free radical-induced damage, become abundant in the liver to facilitate accelerated liver cell renewal [21] . Our study showed that the number of binucleated hepatocytes with enlarged nuclei significantly (P < 0.05) increased in the KT-E group (AA-deficient) compared to the KV-E (AA-supplemented) and WT-E (wild-type) groups (Figure 4). These results indicate that AA inhibited the production of binucleated hepatocytes in the KV-E and WT-E groups through an antioxidant defense mechanism to protect against alcohol-mediated oxidative stress.
Among the mechanisms by which ethanol induces oxidative stress, one central mechanism appears to be the induction of CYP2E1, which is an efficient generator of ROS [26] . Our results revealed that hepatic CYP2E1 expression was reduced in the KV-E group when compared to the other control groups (Figure 5). Ethanol is also a substrate of CYP2E1, which generates highly reactive products, such as acetaldehyde and the 1-hydroxyethyl radical and thus contributes to alcohol-induced liver damage [37] . In the intragastric model of ethanol feeding, a significant increase in microsomal lipid peroxidation was observed, and ethanol-induced liver pathology has been shown to correlate with CYP2E1 upregulation and enhanced lipid peroxidation [38] - [42] . In the present study, mice in the KV-E group showed the highest cytochrome CYP2E1 expression levels when compared to the KV-E and WT-E groups. In turn, elevated levels of CYP2E1 can aggravate acute alcohol-induced liver injury.
5. Conclusion
In conclusion, our results provide strong experimental evidence that ascorbic acid (AA) supplementation in SMP30 KO mice prevented alcohol-induced hepatic cellular damage. SMP30 KO mice were successfully used to elucidate the biological activities of AA and could thus serve as promising mouse models in subsequent studies that aim to investigate the role of AA in vivo. More detail study about ascorbic acid protecting alcohol-induced liver injury is needed.
Acknowledgements
This work is supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), which is funded by the Ministry of Health &Welfare, Republic of Korea (grant number: HI15C0001).
Cite this paper
Cho, E.-M., Ullah, H.M.A., Elfadl, A.K., Chung, M.-J., Kim, S.-K., Kim, Y.D., Lee, E.-J. and Jeong, K.-S. (2017) Ascorbic Acid Attenuates Acute Ethanol-Induced Liver Injury in SMP30 Knockout Mice. Food and Nutrition Sciences, 8, 1114-1126. https://doi.org/10.4236/fns.2017.812083
References
- 1. Unlu, A., et al. (2016) High-Dose Vitamin C and Cancer. Journal of Oncological Science, 1, 10-12.
https://doi.org/10.1016/j.jons.2015.11.010 - 2. Lobo, V., Patil, A., Phatak, A. and Chandra, N. (2010) Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharmacognosy Reviews, 4, 118-126.
https://doi.org/10.4103/0973-7847.70902 - 3. Van der Reest, J. and Gottlieb, E. (2016) Anti-Cancer Effects of Vitamin C Revisited. Cell Research, 26, 269-170. https://doi.org/10.1038/cr.2016.7
- 4. Ivaram, A.G., Suresh, M.V. and Indira, M. (2003) Combined Effect of Ascorbic Acid and Selenium Supplementation Alcohol-Induced Oxidative Stress in Guinea Pigs. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology, 134, 397-401.
https://doi.org/10.1016/S1532-0456(03)00009-7 - 5. Cornelli, U., et al. (2001) Bioavailability an Antioxidant Activity of Some Food Supplements in Men and Women Using The D-Roms Test as a Marker of Oxidative Stress. Journal of Nutrition, 131, 3208-3211.
- 6. Ishigami, A. and Maruyama, N. (2007) Significance of SMP30 in Gerontology. Geriatrics & Gerontology International, 7, 316-325.
https://doi.org/10.1111/j.1447-0594.2007.00420.x - 7. Maruyama, N., Ishigami, A. and Kondo, Y. (2010) Pathophysiological Significance of Senescence Marker Protein-30. Geriatrics & Gerontology International, 10, 88-98.
https://doi.org/10.1111/j.1447-0594.2010.00586.x - 8. Kondo, Y., et al. (2006) Senescence Marker Protein 30 Functions as Gluconolactonase in L-Ascorbic Acid Biosynthesis, and Its Knockout Mice Are Prone to Scurvy. Proceedings of the National Academy of Science, 103, 5723-5728.
https://doi.org/10.1073/pnas.0511225103 - 9. Beier, J.I., Arteel, G.E. and McClain, C.J. (2011) Advances in Alcoholic Liver Disease. Current Gastroenterology Reports, 13, 56-64.
https://doi.org/10.1007/s11894-010-0157-5 - 10. Liu, S.L., et al. (2011) Discovery of Serum Biomarkers of Alcoholic Fatty Liver in a Rodent Model: C-Reactive Protein. Journal of Biomedical Science, 18, 1-10.
https://doi.org/10.1186/1423-0127-18-52 - 11. Choudhury, M., et al. (2010) Evidence for the Role of Oxidative Stress in the Acetylation of Histone H3 by Ethanol in Rat Hepatocytes. Alcohol, 44, 531-540.
https://doi.org/10.1016/j.alcohol.2010.06.003 - 12. Louvet, A. and Mathurin, P. (2015) Alcoholic Liver Disease: Mechanisms of Injury and Targeted Treatment. Nature Reviews Gastroenterology & Hepatology, 12, 231-242.
https://doi.org/10.1038/nrgastro.2015.35 - 13. Cederbaum, A.I. (2012) Alcohol Metabolism. Clinical Liver Disease, 16, 667-685.
https://doi.org/10.1016/j.cld.2012.08.002 - 14. Lieber, C.S. (2004) Alcoholic Fatty Liver: Its Pathogenesis and Mechanism of Progression to Inflammation and Fibrosis. Alcohol, 34, 9-19.
https://doi.org/10.1016/j.alcohol.2004.07.008 - 15. Park, H.Y., et al. (2013) Narirutin Fraction from Citrus Peels Attenuates Alcoholic Liver Disease in Mice. Food & Chemical Toxicology, 555, 637-644.
https://doi.org/10.1016/j.fct.2013.01.060 - 16. Wu, D. and Cederbaum, A.I. (2003) Alcohol, Oxidative Stress, and Free Radical Damage. Alcohol Research & Health, 27, 277-284.
- 17. Cho, Y.E., Im, E.J., Moon, P.G., Mezey, E., Song, B.J. and Baek, M.C. (2017) Increased Liver-Specific Proteins in Circulating Extracellular Vesicles as Potential Biomarkers for Drug- and Alcohol-Induced Liver Injury. PloS One, 12, e0172463.
https://doi.org/10.1371/journal.pone.0172463 - 18. Ishigami, A., et al. (2002) Senescence Marker Protein-30 Knockout Mouse Liver Is Highly Susceptible to Tumor Necrosis Factor-Alpha- and Fas-Mediated Apoptosis. American Journal of Pathology, 161, 1273-1281.
https://doi.org/10.1016/S0002-9440(10)64404-5 - 19. Henaneberger, C., Grantyn, R. and Rothe, T. (2000) Rapid Genotyping of Newborn Gene Mutant Mice. Journal of Newuroscience Methods, 100, 123-126.
https://doi.org/10.1016/S0165-0270(00)00241-7 - 20. Zhou, Z., et al. (2003) A Critical Involvement of Oxidative Stress in Acute Alcohol-Induced Hepatic TNF-Alpha Production. American Journal of Pathology, 163, 1137-1146.
https://doi.org/10.1016/S0002-9440(10)63473-6 - 21. Coodley, E.L. (1971) Enzyme Diagnosis in Hepatic Disease. American Journal of Gastroenterology, 56, 413-419.
- 22. Maharwal, J., Samarth, R.M. and Saini, M.R. (2005) Antioxidative Effect of Rajgira Leaf Extract in Liver of Swiss Albino Mice after Exposure to Different Doses of Gamma Radiation. Phytotherapy Research, 19, 717-720.
- 23. Roman, J., et al. (1999) Differential Role of Ethanol and Acetaldehyde in the Induction of Oxidative Stress in HEP G2 Cells: Effect on Transcription Factors AP-1 and NF-kappaB. Hepatology, 30, 1473-1480.
- 24. Linster, C.L. and Van, S.E. (2007) Vitamin C Biosynthesis, Recycling and Degradation in Mammals. FEBS J, 274, 1-22.
- 25. Yan, X., et al. (2017) Inhibition of Histone Acetylation by Curcumin Reduces Alchol-Induced Fetal Cardiac Apoptosis. Journal of Biomedical Science, 24, 1-9.
- 26. Hoek, J.B. and Pastorino, J.G. (2002) Ethanol, Oxidative Stress, and Cytokine-Induced Liver Cell Injury. Alcohol, 27, 63-68.
- 27. Lu, Y. and Cederbaum, A.I. (2008) CYP2E1 and Oxidative Liver Injury by Alcohol. Free Radical Biology and Medicine, 44, 723-738.
- 28. Schieber, M. and Chandel, N.S. (2014) ROS Function in Redox Signaling and Oxidative Stress. Current Biology, 24, 453-462.
- 29. Knockaert, L., et al. (2011) Mitochondrial CYP2E1 Is Sufficient to Mediate Oxidative Stress and Cytotoxicity Induced by Ethanol and Acetaminophen. Toxicology in Vitro, 25, 475-484.
- 30. Zhang, P., et al. (2014) Berberine Protects Liver from Ethanol-Induced Oxidative Stress and Steatosis in Mice. Food & Chemical Toxicology, 74, 225-232.
https://doi.org/10.1016/j.fct.2014.10.005 - 31. Jordao, J.R., et al. (2004) Effect of an Acute Dose of Ethanol on Lipid Peroxidation in Rats: Action of Vitamin E. Food & Chemical Toxicology, 42, 459-464.
- 32. Koch, O.R., et al. (2004) Oxidative Stress and Antioxidant Defenses in Ethanol-Induced Cell Injury. Molecular Aspects of Medicine, 25, 191-198.
- 33. Tilg, H., Moschen, A.R. and Kaneider, N.C. (2011) Pathways of Liver Injury in Alcoholic Liver Diseases. Journal of Hepatology, 55, 1159-1161.
- 34. Cederbaum, A.I. (2003) Iron and CYP2E1-Dependant Oxidative Stress and Toxicity. Alcohol, 30, 115-120.
- 35. Ekstrom, G. and Ingelman-Sundberg, M. (1989) Rat Liver Microsomal NADPH-Supported Oxidase Activity and Lipid Peroxidation Dependant Onethanol-Inducible Cytochrome P450 (P-450ⅡE1). Biochemical Pharmacology, 38, 1313-1319.
- 36. Giannini, E.G., Roberto, T. and Vincenzo, S. (2005) Liver Enzyme Alteration: A Guide for Clinicians. Canadian Medical Association Journal, 172, 367-379.
- 37. Gowda, S., et al. (2009) A Review on Laboratory Liver Function Tests. The Pan African Medical Journal, 3, 1-11.
- 38. Cederbaum, A.I. (2006) CYP2E1-Biochemical and Toxicological Aspects and Role in Alcohol-Induced Liver Injury. Mount Sinai Journal of Medicine, 73, 657-672.
- 39. Bolt, H.M., Roos, P.H. and Their, R. (2003) The Cytochrom P-450 Isoenzyme CYP2E1 in the Biological Processing of Industrial Chemicals: Consequences for Occupational and Environmental Medicine. International Archives of Occupational and Environmental Health, 76, 174-178.
- 40. Koop, D.R. (1992) Oxidative and Reductive Mechanism by Cytochrome P4502E1. FASEB J, 6, 724-730.
- 41. Lieber, C.S. (1997) Cytochrome P-4502E1: Its Physiological and Pathological Role. Physiological Reviews, 77, 517-544.
- 42. Tanaka, E., Terada, M. and Misawa, S. (2000) CytochromeP450 2E1: Its Clinical and Toxicological Role. Journal of Clinical Pharmacy and Therapeutics, 25, 165-175.