Advances in Nanoparticles
Vol. 2 No. 4 (2013) , Article ID: 39008 , 7 pages DOI:10.4236/anp.2013.24045
Nanonization of Niflumic Acid by Co-Grinding
Department of Pharmaceutical Technology, University of Szeged, Szeged, Hungary
Email: *revesz@pharm.u-szeged.hu
Copyright © 2013 Tímea Szunyogh et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 17, 2013; revised September 26, 2013; accepted October 7, 2013
Keywords: Niflumic Acid; Particle Size Decrease; Nanonization; Co-Grinding
ABSTRACT
The aim of this study was to produce niflumic acid nanoparticles without using an organic solvent, in order to achieve an increased rate of dissolution of the final products. Co-grinding with excipients was used to decrease the particle size. Poloxamer 188 (P) and mannitol (M) applied as co-grinding materials stabilized the system, preventing aggregation of the nanocrystals. The morphology and particle size distribution of the products were visualized by using scanning electron microscopy and laser diffraction. The crystalline states of the samples were investigated by differential scanning calorimetry and X-ray powder diffraction. The rate of dissolution of niflumic acid was measured with a paddle method from simulated media. It was concluded that the particles produced were in the nanometer range (the mean particle size was ~250 nm) and the nanoparticles maintained their crystallinity during the process. The rate of dissolution of the coground sample was significantly improved.
1. Introduction
The formulation of poorly water-soluble drug materials is currently a major challenge for the pharmaceutical industry. In some cases, the production of microand nanosized crystals of active ingredients through novel methods can solve solubility problems, resulting in improved drug release and enhanced bioavailability. This is particularly important as concerns drugs with low solubility and high permeability in class II of the Biopharmaceutical Classification System (BCS) [1]. The Noyes-Whitney equation implies that a decrease in particle size will lead to an increase in the surface area, which can improve the drug dissolution velocity [2]. The processes of the dissolution and recrystallization of drug particles eventually come into equilibrium. Crystals in the nanometer range exhibit a higher dissolution pressure than that of microcrystals, and their saturation solubility is therefore also higher [3].
The various methods used to decrease particle size into the microor nanosize range can be divided into two main categories, as bottom-up and top-down techniques. These two categories are not separated sharply because use of the top-down together with the bottom-up is important to control the particle size [4-8]. In the bottom-up approach, the drug is first dissolved in a solvent, and the resulting solution is then added to a non-solvent, as a consequence of which the drug precipitates. Examples include supercritical fluid technology [9], evaporation precipitation from aqueous solution [10], melt emulsification, solvent diffusion and solvent evaporation. In the frequently used top-down methods, the raw material is progressively broken down until microor nanosized particles are produced. This particularly involves grinding [11]. In wet-grinding, the grinding medium is water or a suitable buffer; the drug and a stabilizer are used together. Dry-grinding is often used to decrease particle size in the absence of water or any organic solvent, but the particle size is limited because of the aggregation of the particles [12]. The use of one or more excipients can prevent the particles from undergoing aggregation. In co-grinding processes, the grinding chamber is charged with pearls or balls, a drug powder and a stabilizer. The pearls are rotated and generate strong shear forces which disintegrate the drug powder into nanoparticles. The physicochemical properties of the particles produced depend on the number of pearls, the amounts of drug and stabilizer, and the duration and the speed of grinding. This is a useful method because it involves low costs and can be carried out easily and economically, without any organic solvent. It can allow rapid production [13,14].
The model drug used in our present work was niflumic acid (NIF), which is an important nonsteroidal anti-inflammatory drug, used to treat certain kinds of rheumatic diseases, for example rheumatoid arthritis, arthrosis and decreased joint diseases. It is a BCS II drug, with poor solubility and high permeability. Although it is used widely, it has some side-effects, such as nausea and vomiting [15,16].
Some results have been published earlier in connection with decrease of the particle size of NIF to the microand nanometer range with the aim of achieving faster dissolution and higher solubility. Several techniques have been used to prepare micrometer-sized NIF particles with excipients, e.g. use of a ternary system of NIF, hydroxypropyl-β-cyclodextrin (HPβCD) and polyvinylpirrolidone (PVP K-25) in different ratios. Co-evaporation has been used to improve the dissolution rate [17,18]. In other work, PVP K-25 was applied to prepare physical mixtures and solvent evaporated methods in order to extend the rate of dissolution of NIF [19,20].
In our own previous study, bottom-up and top-down methods were used to increase the extent of dissolution of NIF. The bottom-up solvent diffusion method was applied to change the crystal size and distribution and the physicochemical behavior of NIF. In this method, excipients are dissolved in water and NIF is also dissolved in a suitable organic solvent (ethyl acetate). In the solvent evaporation method, NIF was dissolved in acetone, which is immiscible with water. The mean particle size was only 5 µm and the dissolution rate was therefore significantly improved for both products [21]. Wet grinding as a top-down method was applied with excipients such as mannitol and Poloxamer188. This method also resulted in micro particles [22].
In our earlier work, the processibility of nanosized NIF was investigated. The first chosen process was electrospray crystallization [23]. This is a novel bottom-up method, where acetone is used as an organic solvent. The formulated particles have a mean size of ~500 nm, and range from amorphous to a crystalline state. The sample produced was mixed with excipients after the crystallization process. It was found that the rate of dissolution of the physical mixture was improved in simulated gastric juice and in intestinal juice, but it was not perfect. The amount of NIF dissolved was increased to 41% after 15 min and to 73% in 1 h [24,25]. The nanoparticles of NIF did not result in a sufficiently high dissolution pressure because of the cohesion forces and the necessary effect of the increased surface area was not attained.
The goal of our present research work was to produce NIF nanoparticles smaller than 500 nm, and with a higher dissolution rate than previously. Co-grinding without an organic solvent (dry-grinding) was used to produce a stable, nanocrystalline form of NIF. Poloxamer 188 (P) and mannitol (M) as co-grinding materials were used to stabilize the system against aggregation of the nanocrystals. The physicochemical properties of the nanonized products were compared with those of the starting physical mixture and the extents of dissolution of NIF were investigated.
2. Materials and Methods
2.1. Materials
NIF(2-{[3-(trifluoromethyl)phenyl]amino}-3-pyridinecarboxylic acid) with a mean particle size of ~80 µm was from Gedeon Richter Pharmaceutical Factory, Budapest, Hungary (indicated as Raw-NIF in the text). β-D-Mannitol (M) was purchased from Hungaropharma Plc. (Budapest, Hungary), and Poloxamer 188 (polyethylenepolypropylene glycol) (P) from Fluka (Ljubljana, Slovenia).
2.2. Preparations
2.2.1. Preparation of the Sample by Grinding
The Raw-NIF was ground without excipients (Mill-NIF) with a Planetary Monomill PM 100 (Retsch Technology GmbH, Haan, Germany). The grinding was performed for 2 h in a 50 cm3 stainless steel milling drum containing 10 stainless steel balls 10 mm in diameter.
2.2.2. Preparation of the Sample by Co-Grinding
Because of the possibility of aggregation, Raw-NIF was ground together with the calculated amount of M as carrier and P as additive. The product is referred to in the text as NIF-M-P co-grinding. The presence of M as carrier ensures the homogeneous distribution of the NIF in the system and prevents the aggregation of NIF crystals, while P as a stabilizer helps by wetting to arrest the aggregation of the drug. For comparison, the physical mixture of the components was used (NIF-M-P physical mixture), which was prepared from the calculated amounts of Raw-NIF and carriers (M and P) with a Turbula mixer (Willy A. Bachofen Machinenfabrik Basel, Switzerland) at 50 rpm for 10 min. The compositions of the samples are presented in Table 1.

Table 1. Compositions of the samples (w/w).
2.3. Micrometric Properties
2.3.1. Particle Size and Distribution
The volume particle-size distribution of Raw-NIF was measured by laser diffraction (Mastersizer S, Malvern Instruments Ltd., Worcestershire, UK), with the following parameters: 300RF lens; small volume dispersion unit (2000 rpm); the refractive index for the dispersed particles was 1.510 and that for the dispersion medium (a saturated aqueous solution of NIF) was 1.330. The water-soluble components dissolved readily in the dispersion medium, and for the other samples therefore, only the NIF was measured.
D (V; 0.1) is the particle diameter at which 10% of the particles in the volume have a diameter below this value. The volume median diameter D (V; 0.5) is the diameter at which 50% of the particles have a diameter above and 50% have a diameter below this value. D (V; 0.9), means that 90% of the volume is distributed below this particle diameter.
2.3.2. Particle Morphology
The shape and surface characteristics of the particles in the various samples were visualized by using a scanning electron microscope (SEM) (Hitachi S4700, Hitachi Scientific Ltd., Tokyo, Japan). Briefly, the samples were sputter-coated with gold-palladium under an argon atmosphere, using a gold sputter module in a high-vacuum evaporator, and the samples were examined with the SEM set at 15 kV. The air pressure was 1.3 - 13.0 mPa. NIF(2-{[3-(trifluoromethyl)phenyl]amino}-3-pyridinecarboxylic acid) with a mean particle size of ~80 µm was from Gedeon Richter Pharmaceutical Factory, Budapest, Hungary (indicated as Raw-NIF in the text).
2.4. Structural Parameters
2.4.1. Thermoanalytical Measurements
The Mettler Toledo thermal analysis system with the STARe thermal analysis program V9.1 (Mettler Inc., Schwerzenbach, Switzerland) was applied to characterize the structures of the products. The DSC (differential scanning calorimetry) and TG (thermogravimetry) measurements were made by examining approximately 2 - 5 mg of material in the temperature range between 25˚C and 300˚C. The heating rate was 5˚C min−1. Argon was used as carrier gas during the DSC investigations, at a flow rate of 10 l h−1.
In Equation (1), the crystallinity index for NIF (CINIF) was calculated from the heats of fusion:
 (1)
(1)
where  = normalized enthalpy (J·g−1) of NIF, and
= normalized enthalpy (J·g−1) of NIF, and  = normalized enthalpy (J·g−1) of Raw-NIF [26].
= normalized enthalpy (J·g−1) of Raw-NIF [26].
2.4.2. X-Ray Powder Diffraction (XRPD)
The physical states of the Raw-NIF and the excipients in the different samples were evaluated by XRPD with a Miniflex II X-ray Diffractometer (Rigaku Co., Tokyo, Japan), where the tube anode was Cu with Kα = 1.5405. Patterns were collected at a tube voltage of 30 kV and a tube current of 15 mA in step scan mode (4˚ min−1). The instrument was calibrated by using Si.
2.5. Physicochemical Properties
2.5.1. Drug Content
The drug contents in the NIF-M-P physical mixture and NIF-M-P co-grinding samples were determined by dissolving an amount of dried sample which contained 10 mg of NIF in 10 ml of intestinal juice (pH = 6.8 ± 0.1). The solution was then stirred with a magnetic stirrer (400 rpm) at 37˚C for 24 h. After this, the solution was filtered and analyzed spectrophotometrically (ATI-Unicam UVVis spectrophotometer) at 288 nm. Each sample was prepared and analyzed in triplicate.
2.5.2. In Vitro Dissolution Testing
The dissolution profiles of product samples containing 28 mg of NIF (corresponding to the therapeutic dose) were examined by a paddle method. The medium was 100 ml of gastric juice (pH = 1.2) or intestinal juice (pH = 6.8). The rotation speed of the paddles was 100 rpm and sampling was performed up to 120 min. After filtration (the pore size in filtration was 0.22 µm) with the application of a Millex-GV syringe-driven filter unit (Millipore Corporation, Bedford, USA) and dilution, the NIF contents of the samples were determined spectrophotometrically at 256 (gastric juice) or 288 nm (intestinal juice). Statistical tests to compare the dissolution data were performed by ANOVA. The difference between samples was deemed statistically significant if the 95% confidence intervals for the means did not overlap (p < 0.05).
3. Results and Discussion
3.1. Particle Size Distribution
The particle size analysis revealed that the crystals of Raw-NIF had a mean particle size of approximately 81 µm. After grinding without any excipients, the mean particle size of Mill-NIF decreased, to give a D (V; 0.5) value of 16.65 µm and aggregation was observed. In the NIF-M-P physical mixture, the size of the NIF crystals decreased to 62.58 µm because of the disintegrating effect in the Turbula mixer. After application of the excipients during the grinding process, the particle size of NIF was nearly 200 nm. The excipients (M and P) clearly prevented aggregation of the nanoparticles. Use of the excipients during the grinding process is therefore of fundamental importance (Table 2).
3.2. Particle Morphology
The changes in the habit of the NIF crystals were revealed by the SEM pictures (Figure 1).
The Raw-NIF crystals had a smooth surface with prismatic form, with a mean particle size of ~81 µm. In the case of the Mill-NIF, somewhat more spherical NIF crystals were found. It may be stated that there were some nanonized crystals present, but these particles were clustered into aggregates because of the high cohesion forces. Excipients are therefore needed during the milling process. The characteristic morphology of the Raw-NIF was also seen in the NIF-M-P physical mixture. In the NIFM-P co-grinding sample, submicron-sized NIF crystals measuring ~200 nm were produced. As M is a highly water-soluble compound, it could create a highly hydrophilic environment around the NIF nanoparticles and additionally the particle size of M decreased. The starting M had D (V; 0.5) = 36.37 µm.
3.3. Thermoanalytical Behavior
DSC was employed to investigate the melting points of the Raw-NIF and the samples and the crystallinity of

Table 2. Particle size distribution of NIF and NIF in samples.

Figure 1. SEM pictures of Raw-NIF and the products.
Raw-NIF and Mill-NIF. Single sharp endothermic peaks were observed (Figure 2), corresponding to the melting points of Raw-NIF (202.27˚C) and Mill-NIF (202.37˚C), i.e. nearly identical. The reason is the aggregation of the NIF nanocrystals in the Mill-NIF. The melting point of nanonized crystals is usually lower because their heat flow is faster than that of the microcrystals. The calculated crystallinity index for Mill-NIF was 99.77% (the reference value was 100% for Raw-NIF). It can be concluded that the grinding process did not influence the crystallinity of Mill-NIF. It is known that crystalline forms are more stable than amorphous material.
Figure 3 demonstrates three endothermic peaks for the NIF-M-P physical mixture: the melting point of p at 55˚C, the melting point of M at 166.4˚C, and the melting point of NIF at ~200˚C. For this sample, the melting point of NIF was decreased. For the co-ground sample, the melting point of NIF has almost disappeared, the reason probably being that the particle size has decreased and the NIF has dissolved into the molten M.
3.4. X-Ray Powder Diffraction
XRPD was used to examine the crystalline phase of the products. The pattern for Mill-NIF (Figure 4), exhibited the characteristic peaks of Raw-NIF at 2θ values of 8.2, 12.88, 16.16, 23.18 and 25.4. It can be seen that the intensities of the main peak at 2θ = 23.18 was decreased for the Mill-NIF sample because of the aggregation of the nanocrystals, but the sample was in crystalline form.
Comparison of the XRPD patterns of the products containing excipients revealed that the characteristic peaks of NIF-M-P physical mixture and NIF-M-P co-grinding were in the same positions, but with different peak intensities. The reason is the decrease in particle size and the presence of M, which covers the surface of the NIF, resulting in a reduced peak intensity. During the co-grinding process, the structure of the M is also modified from the β to the α and δ modifications (2θ = 11.48) (Figure 5).
3.5. Drug Content and in Vitro Dissolution Testing
NIF displays amphoteric behaviour, with isoelectric point at pH = 3.3, where the molecule is predominantly present in zwitterionic form. The dissolution of NIF is pH-dependent.
The drug content of the final products (NIF-M-P physical mixture and NIF-M-P co-grinding) was 98 ± 2%, in comparison with 100% for Raw-NIF.
The dissolution of Raw-NIF in simulated intestinal juice is better than that in simulated gastric juice (Figures 6 and 7). In the gastric juice, no significant differences were measured between the dissolution rates of
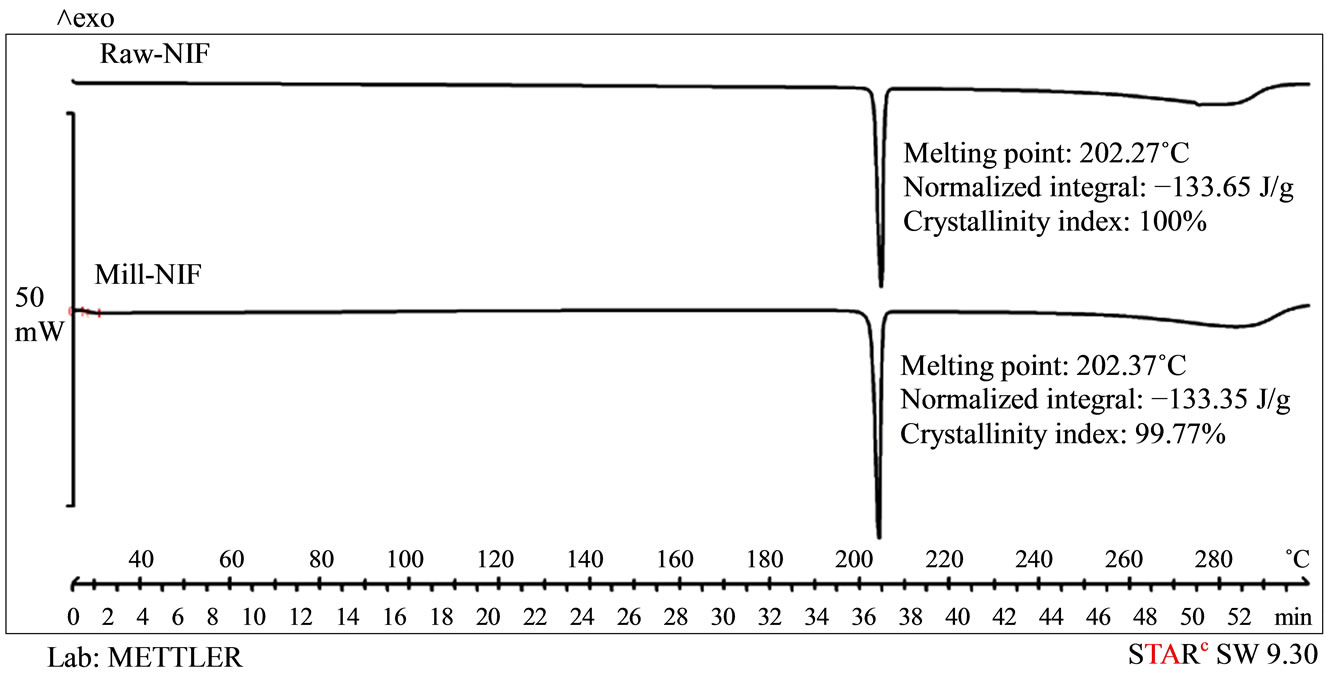
Figure 2. DSC curves of Raw-NIF and Mill-NIF.
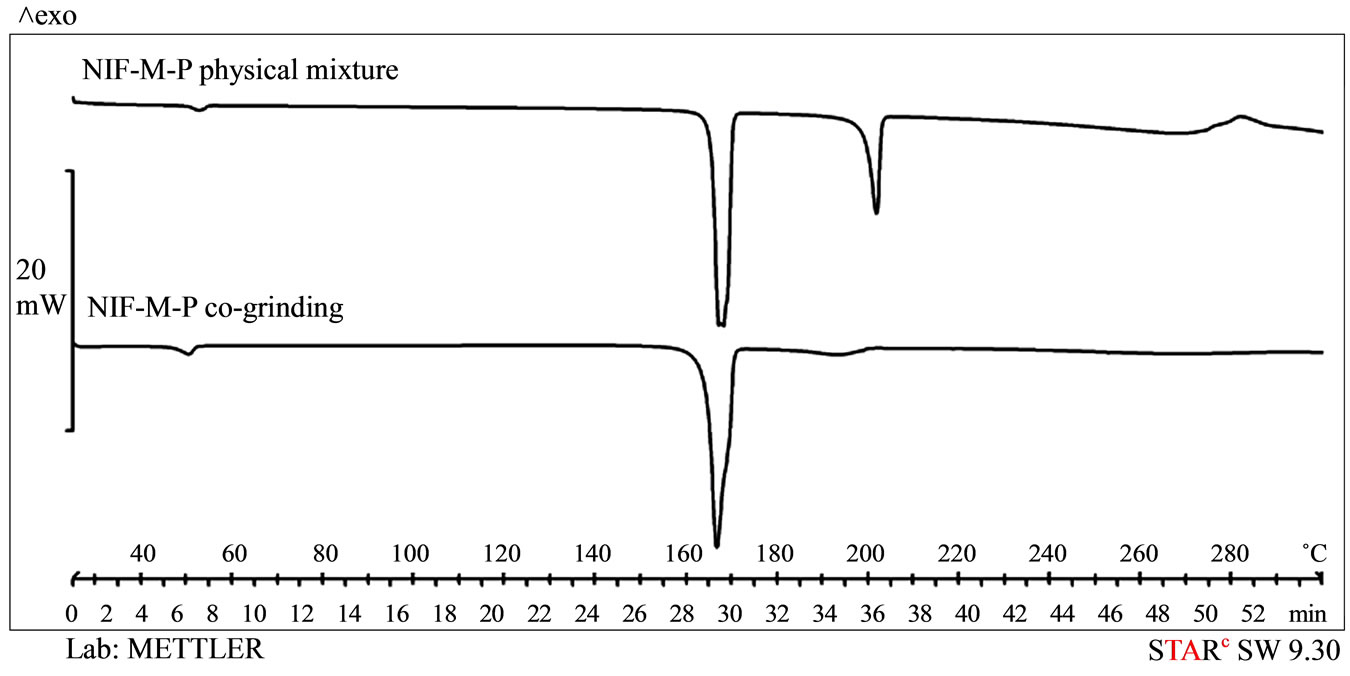
Figure 3. DSC curves of the samples made by using excipients.
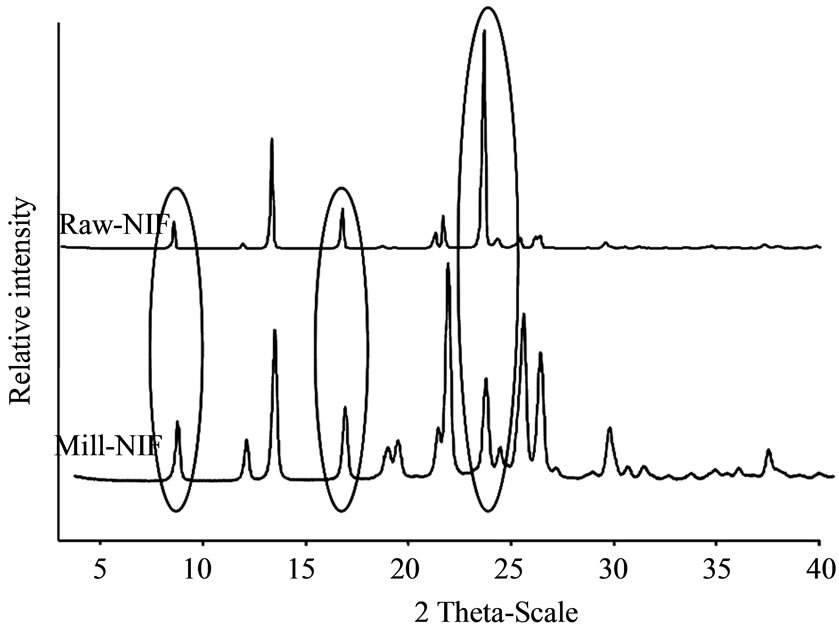
Figure 4. XRPD patterns of raw NIF and Mill-NIF.
Raw-NIF, Mill-NIF and NIF-M-P physical mixture. It can be concluded that neither the excipients nor the cho-

Figure 5. XRPD patterns of the references.
sen process influenced the extent of dissolution of NIF. Although Mill-NIF included nanosized particles, they clustered into aggregates, and there was therefore not
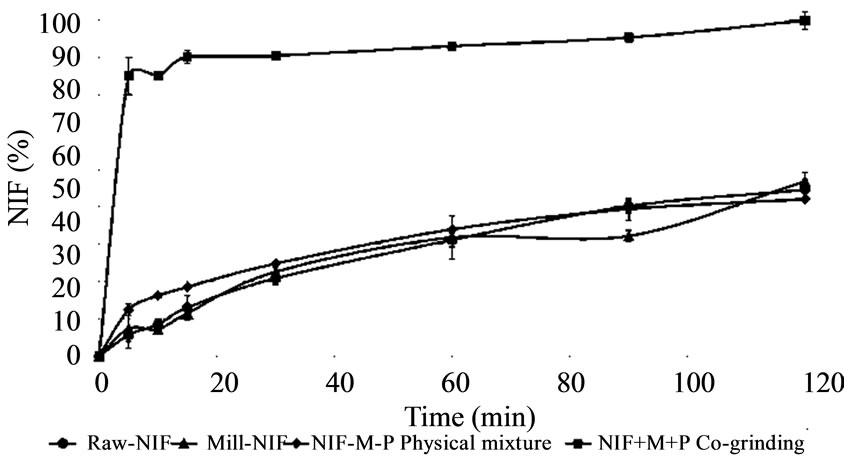
Figure 6. In vitro dissolution in gastric juice (pH = 1.2).

Figure 7. In vitro dissolution in intestinal juice (pH = 6.8).
sufficient surface to ensure an increase in the dissolution rate. In contrast, the presence of both M and P and the co-grinding process (NIF-M-P co-grinding) had a great effect on the dissolution rate: 70% of the NIF dissolved within 5 min. Although dissolution of 100% of the drug required 140 min, the dissolution profile in gastric juice, with a very fast initial dissolution phase, predicts a fast effect in pain therapy.
For all of the samples, the dissolution in intestinal juice, was faster all of the samples, than that in gastric medium. The solubility of NIF in gastric juice was 0.143 mg·ml−1 and that 2.125 mg·ml−1 was in intestinal juice. However, in the presence of the excipients, higher dissolution rates were achieved, and 60% of the NIF was dissolved in the first 10 min. It could therefore be concluded that the excipients influenced the dissolution rate. On combination of the excipients and the grinding process, the rate of dissolution was much better, nearly 100% of the NIF dissolving in 20 min.
4. Conclusions
Nanocrystals of NIF were produced in a co-grinding process in order to increase the rate of dissolution of the final products. P and M were used as co-grinding materials to stabilize the products against aggregation of the nanocrystals.
The co-grinding with the excipients resulted in nanoparticles with a mean particle size of 250 nm. Thermoanalytical and XRPD investigations confirmed that the nanoparticles maintained their crystallinity during the grinding process. M and P as excipients are very important from the aspect of the particle size reduction and the distribution of nanonized NIF in the solid system. The nanonized NIF (NIF-M-P co-grinding) dissolved significantly faster than the other products (Raw-NIF, Mill-NIF, NIF-M-P physical mixture) in gastric juice. The dissolution in intestinal juice was faster for all samples than that in gastric medium, but the product of NIF-M-P co-grinding exhibited the fastest and most extensive dissolution. Co-grinding with M and P can be suggested for the nanonization of NIF in order to achieve fast dissolution in the gastric medium.
It should be added that the co-grinding process involves fast dry-technology (without solvents, and without a mixing process for the nanonized drug and excipients). The critical parameters of the process can be characterized well for design of industrial products (scaling-up process). The dry-technology results in stable solid particles with crystalline behavior. Our results indicate that this technology can be recommended for the development of original and generic dosage forms to increase the bioavailability of different poorly water-soluble drugs.
5. Acknowledgements
TÁMOP-4.2.2/B-09/1/KONV-2010-0005, “Creating the Center of Excellence at the University of Szeged”, is supported by the European Union and co-financed by the European Regional Development Fund.
REFERENCES
- Y. Kawabata, K. Wada, M. Nakatani, S. Yamada and S. Onoue, “Formulation Design for Poorly Water-Soluble Drugs Based on Biopharmaceutics Classification System: Basic Approaches and Practical Applications,” International Journal of Pharmaceutics, Vol. 420, No. 1, 2011, pp. 1-10. http://dx.doi.org/10.1016/j.ijpharm.2011.08.032
- A. A. Noyes and W. R. Whitney, “The Rate of Solution of Solid Substances in Their Own Solution,” Journal of the American Chemical Society, Vol. 19, No. 12, 1897, pp. 930-934. http://dx.doi.org/10.1021/ja02086a003
- J-U. A. H. Junghans and R. H. Müller, “Nanocrystals Technology, Drug Delivery and Clinical Applications,” International Journal of Nanomedicine, Vol. 3, No. 3, 2008, pp. 295-309. http://dx.doi.org/10.2147/IJN.S595
- G. G. Liversidge and P. Conzentino, “Drug Particle Size Reduction for Decreasing Gastric Irritancy and Enhancing Absorption of Naproxen in Rats,” International Journal of Pharmaceutics, Vol. 125, No. 2, 1995, pp. 309-313. http://dx.doi.org/10.1016/0378-5173(95)00148-C
- P. Liu, X. Rong, J. Laru, J. Veen, B. Kiesvaara, J. Hiruonen, T. Laaksonen and L. Peltonen, “Nanosuspensions of Poorly Soluble Drugs: Preparation and Development by Wet Milling,” International Journal of Pharmaceutics, Vol. 411, No. 1-2, 2011, pp. 215-222. http://dx.doi.org/10.1016/j.ijpharm.2011.03.050
- R. H. Müller and A. Akkar, “Drug Nanocrystals of poorly Soluble Drugs,” Encyclopedia of Nanoscience and Nanotechnology, Vol. 62, No. 1, 2004, pp. 627-638.
- P. Kocbek, S. Baumgartner and J. Kristl, “Preparation and Evaluation of Nanosuspension for Enhancing the Dissolution of Poorly Soluble Drugs,” International Journal of Pharmaceutics, Vol. 312, No. 1-2, 2006, pp. 179-186. http://dx.doi.org/10.1016/j.ijpharm.2006.01.008
- J. Hecq, M. Deleers, D. Fanara, H. Vranckx and K. Amighi, “Preparation and Characterization of Nanocrystals for Solubility and Dissolution Rate Enhancement of Nifedipine, ” International Journal of Pharmaceutics, Vol. 299, No. 1-2, 2005, pp. 167-177. http://dx.doi.org/10.1016/j.ijpharm.2005.05.014
- T. Yasuji, H. Takeuchi and Y. Kawashima, “Particle Design of Poorly Water-Soluble Substances Using Supercritical Fluid Technologies,” Advanced Drug Delivery Reviews, Vol. 60, No. 3, 2008, pp. 388-398. http://dx.doi.org/10.1016/j.addr.2007.03.025
- M. Sarkari, J. Brown, X. Chen, S. Swinnea, R. O. Williams and K. P. Johnston, “Enhanced Drug Dissolution Using Evaporative Precipitation into Aqueous Solution,” International Journal of Pharmaceutics, Vol. 243, No. 1-2, 2002, pp. 17-31. http://dx.doi.org/10.1016/S0378-5173(02)00072-8
- J. Chingunpituk, “Nanosuspension Technology for Drug Delivery,” Walailak Journal of Science and Technology, Vol. 4, No. 2, 2007, pp. 139-153.
- R. Ravichandran, “Nanotechnology-Based Drug Delivery System,” Nanobiotechnology, Vol. 5, No. 1-4, 2009, pp. 17-33. http://dx.doi.org/10.1007/s12030-009-9028-2
- L. Kürti, Á. Kukovecz, G. Kozma, R. Ambrus, M. A. Deli and P. Szabó-Révész, “Study of the Parameters Influencing the Co-Gringing Process for the Production Meloxicam Nanoparticles,” Powder Technology, Vol. 212, No. 1, 2012, pp. 210-217. http://dx.doi.org/10.1016/j.powtec.2011.05.018
- Y. S. Thorat, I. D. Gonjari and A. H. Hosani, “Solubility Enhancement Techniques: A Review on Conventional and Novel Approaches,” International Journal of Pharmacy and Pharmaceutical Sciences, Vol. 2, No. 10, 2011, pp. 2501-2513.
- S. Budavari, “The Merck Index,” 11th Edition, Merck & Co., Rahway, New York, 1989.
- M. William, “The Extra Pharmacopoeia,” 31st Edition, Royal Pharmaceutical Society, London, 1996.
- M. Kata, R. Ambrus and Z. Aigner, “Preparation and Investigation of Inclusion Complexes Containing Niflumic Acid and Cyclodextrins,” Journal of Inclusion Phenomena and Macrocyclic Chemistry, Vol. 44, No. 1-4, 2002, pp. 123-126. http://dx.doi.org/10.1023/A:1023074025175
- R. Ambrus, Z. Aigner, L. Catenacci, G. Bettinetti, P. Szabó-Révész and M. Sorrenti, “Physico-Chemical Characterization and Dissolution Properties of Niflumic AcidCyclodextrin-PVP Ternary Systems,” Journal of Thermal Analysis and Calorimetry, Vol. 104, No. 1, 2011, pp. 291-297. http://dx.doi.org/10.1007/s10973-010-1069-1
- R. Ambrus, Z. Aigner, C. Dehelean, C. Soica and P. Szabó-Révész, “Physico-Chemical Studies on Solid Dispersions of Niflumic Acid Prepared with PVP,” Revista de Chimie, Vol. 58, No. 1, 2007, pp. 60-64.
- R. Ambrus, Z. Aigner, C. Soica, C. Peev and P. Szabó- Révész, “Amorphisation of Niflumic Acid with Polyvinylpyrrolidone Prepared Solid Dispersion to Reach Rapid Drug Release,” Revista de Chimie, Vol. 58, No. 2, 2007, pp. 206-209.
- T. Szunyogh, R. Ambrus and P. Szabó-Révész, “Formation of Niflumic Acid Particle Size by Solvent Diffusion and Solvent Evaporation as Precipitation Method,” Journal of Drug Delivery Science and Technology, Vol. 22, No. 4, 2012, pp. 307-312.
- T. Szunyogh, R. Ambrus and P. Szabó-Révész, “Importance of Particle Size Decrease in the Preformulation,” Acta Pharmaceutica Hungarica, Vol. 81, No. 1, 2011, pp. 29-36.
- A. Jaworek, “Microand Nanoparticle Production by Electrospraying,” Powder Technology, Vol. 176, No. 1, 2007, pp. 18-35. http://dx.doi.org/10.1016/j.powtec.2007.01.035
- R. Ambrus, N. Radacsi, T. Szunyogh, A. E. D. M. Van Der Heijden, J. H. Ter Horst and P. Szabó-Révész, “Analysis of Submicron-Sized Niflumic Acid Crystals Prepared by Electrospray Crystallization,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 76, 2013, pp. 1-7. http://dx.doi.org/10.1016/j.jpba.2012.12.001
- N. Radacsi, R. Ambrus, T. Szunyogh, A. I. Stankiewicz, A.E.D.M van der Heijden and J. H. ter Horst, “Electrospray Crystallization for Nano-Sized Pharmaceuticals with Improved Properties,” Crystal Growth & Design, Vol. 12, No. 7, 2012, pp. 3514-352. http://dx.doi.org/10.1021/cg300285w
- M. Wagner, “Thermal Analysis in Practice,” DSC Evaluations, Schwerzenbach, 2009, pp. 90-141.
NOTES
*Corresponding author.