Crystal Structure Theory and Applications
Vol.2 No.2(2013), Article ID:33493,5 pages DOI:10.4236/csta.2013.22012
Synthesis, Spectroscopic and Crystal Structure Studies on Ethyl 5,7-Dimethyl Coumarin-4-Acetate
1Department of Chemistry, Karnatak University, Dharwad, India
2M S Ramaiah Institute of Technology, Department of Physics, Bangalore, India
Email: *manohar274@gmail.com
Copyright © 2013 Kiran K. Pujar et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received November 1, 2012; revised January 3, 2013; accepted January 11, 2013
Keywords: Coumarin-4-Acetic acid Ethyl Ester; X-Ray Diffraction
ABSTRACT
Ethyl ester of 5,7-dimethyl coumarin-4-acetic acid has been synthesized from 3,5-Xylenol in a two step sequence of reaction involving Pechmann cyclisation and acid catalyzed esterification. The title compound 2 crystallizes in Monoclinic form, space group P 1 21/c 1, with a = 8.6248(4) Ǻ, b = 18.9103(8) Ǻ, c = 8.4204(4) Ǻ, β = 101.241(2), V = 1347.00(11) Ǻ, D cal = 1.283 Mg/cm3, Z = 4. The molecule is stabilized by intermolecular C-H … O bonds.
1. Introduction
Coumarins are a group of naturally occurring lactones with wide ranging biological activities [1]. Varieties of 4-substituted coumarins possessing hydroxyl [2], aminoalkyl [3] arylaminomethyl [4] sulphonamido [5] and aryloxymethyl [6] groups have exhibited anti coagulant, anti microbial and anti inflammatory activities. Coumarin 4-acetic acids have been found to exhibit good inflammation inhibiting activity in animal models [7]. They have been employed as key intermediates in the design and synthesis of polycyclic coumarins related to Protoberberine alkaloids [8]. The reactivity of C4-methylene group has been employed for the construction of many 4-substituted bi-heterocyclic coumarins. Solid state conformational studies on coumarin 4-acetic acid [9], ester [10,11] and dithionate [12] have been reported recently. It needs to be emphasized that the crystallographic study on esters is of considerable biological interest [13] since it determines the preferred orientation of the rotamer (Figure 1), in turn the direction of the electron pair on

Figure 1. General structure for Rotamers of Esters.
alkyl oxygen. In view of the biological significance of 4-substituted coumarins and interest associated with the geometry of esters the title compound has been synthesized, charac terized and studied for its solid state conformation by X-ray studies.
2. Experimental
2.1. Synthesis of the Title Compound
Compound 1 has been prepared by the reaction of 3,5-xylenol and citric acid by using sulphuric acid as condensing agent according to general procedure reported for coumarin 4-acetic acids [14]. Compound 1 was further converted into its ester (compound 2) by refluxing in dry ethanol with catalytic quantity of sulphuric acid by Fischer esterification. The resulting solution was poured into crushed ice, the precipitate was filtered off, washed with 5% NaHCO3, and with water and recrystallized from ethanol. (Figure 2) M.P. 123-125.C, yield 95%. The authenticity of the compound has been established by UV, IR, 1H, 13C NMR, and Mass spectra. Crystals suitable for diffraction study were grown by a slow evaporation technique using 1:1 mixture of ethanol and dioxan at room temperature.
2.2. Characterization and Physical Measurements
The melting point was determined in open capillaries and is uncorrected. The IR absorption spectrum was obtained by KBr pellet using a FT IR spectrometer. UV-vis spectrum was recorded in the range of 200 nm to 800 nm using a Lambda 35 Perkin-Elmer spectrophotometer. The 1H and 13C NMR spectra were determined on a Bruker Avance 400 model at 400 and 200 MHz respectively. Solvents were dried by refluxing with the appropriate drying agents and distilled before use.
2.3. X-Ray Crystallography
For the crystal structure determination, the single-crystal of the compound C15H16O4 was used for data collection. The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P 1 21/c 1, with Z = 4 for the formula unit C15H16O4. The final anisotropic full-matrix least-squares refinement on F2 with 176 variables converged at R1 = 4.03%, for the observed data and w R2 = 12.44% for all data. The goodness-of-fit was 1.008. The largest peak in the final difference electron density synthesis was 0.148 e−/Ǻ and the largest hole was −0.136 e−/Ǻ with an RMS deviation of 0.029 e−/Ǻ. On the basis of the final model, the calculated density was 1.283 g/cm3 and F(000), 552 e−/Ǻ. Molecular structure of the compound showing the atomic numbering scheme is shown in Figure 3. The crystallographic parameters of the compound were presented in Table 1. Selected bond distances and bond angles are listed in Table 2 and selected Hydrogen-bonding parameters listed in Table 3.
3. Results and Discussion
Title compound crystallizes in the monoclinic centrosymmetric space group P 1 21/c 1with Z = 4. The structure of the compound consists of ethyl ester group. Selected bond lengths and bond distances for the title com ding parameters are listed in Table 3.
3.1 Molecular Skeleton
Coumarin ring is planar and carbethoxy group attached to the C4-CH2 is out of plane by an angle of 11.26. The methyl group oriented at an angle of 90˚ to the mean plane of the bicyclic system. The ester carbonyl is oriented perpendicular to the C3-C4 double bond. The molecule adopts the S-cis arrangement across the alkyl-oxygen bond as revealed in the ORTEP diagram (Figure 3). The exocyclic Oxygen is involved in intermolecular hydrogen bonding (O4····H-C1) with the CH3 group of ester. A weak hydrogen bonding (O2····.H-C4) is also observed between the C4-CH2 and the carbonyl oxygen of ester. The molecular packing diagram (Figure 4) shows hydrogen bonding and the crystalline state is stabilized by vander waals forces of attraction. The S-cis arrangement is also supported by the dihedral angle of 1.8 between C2-O1-C3-O2 indicating an eclipsed arrangement (Table 3).
3.2. Bond Lengths
Bond lengths have been discussed in Table 2. The aromatic C-C bond lengths are in the range of 1.30 - 1.40 Å. The ester C13-O4 bond shows a bond length of 1.20 Å which is similar to the lactone distance of 1.19 Å between O2-C3 .The alkyl oxygen O1-C3 bond is 1.32 Å whereas the corresponding bond of O3-C13 bond is slightly longer by 1.35 Å.
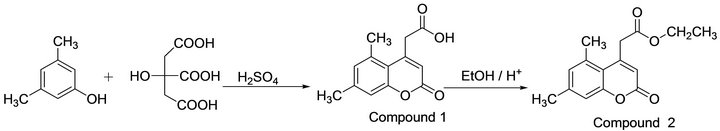
Figure 2. Chemical structure and synthetic pathway of the title compound.
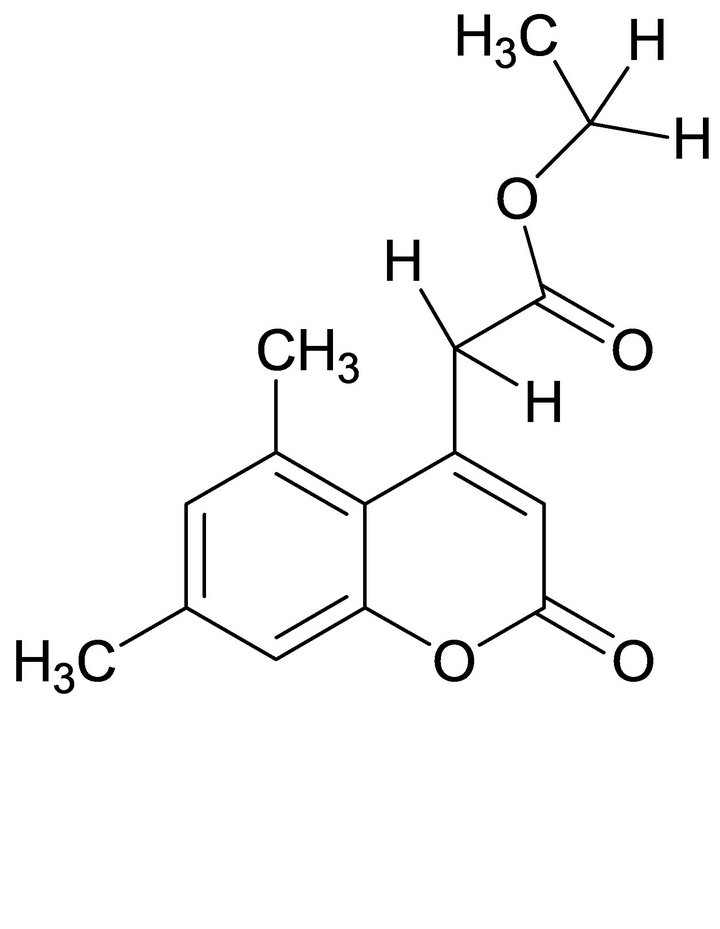

Figure 3. ORTEP plot of molecule with displacement ellipsoids drawn at 50% probability Covalent structure of compound 2.
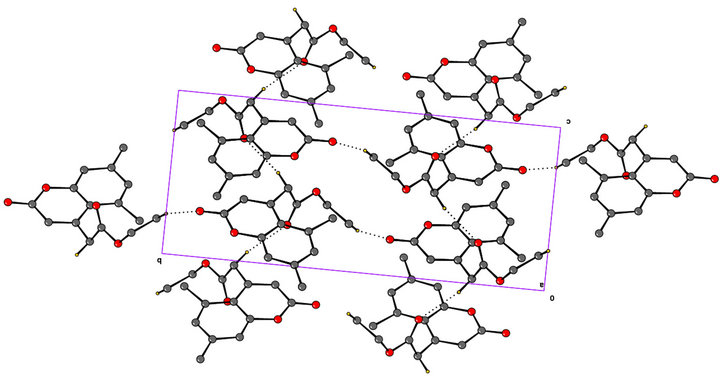
Figure 4. Packing diagram dotted lines indicate C-H····O hydrogen bonds of the molecule viewed down a-axis.

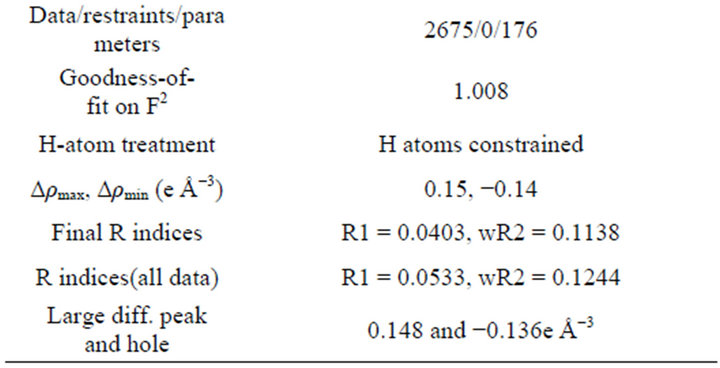
Table 1. Crystallographic data and structure refinement parameters.
3.3. Bond Angles
Bond angles have been discussed in Table 2. All the aromatic C-C-C bond angles are in the expected range
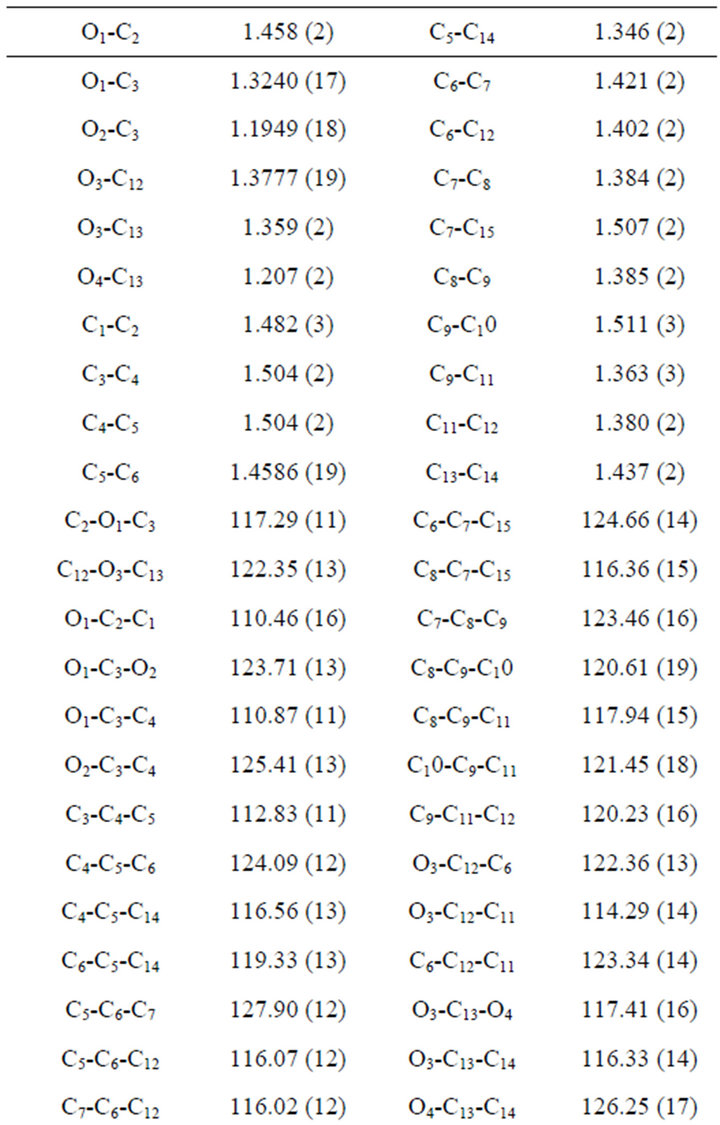

Table 2. Selected bond lengths and bond angles (Å, ˚).

Table 3. Selected hydrogen-bond parameters.
carbon. The angles O2-C3-O1 and O2-C3-C4 are 123.7˚ and 125.4˚ whereas the O1-C3-C4 angle is significantly reduced to 110.8˚. The values supporting the fractional (sp) hybridized orbitals are involved in bonding.
3.4. Spectral Studies
UV—Visible spectrum—The UV spectrum of the title compound 2 in DMSO shows a strong band at 297 nm whereas fluorescence band appears at 387 nm which shows emission over a long range.
FT IR spectrum—The FT IR spectrum of the title compound 2 shows the strong characteristic absorption band at 1725 cm−1 region due to lactone carbonyl stretching frequency and band at 1615 cm−1 observed due to C = C stretching.
1H NMR spectrum—The structure of the title compound 2 was further characterized by NMR absorption. Important structural information can be obtained from its NMR Spectra. 1H NMR(400 MHz, DMSO-d6) δ ppm: 2.34 (s, 3H, C5-CH3 ), 2.54 (s, 3H, C7-CH3 ), 1.15 (t, 3H, CH3-of ester, J = 7.2Hz), 4.15 (q, 4H, C4-CH2 and CH2 of ester J = 7.2Hz) 6.39 (s, 1H, C3-H), 7.0 (s, 1H, C6-H), 7.1 (s, 1H, C8-H)
13C NMR spectrum: 13C NMR spectrum (100 MHz DMSO-d6 ) (δ ppm) 14-CH3, 20 C7-CH3, 22 C5-CH3, 40 C4-CH2, 61 O-CH2, 115, 118, 130, 136, 142, 150, 155 these shifts correspond to aromatic carbons. Low intensity signals at 159, 170 are for carbonyl carbons of coumarin and ester respectively.
3.5. Crystal Structure
A specimen of C15H16O4 was used for the X-ray crystallographic analysis. The X-ray intensity data were measured. The total exposure time was 2.50 hours. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using a monoclinic unit cell yielded a total of 12192 reflections to a maximum θ angle of 26.07˚ (0.81 Å resolution), of which 2675 were independent (average redundancy 4.558, completeness = 99.9%, Rint = 2.13%, Rsig = 1.71%) and 2113 (78.99%) were greater than 2σ(F2). The final cell constants of a = 8.6248 (4) Å, b = 18.9103 (8) Å, c = 8.4204 (4) Å, β = 101.241 (2)˚, volume = 1347.00 (11) Å3, are based upon the refinement of the XYZ-centroids of 3786 reflections above 20 σ(I) with 5.382˚ < 2θ < 51.85˚. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.920. The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P 1 21/c 1, with Z = 4 for the formula unit, C15H16O4. The final anisotropic full-matrix least-squares refinement on F2 with 176 variables converged at R1 = 4.03%, for the observed data and wR2 = 12.44% for all data. The goodness-of-fit was 1.008. The largest peak in the final difference electron density synthesis was 0.148 e−/Å3 and the largest hole was −0.136 e−/Å3 with an RMS deviation of 0.029 e−/Å3. On the basis of the final model, the calculated density was 1.283 g/cm3 and F(000), 552 e−.
4. Conclusion
It can be concluded that the newly synthesized 5, 7-dimethyl coumarin-4-acetic ethyl ester exists mainly in the S-cis arrangement in the solid state. The packing diagram revealed the existence of only S-cis isomer which is stabilized by C-H····O bonds.
5. Acknowledgements
Fellowship from SC-ST cell Karnatak University, Dharwad is gratefully acknowledged.
REFERENCES
- M. V. Kulkarni, G. M. Kulkarni, C. H. Lin and C. M. Sun, “Coumarins and 1-Azacoumarins as Biodynamic Agents,” Current Medicinal Chemistry, Vol. 13, No. 23, 2006, pp. 2795-2818. doi:10.2174/092986706778521968
- S. Roseman, C. F. Heubuer, R. Pankratz and K. P. Link, “Studies on 4-Hydroxy Coumarins. XVI. The Metabolism of 4-Hydroxy Coumarins in the Dog,” Journal of the American Chemical Society, Vol. 76, No. 6, 1954, pp. 1650- 1652. doi:10.1021/ja01635a055
- P. Laurin, D. Ferroud, L. Schiol, M. Klich, C. DupuisHamelin, P. Mauvais, P. Lassaigne, A. Bonnefoy and B. Musicki, “Structure-Activity Relationship in Two Series of Aminoalkyl Substituted Coumarin Inhibitors of Gyrase B,” Bioorganic and Medicinal Chemistry Letters, Vol. 9, No. 19, 1999, pp. 2875-2880.
- M. V. Kulkarni and V. D. Patil, “Studies on Coumarins (Part 1),” Archiv der Pharmazie, Vol. 314, No. 8, 1981, pp. 708-711. doi:10.1002/ardp.19813140810
- S. S. Hanmantgad, M. V. Kulkarni and V. D. Patil, “Synthesis and Biological Activity of Some 4-(SulphonamidoMethyl)-Coumarins,” Indian Journal of Chemistry, Vol. 24B, No. 4, 1985, pp. 459-461.
- M. Ghate and M. V. Kulkarni, “Synthesis of Vanillin Ethers from 4-Bromomethyl Coumarins as Anti-Inflammatory Agents,” European Journal of Medicinal Chemistry, Vol. 38, No. 3, 2003, pp. 297-302. doi:10.1016/S0223-5234(03)00016-3
- M. Basanagouda, V. B. Jadhav, M. V. Kulkarni and R. Nagendra Rao, “Computer Aided Prediction of Biological Activity Spectra: Study of Correlation between Predicted and Observed Activities for Coumarin-4-Acetic Acids,” Indian Journal of Pharmaceutical Sciences, Vol. 73, No. 1, 2011, pp. 88-92.
- V. B. Jadhav, S. K. Nayak, T. N. Row and M. V. Kulkarni, “Synthesis, Structure and DNA Cleavage Studies of Coumarin Analogues of Tetrahydroisoquinoline and Protoberberine Alkaloids,” European Journal of Medicinal Chemistry, Vol. 45, No. 9, 2010, pp. 3575-3580.
- K. Sivakumar, F. Jesurethinam, K. Subramanian and S. Natarajan, “7-Hydroxy-4-Coumarin Acetic Acid Monohydrate,” Acta Crystallographica Section C, Vol. 46, 1990, pp. 1663-1665. doi:10.1107/S0108270189011522
- S. Naveen, S. M. Anandwalkar, et al., “7,8-Dimethyl Coumarin 4-Acetic Acid Ethyl Esters,” Analytical Sciences, Vol. 22, 2006, pp. X173-X174.
- I. Fujii, Y. Mano and N. Hirayama, “Crystal of 4-Ethoxycoumarin” Analytical Science, Vol. 21, 2005, pp. x7-x8
- N. M. Mahabaleshwaraiah, K. M. Kumar, O. Kotresh, W. F. A. Al-Eryani and H. C. Devarajgowda, “(6-Methoxy-2- Oxo-2H-Chromen-4-yl)Methylpyrrolidine-1-Carbodithiate,” Acta Crystallographica, Vol. E68, No. 5, 2012, p. 1566.
- P. Deslongchamps, U. O. Cheriyan, J. P. Pradere, P. Soucy and R. J. Taillefer, “Hydrolysis and Isomerization of syn Unsymmetrical N,N-Dialkylated Immidate Salts. Experimental Evidence for Conformational Changes and for Stereo Electronically Controlled Cleaves in Hemi-Orthoamide Tetrahedral Intermediates,” Nouveau Journal de Chimie, Vol. 3, No. 2, 1979, pp. 343-350.
- S. C. Laskowski and R. O. Clinton, “Coumarins. II. Derivative of Coumarins-3- and -4-Acetic Acids,” Journal of the American Chemical Society, Vol. 72, No. 9, 1950, pp. 3987-3991. doi:10.1021/ja01165a043
NOTES
*Corresponding author.