Pharmacology & Pharmacy
Vol.4 No.2(2013), Article ID:29744,6 pages DOI:10.4236/pp.2013.42025
Effect of Ap4A, UTP and Salbutamol on Mucociliary Clearance in a Mouse Model of Cystic Fibrosis (in Situ)
![]()
Department of Pharmacology, Institute of Medicinal Chemistry, University of Muenster, Muenster, Germany.
Email: *verspoh@uni-muenster.de
Copyright © 2013 Frank Begrow, Eugen J. Verspohl. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 21st, 2013; revised February 18th, 2013; accepted March 31st, 2013
Keywords: Cystic Fibrosis; Mucociliary Clearance in Situ; Ap4A; UTP
ABSTRACT
Cystic fibrosis is a life-threatening, wide spread genetic disease diagnosed in 1 to 3000 livebirths of the Caucasian population. Here a mouse model for this disease is described and optimized using the CFTR-channel selective inhibitor CFTR(inh 172). The target parameter was mucociliary clearance measured using microdialysis of the transported fluorescent dye rhodamine in the mouse trachea in situ. The impact of Ap4A (diadenosine tetraphosphate) as a potential drug was investigated. Its inhalation was effective at low concentrations; established compounds such as Salbutamol and UTP increased mucociliary clearance as well. Our data show a functioning model of cystic fibrosis and the effectiveness of the newly tested Ap4A.
1. Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is important for ion regulation. It represents a Cl− channel expressed in epithelial cells in e.g. airways, intestine, pancreas and testis. CFTR is also a regulator of other ion channels and other proteins [1]. Mutations of CFTR gene may cause cystic fibrosis (CF), the most common lethal autosomal recessive genetic disease in Caucasian population with a frequency of approx. 1 in 3000 livebirths [1] and 40,000 diseases in the EU. As observed in human CF patients and CF mouse models CFTR has an impact on lung, intestinal and on pancreatic fluid transport and on male fertility. The disturbed Cl− transport leads to increased salt concentrations in the respiratory system which reduces the function of antibacterial molecules from epithelial cells such as defensins. CF lung disease (mutation) is characterized by persistent pulmonary infection and mucus plugging of the airways initiated by the failure of solute transport across the airway epithelium [2].
CF treatment with mucoactive agents comprises three approaches: 1) expectorants, which add water to the airway; 2) ion transport modifiers, which promote ion and water transport across airway epithelium; and 3) mucokinetics, which improve cough-mediated clearance by increasing airflow or reducing sputum adhesivity [3]. The major aim, however, should be strategies correcting the ion transport deficiency of CF. Airway surfaces hydration can be obtained by stimulating secretion (through activation of the CFTR and/or calcium-activated chloride channels), and/or inhibiting fluid absorption (through the epithelial sodium channel) which finally results in stimulating mucociliary clearance which would lower secondary lung infection.
Extracellular nucleotides regulate ion transport, ciliary beat frequency, mucociliary clearance and improve mucus (mucosal) hydration on the surface of the airways by acting on P2 nucleotide receptors, in particular the P2Y2 receptor [4]. As demonstrated by in situ hybridization, the P2Y2 receptor mRNA is located in lung epithelial cells and not in smooth muscle or stromal tissue [5]. P2Y2 receptor agonists bypass the defective CFTR chloride channel by activating an alternative chloride channel [6]. They also inhibit sodium absorption, restore chloride conductance and rehydrate the CF airway surface liquid. Typical effective P2Y2 receptor agonists are UTP, ATP, UTPγS and ATPγS. UTP has an effect on ciliary beat frequency [7,8], also acts via P2Y4 receptors (PPADS is an antagonist) and has a strong effect on Ca2+ and Cl− currents [9]. Close to the pharmacological profile of UTP is Denufosol tetrasodium (INS37217) [6,10,11] which is going to be clinically developed but may be not much more effective than placebo with respect to lung function. A single inhalative administration of metabolically stable denufosol enhanced mucus transport for at least 8 h [10] being an advantage over other P2Y2 agonists.
Some novel agents and procedures to treat cystic fibrosis are summarized [12]: anti-infective drugs to be nebulized, hyperosmolaric agents, CFTR-assist drugs (they assist or repair the CFTR protein), ENaC (epithelial sodium channel) blockers and EPAC (cAMP-derivative stimulating like 8-pCPT-2’-O-Me-cAMP). A drug named Moli1901 was shown to increase Cl− secretion via an alternative chloride channel [13]. P2Y6 agonists [14] increase Ca2+ and thereby CFTR-dependent Cl− secretion. A3 receptor agonists were shown to enhance mucociliary clearance probably through Ca2+-mediated stimulation of ciliary motility of airway epithelium [15]. Anion and liquid secretion are essential for normal mucociliary transport defined by using bumetanide and dimethylamiloride [16]. Also a new causal therapy was recently established: ivacaftor, a selective potentiator of CFTR protein if a G551D mutation of the CFTR gene is the reason.
Not many CF animal models exist which show the typical symptoms of impaired mucociliary clearance. We decided to use the CFTR-channel selective inhibitor CFTR(inh 172), a thiazolidinone, being identified by high-throughput screening tested for blocking cholera toxin-induced intestinal fluid secretion [17,18]. By blocking the CFTR-chloride channel in the airways, CFTR(inh 172) can mimic the symptoms of the defective CFTR in cystic fibrosis [19].
The aim of this study was to use and optimize the CFTR inhibitor CFTR(inh 172) for a mouse model of CF and to test Ap4A in this model of CF as a P2X1-, P2X2-, P2X3-receptor agonist.
2. Materials and Methods
2.1. Compounds
The CFTR(inh 172) was from Sigma-Aldrich, Steinheim, Germany. Ap4A, UTP, Salbutamol and D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) were purchased from Sigma-Aldrich.
Solubility of Compounds
CFRT(inh 172) may be used orally or i.p.; solubility problems must be overcome by e.g. dissolving it in DMSO (mice get 4 ml/kg b.w. DMSO in total) or in TPGS (D-α-Tocopherol polyethylene glycol 1000 succinate) [18] for either i.p. or oral application with an orogastric feeding needle. In all experiments the drug was given three times (24 h, 12 h and 3 h) before starting the experiment. For control experiments we used TPGS and DMSO alone, respectively.
Ap4A, UTP and Salbutamol were dissolved in 0.12% saline and nebulized using a Pari-Turbo-Boy (PARI GmbH, Starnberg, Germany) with a custom made mouse-adapter. Mice inhaled the mentioned compounds for 10 minutes before starting each experiment. For control experiments 0.12% saline was nebulized. It is known that approximately 7 mg of a 40 mg nebulizer load is deposited in the lungs of a human [6].
2.2. Animals
To study the mucociliary clearance in situ mice from a C57BL/6 strain (Charles River Laboratories, Sulzfeld, Germany) were used. Mice were allowed food and water ad libitum. All experiments were approved by the German animal welfare committee (8.87/5010.37.09.245).
2.3. Mucociliary Clearance and in Situ Microdialysis
Mucociliary clearance was determined as recently described [20]. To measure the mucociliary transport, the mice were anesthetized with Avertin® (0.4 g/kg tribromethanol and 0.4 mL/kg amylalcohol) and body temperature was maintained at 37˚C by a heating pad in combination with a thermo-controller and a temperature probe (CMA Microdialysis, Solna, Sweden). For measurements of the tracheal mucociliary clearance, the trachea was unveiled carefully and a small incision was made directly beyond the larynx. A microcapillary tube (DETAKTA, 22,851 Norderstedt, Germany) with a diameter of 80 µm was loaded with Rhodamine 123 fluorescent dye (15 nL/g body weight) and introduced 16 mm into the trachea. The tube was connected to a micro syringe, so that the dye could easily be placed by pressing 500 nL air into the tube. To prevent a capillary suction effect of the surface of the tube, resulting in immediate detection of the dye, a small silicon sphere (<100 µm) was placed at the edge of the tube. Leaving the tube in its place, the tip of the microdialysis probe was inserted 4 mm through the same incision into the trachea and fixed in its position by a custom made retaining jig. It was important, not to let air come through the incision by the breathing animal, getting the airways dry and resulting in no measurable transport of the dye. Hence the incision has to be as small as possible, fitting probe and tube. After placing the dye, the mucociliary transport velocity could be calculated from the time, the dye needed to travel the defined distance of 12 mm through the trachea, to reach the tip of the microdialysis probe.
For all experiments, CMA/20/04 PC probes and a CMA/102 microdialysis pump (CMA Microdialysis, Solna, Sweden) were used. The probe was perfused by phosphate buffered saline at a flow rate of constant 4 µL/min. After deposition of the fluorescent dye, the dialysate was collected in intervals of 15 s in 96-well Nunc plates with a conical bottom, to take account of the small sample volume of 1 µL per well. The experiment was finished after 24 min, when each of the 96 wells was filled with 1 µL dialysate. The plate was then rapidly inserted into a FluoStar Galaxy fluorescence microplate reader (BMG Lab Tech, 07743 Jena, Germany). The fluorescence intensity recorded as counts in each well was obtained as an equivalent of the dye concentration at an excitation wavelength of 485 nm and an emission wavelength of 520 nm. In earlier test experiments recovery was independent of the dye concentration and it amounted 10% - 11% (data not shown). The death time of the probe with its tubes was obtained by placing the probe directly in a container with rhodamine dye and starting the perfusion immediately. In every performed experiment the dye was detectable in the 5th well (the 5th 15 s time interval), so the time to get the death volume out, here was 60 s. This 60 s were subtracted from each calculated dye travelingtime.
The data was collected using a standard personal computer with FluoStar Galaxy software (BMG Lab Tech, Jena, Germany). The first appearance of fluorescent dye in the MCT measurements was detected by averaging the mean background dye concentration and using the first measuring point, which was three SD above this mean background. More details were recently described [21].
2.4. Statistical Analysis
The results are expressed as mean ± SEM of a given number of independent experiments. For statistical evaluation multiple comparisons of means were carried out by one-way analysis of variance followed by a post-hoc test (Student’s t-test).
3. Results
3.1. Elaboration of Optimum Conditions for Use of CFTR(inh 172)
Figure 1 shows the effect of inhaled UTP (10 mM) and Ap4A (5 mM) on mucociliary transport (MCT) in healthy mice. The data indicate that both compounds are effective in accelerating the mucociliary transport velocity (compare data shown later for CFTR-inhibitor pretreated mice).
Next mice were investigated which were pretreated with the CFTR(inh 172) to induce a model of CF. Both i.p. and oral administration of the inhibitor were used in order to find out optimum conditions.
Figure 2 shows a basic experiment indicating that i.p. administration of the CFTR(inh 172) acutely restrains mucociliary transport. A higher dose of 3 times 1 mg i.p.
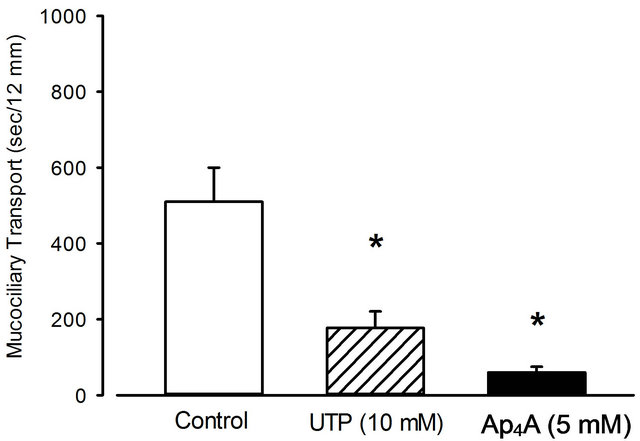
Figure 1. Effect of nebulized UTP and Ap4A on mucociliary transport. The compounds were nebulized for 10 minutes at a concentration of 10 and 5 mM, respectively. 0.12% saline was used for control. Mean + SEM of 3 to 5 experiments. *p < 0.05 vs. control.

Figure 2. Effect of i.p. administration of CFTR(inh 172) on mucociliary transport. Mean + SEM of 4 experiments. *p < 0.05 vs. control.
within 24 hours did not improve the results (data not shown). DMSO (4 mg/kg∙per∙day) alone had a slightly accelerating effect compared to saline control (see Figure 1).
In order to possibly optimize the effect of the CFTR inhibitor, the compound was given orally. For initial experiments an i.p. dose of 250 µg/kg was given as used by others for another target (small intestine) [17] was given i.p. When 16 mg/kg b.w. of the inhibitor were used 3 times a day or 5 times within two days perorally, there was an even greater effect as shown in Figure 3 compared to the i.p. administration shown before in Figure 2. Please note that the effect of the control is different when either saline or TPGS (D-α-Tocopherol polyethylene glycol 1000 succinate) was used: TPGS itself increased mucociliary clearance giving the CFTR inhibitor a more pronounced effect compared to control. The dose and time schedule of application were even increased to
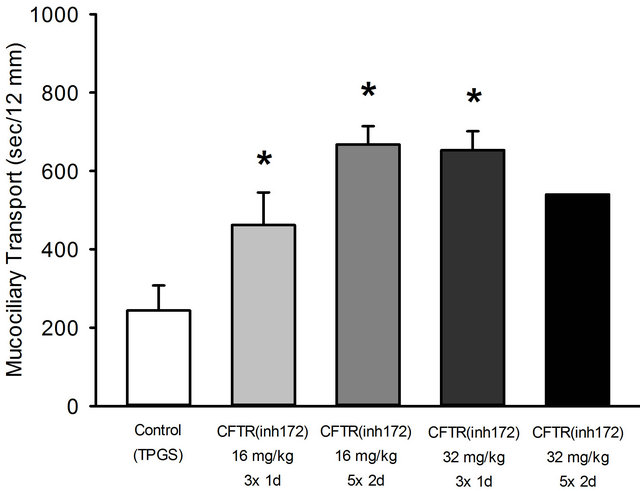
Figure 3. Effect of various doses and time intervals of application of CFTR(inh 172) and solvent TPGS as control on mucociliary clearance. The doses were given 3 times within 24 hours or 5 times within 48 hours. Mean + SEM of 4 - 5 experiments. (Note: Since the highest dose given 5 times did not improve the effect, only one experiment was performed. *p < 0.05 vs. control.
enlarge the effect as shown in Figure 3. A dose-dependency is obvious, although administration for more than 3 times did not improve the data at the high dose. The results were the basis for a decision with respect to the dose in further experiments: a dose of 3 times 32 mg/kg b.w. per day was chosen to be most effective in decelerating the mucociliary transport, while applicating the equipotent dose of 16 mg/kg for two days would be more time-consuming. Corresponding control experiments with TPGS were performed by applicating TPGS three times and five times per day respectively. Figure 3 presents only results of the three time TPGS dosing, since a five times dosing per day led to exactly the same result (data not shown).
3.2. Ap4A Effect
Salbutamol is known to accelerate ciliary beat frequency and thereby improving MCT [22]. It is of interest whether this compound is also effective in mice pretreated with the CFTR(inh 172) under these conditions. Salbutamol and UTP were able to rather completely antagonize the effect of the CFTR-inhibitor, i.e. to reverse the inhibition of mucociliary clearance (Figure 4). Ap4A at the indicated concentration was as effective as Salbutamol or UTP (Figure 4) (please note that only one effective concentration was used).
4. Discussion
The aim of the study was to establish a new in situ mouse model for tracheal CF symptoms using a CFTR-inhibitor (CFTR(inh 172)) as a small molecule temporarily block-

Figure 4. Effect of Salbutamol, UTP and Ap4A on mucociliary clearance at the indicated doses under pretreatment with CFTR(inh 172). The CFTR inhibitor was given 3 times in a dose of 32 mg/kg b.w. within 24 hours. Mean + SEM of 4 - 5 experiments. After the pretreatment with the CFTR inhibitor, Salbutamol, UTP or Ap4A were acutely inhaled (10 min before starting the experiment) by the mice. Control experiment: pretreatment with TPGS plus NaCl 0.12% per inhalation. *p < 0.05 vs. NaCl.
ing the CFTR Cl− channel. The conditions of the model were optimized by varying doses, time schedule and the mode of application. The optimum turned out to be its application 3 times 32 mg/kg b.w. in 24 hours intragastrally. The CFTR(inh 172) was able to slow down the mucociliary transport more than two fold in dose response experiments and this effect could be totally abolished by Salbutamol, which is well known to accelerate MCT [23,24]. Thus this model proves to be working.
Other models for testing new therapies of CF exist which possess major drawbacks, there are the models based on transgenic animals: ΔF508 Transgenic CFTR(-/-) a misfolded CFTR protein or complete null CF-mice [25,26]. Other ΔF508 mouse models use special mutations of the CFTR-protein. Transgenic animal models may be useful for the testing of special gene therapies considering the different mutations in the CFTR gene. The disadvantage of most of these mutant or knock out mice models is that the modification or deletion of the CFTR channel often provokes a lethal outcome. So the pups for example suffer from obstructions of inspissated meconium in the small intestine with a mortality of 50% - 95%. The small number of animals which reach maturity often did not show any difference in the mucociliary transport compared to wildtype animals. Maybe there is an activation of compensatory mechanisms leading to a survival of the fittest, with only mild or no measurable symptoms of CF disease. Even other models may only partly mirror the CF disease situation since pathophysiological theories are complex [27]: CFTR is not or only partly present in a mutated form; there also exists the possibility of sodium channel overexpression [28]. Other models would be: using the CFTR-blocker SP-303 or using DIDS with respect to the Cl− channel [29].
Another approach could be mitiglinide used for the treatment of type 2 diabetes which inhibits CFTR-channels in excised inside-out membrane patches from C127 cells expressing wild-type human CFTR [30]. Mitiglinide may inhibit CFTR by occluding the channel pore and preventing Cl− permeation with expectable effects on the mucociliary transport.
The model used appears to be useful with respect to distinct compounds: UTP and Ap4A, both acting via purinergic receptors, improved MCT as effective as Salbutamol, perhaps by bypassing the blocked CFTRchannel through the activation of alternative Cl− channels and rehydrating the periciliary liquid layer leading to a better ciliary function. The model used may only partly mirror the CF disease situation, but it can be of great value for rapid in situ testing of new compounds improving the tracheal mucus transport function. Ap4A possibly is equieffective as UTP. Ap4A has been shown to have positive and negative effects with respect to be an antagonist against cardiovascular problems (inhibition of thrombycyte aggregation) [31] and to be negative with respect to diabetes [32]. Data of this model may be relevant: There exists an allimetric correlation between tracheal mucous velocity and body weight [33]; thus data obtained from small animals can be translated for other animals or humans.
5. Conclusion
By using CFTR(inh 172) it was possible to show some typical tracheal CF symptoms for the in vivo/in situ testing of compounds which could possibly improve the ciliary transport function. CFTR(inh 172) shows a temporary dose dependent decrease in mucociliary transport in adult mice. The effectiveness of established compounds and of the newly tested compound Ap4A was demonstrated.
REFERENCES
- B. Marcet and J. M. Boeynaems, “Relationships between Cystic Fibrosis Transmembrane Conductance Regulator, Extracellular Nucleotides and Cystic Fibrosis,” Pharmacology & Therapeutics, Vol. 112, No. 3, 2006, pp. 719- 732. doi:10.1016/j.pharmthera.2006.05.010
- M. T. Clunes and R. C. Boucher, “Front-Runners for Pharmacotherapeutic Correction of the Airway Ion Transport Defect in Cystic Fibrosis,” Current Opinion in Pharmacology, Vol. 8, No. 3, 2008, pp. 292-299. doi:10.1016/j.coph.2008.04.006
- P. T. Bye and M. R. Elkins, “Other Mucoactive Agents for Cystic Fibrosis,” Paediatric Respiratory Reviews, Vol. 8, No. 1, 2007, pp. 30-39. doi:10.1016/j.prrv.2007.02.008
- D. Kellerman, et al., “Inhaled P2Y2 Receptor Agonists as a Treatment for Patients with Cystic Fibrosis Lung Disease,” Advanced Drug Delivery Reviews, Vol. 54, No. 11, 2002, pp. 1463-1474. doi:10.1016/S0169-409X(02)00154-0
- B. R. Yerxa, et al., “Potency and Duration of Action of Synthetic P2Y2 Receptor Agonists on Schirmer Scores in Rabbits,” Advances in Experimental Medicine and Biology, Vol. 506, 2002, pp. 261-265.
- D. Kellerman, et al., “Denufosol: A Review of Studies with Inhaled P2Y(2) Agonists that Led to Phase 3,” Pulmonary Pharmacology & Therapeutics, Vol. 21, No. 4, 2008, pp. 600-607. doi:10.1016/j.pupt.2007.12.003
- M. R. Knowles, et al., “Extracellular ATP and UTP Induce Chloride Secretion in Nasal Epithelia of Cystic Fibrosis Patients and Normal Subjects in Vivo,” Chest, Vol. 101, No. 3, 1992, pp. 60-63. doi:10.1378/chest.101.3_Supplement.60S
- M. R. Knowles, et al., “Pharmacological Treatment of Abnormal Ion Transport in the Airway Epithelium in Cystic Fibrosis,” Chest, Vol. 107, No. 2, 1995, pp. 71-76. doi:10.1378/chest.107.2_Supplement.71S
- S. Yamamoto, et al., “Regulation of Extracellular UTPActivated Cl− Current by P2Y-PLC-PKC Signaling and ATP Hydrolysis in Mouse Ventricular Myocytes,” The Journal of Physiology, Vol. 57, No. 2, 2007, pp. 85-94. doi:10.2170/physiolsci.RP011406
- B. R. Yerxa, et al., “Pharmacology of INS37217 [P(1)- (Uridine 5')-P(4)-(2'-deoxycytidine 5')tetraphosphate, Tetrasodium Salt], a Next-Generation P2Y(2) Receptor Agonist for the Treatment of Cystic Fibrosis,” Journal of Pharmacology and Experimental Therapeutics, Vol. 302, No. 3, 2002, pp. 871-880. doi:10.1124/jpet.102.035485
- R. R. Deterding, et al., “Phase 2 Randomized Safety and Efficacy Trial of Nebulized Denufosol Tetrasodium in Cystic Fibrosis,” American Journal of Respiratory and Critical Care Medicine, Vol. 176, No. 4, 2007, pp. 362- 369. doi:10.1164/rccm.200608-1238OC
- S. Storey and G. Wald, “Novel Agents in Cystic Fibrosis,” Nature Reviews Drug Discovery, Vol. 7, 2008, pp. 555-556. doi:10.1038/nrd2603
- F. Ratjen, “New Pulmonary Therapies for Cystic Fibrosis,” Current Opinion in Pulmonary Medicine, Vol. 13, No. 6, 2007, pp. 541-546. doi:10.1097/MCP.0b013e3282efbc56
- R. Schreiber and K. Kunzelmann, “Purinergic P2Y6 Receptors Induce Ca2+ and CFTR Dependent Cl− Secretion in Mouse Trachea,” Cell Physiol Biochem, Vol. 16, 2005, pp. 99-108. doi:10.1159/000087736
- M. Taira, et al., “Adenosine A3 Receptor-Mediated Potention of Mucociliary Transport and Epithelial Ciliary Motility,” American Journal of Physiology—Lung Cellular and Molecular Physiology, Vol. 282, 2002, pp. 556- 562.
- S. T. Ballard, et al., “Liquid Secretion Inhibitors Reduce Mucociliary Transport in Glandular Airways,” American Journal of Physiology—Lung Cellular and Molecular Physiology, Vol. 283, 2002, pp. 329-335.
- T. Ma, et al., “Thiazolidinone CFTR Inhibitor Identified by High-Throughput Screening Blocks Cholera ToxinInduced Intestinal Fluid Secretion,” Journal of Clinical Investigation, Vol. 110, 2002, pp. 1651-1658.
- J. R. Thiagarajah, et al., “Prevention of Toxin-Induced Intestinal Ion and Fluid Secretion by a Small-Molecule CFTR Inhibitor,” Gastroenterology, Vol. 123, No. 2, 2004, pp. 511-519. doi:10.1053/j.gastro.2003.11.005
- A. Perez, et al., “CFTR Inhibition Mimics the Cystic Fibrosis Inflammatory Profile,” American Journal of Physiology—Lung Cellular and Molecular Physiology, Vol. 292, No. 2, 2006, pp. 383-395. doi:10.1152/ajplung.00403.2005
- B. R. Grubb, et al., “Mucociliary Transport Determined by in Vivo Microdialysis In the Airways of Normal and CF Mice,” American Journal of Physiology—Lung Cellular and Molecular Physiology, Vol. 286, No. 3, 2004, pp. 588-595. doi:10.1152/ajplung.00302.2003
- N. Wienkötter, et al., “The Effect of Thyme Extract on Beta2-Receptors and Mucociliary Clearance,” Planta Medica, Vol. 73, No. 7, 2007, pp. 629-635. doi:10.1055/s-2007-981535
- F. Begrow, et al., “Effect of Myrtol Standardized and Other Substances on the Respiratory Tract: Ciliary Beat Frequency and Mucociliary Clearance as Parameters,” Advances in Therapy, Vol. 29, No. 4, 2012, pp. 350-358. doi:10.1007/s12325-012-0014-z
- A. T. Kimoto, et al., “A New, Simple Method of Measuring Mucociliary Clearance in Guinea—Pigs,” Pulmonary Pharmacology & Therapeutics, Vol. 12, No. 1, 1999, pp. 49-54. doi:10.1006/pupt.1999.0169
- H. Engler and I. Szelenyi, “Tracheal Phenol Red Secretion, a New Method for Screening Mucosecretolytic Compounds,” Journal of Pharmacological Methods, Vol. 11, No. 3, 1984, pp. 151-157. doi:10.1016/0160-5402(84)90033-0
- W. H. Colledge, et al., “Generation and Characterization of a DeltaF508 Cystic Fibrosis Mouse Model,” Nature Genetics, Vol. 10, 1995, pp. 445-452. doi:10.1038/ng0895-445
- J. N. Snouwaert, et al., “An Animal Model for Cystic Fibrosis Made by Gene Targeting,” Science, Vol. 257, No. 5073, 1992, pp. 1083-1088. doi:10.1126/science.257.5073.1083
- K. A. Becker, et al., “Zystische Fibrose—Pathophysiologische Konzepte,” Biospektrum, Vol. 15, 2009, pp. 383- 385.
- M. Mall, et al., “Increased Airway Epithelial Na+ Absorption Produces Cystic Fibrosis-Like Lung Disease in Mice,” Nature Medicine, Vol. 10, 2004, pp. 487-493. doi:10.1038/nm1028
- G. Adam, et al., “Increase in Intracellular Cl− Concentration by cAMPand Ca2+-Dependent Stimulation of M1 Collecting Duct Cells,” Pflugers Archiv, Vol. 449, No. 5, 2005, pp. 470-478. doi:10.1007/s00424-004-1356-4
- Z. Cai, et al., “Inhibition of Heterologously Expressed Cystic Fibrosis Transmembane Conductance Regulator Cl− Channels by Non-Sulphonylurea Hypoglycaemic Agents,” British Journal of Pharmacology, Vol. 128, No. 1, 1999, pp. 108-118. doi:10.1038/sj.bjp.0702748
- P. C. Zamecnik, et al., “Analogues of Diadenosine 5’,5’’’-P1,P4-Tetraphosphate (AP4A) as Potential AntiPlatelet-Aggregation Agents,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 89, No. 6, 1992, pp. 2370-2373. doi:10.1073/pnas.89.6.2370
- E. J. Verspohl and B. Johannwille, “Diadenosine Polyphosphates in Insulin Secreting Cells: Their Interaction with Specific Receptors and Their Degradation,” Diabetes, Vol. 47, No. 11, 1998, pp. 1727-1734. doi:10.2337/diabetes.47.11.1727
- S. A. Felicetti, et al., “Comparison of Tracheal Mucous Transport in Rats, Guinea Pigs, Rabbits, and Dogs,” Journal of Applied Physiology, Vol. 51, No. 6, 1981, pp. 1612-1617.
Abbreviations
Ap4A: Diadenosine tetraphosphate UTP: Uridine triphosphate DIDS: 4,4’-Diisothiocyano-2,2’-stilbenedisulfonic acid
NOTES
*Corresponding author.