Pharmacology & Pharmacy
Vol.3 No.2(2012), Article ID:18716,12 pages DOI:10.4236/pp.2012.32024
LPS-Induced Proliferation and Chemokine Secretion from BEAS-2B Cells
![]()
Department of Pharmacology, Institute of Pharmaceutical and Medicinal Chemistry, University of Münster, Münster, Germany.
Email: *verspoh@uni-muenster.de
Received January 14th, 2012; revised February 26th, 2012; accepted March 7th, 2012
Keywords: LPS; Chemokines; BEAS-2B Cells
ABSTRACT
The surface antigen CD14 plays an important role in innate immunity, serving as a pattern recognition receptor for lipopolysaccharides (LPS). The aim of this study was to investigate the proliferation, NFκB activation, and chemokine secretion of BEAS-2B cells, a human bronchial epithelial cell line, after LPS stimulation, and some details of involved signaling. The presence of CD14 was investigated by flow cytometry. Cell proliferation was measured with a [3H]-thymidine incorporation assay. sCD14, RANTES, and IL-8 concentrations in cell supernatants were measured by ELISA. BEAS-2B cells express CD14 on their surface and secrete soluble CD14 into the supernatant. Cells react on LPS with increased proliferation, activation of NFκB, and the secretion of the pro-inflammatory chemotactic cytokines IL-8 and RANTES, which proves the functionality of the CD14 receptor. Neither CD14 nor sCD14 are regulated by LPS. Specific inhibitors of various intracellular signaling pathways diminish the LPS-induced proliferation and IL-8 secretion: Thus MAP-Kinases p38 and JNK, tyrosine kinases, and PI3-kinase are involved in the signaling cascade from the LPS-CD14-complex on the cell surface to the increased cell proliferation and expression of IL-8; furthermore, ERK 1/2, IRAK 1/4, and the NFκB pathway are involved in the latter. The data show the existence and functionality of CD14 receptors on BEAS-2B cells and elucidate the signaling pathways involved. LPS is able to increase cell proliferation, various cytokines which are dependent on endogenous CD14. Three MAPK pathways, PI3 kinase and tyrosine kinase may be involved. Also CD14 is present/involved which was controversial.
1. Introduction
Lipopolysaccharides (LPS), present in the outer membrane of Gram-negative bacteria, provoke a response of innate immunity by the host, e.g. the expression of proinflammatory cytokines and chemokines [1]. However, an overreaction to LPS can cause sepsis or septic shock and eventually may lead to death. Together with Toll-like receptor 4 (TLR 4) [2], the surface antigen CD14 serves as a pattern recognition receptor for LPS, therefore being essential for cell reaction to LPS [3]. TLR4 is constitutively expressed [2,4] and its relevance was demonstrated using tissues from TLR4−/− animals [2]; however; TLR4 is not expressed on the surface of BEAS-2B cells, but only in the cytosol [2]. The leucine-rich glycoprotein CD14 (53 - 55 kDa) is linked to the cell membrane by a glycosyl-phosphatidylinositol (GPI) tail [5], but can also be secreted as a soluble form (sCD14) which serves as the LPS receptor for CD14-negative cells [6-8]. Although earlier studies report that CD14 is only expressed by cells of the myeloid lineage and these cells enable other cell types to react to LPS by shedding soluble CD14 [9], it was also found on human vein endothelial and intestinal epithelial cells and various other cell types [10-12].
The bronchial epithelium contributes to innate immunity by secreting pro-inflammatory cytokines and chemokines [13]. Thus the lung and bronchial system does not rely on mucociliary clearance and phagocyte activity alone. It has been described that sCD14 induces the production of IL-6 and IL-8 in human bronchial epithelial cells and the effect is augmented by LPS, but no membrane bound CD14 (mCD14) was found on these cells [14]. In contrast, Becker et al. describe the presence of mCD14 on human tracheobronchial epithelial cells [15]. It is, however, not known whether BEAS-2B cells, a human bronchial epithelial cell line, react to LPS in the absence of serum containing sCD14 or express CD14 receptors, even though the existence of TLR 4 receptors was recently demonstrated [16].
The aim of the present study was to prove the presence of CD14 on BEAS-2B cells and to investigate its possible reaction to LPS, concerning cell proliferation, NFκB activation, and the release of two pro-inflammatory chemokines IL-8 and RANTES. We aimed to demonstrate intracellular signaling pathways leading to proliferation and IL-8 secretion by investigating the influence of members of the MAPK family (ERK 1/2, p38 and JNK), tyrosine kinases, PI3 kinase, IRAK (interleukin 1 receptor associated kinase) and NFκB.
2. Materials and Methods
2.1. Chemicals
LPS (from Pseudomonas aeruginosa, serotype 10, purified by phenol extraction), PD 98059, SB 202190, IRAK-1/4-Inhibitor, LY 294002 and polymyxin B (6000 USP units per mg) were obtained from Sigma Aldrich, Steinheim, Germany. Genistein, SB 225002, and SP 600125 were from Enzo Lifesciences, Loerrach, Germany. SN 50 was obtained from Merck Chemicals, Darmstadt, Germany. The anti-CD14 antibodies (clone 18D- 11, APC-conjugated for FACS analysis and unconjugated for functional ELISA analysis) and the APC-conjugated IgG isotype controls were obtained from Immunotools, Friesoythe, Germany. For the proliferation assay, [3H]-thymidine was purchased from Perkin Elmer, Boston, USA, and scintillation cocktail Unisafe 1 from Zinsser Analytik, Frankfurt, Germany.
2.2. Cell Culture
BEAS-2B is a human bronchial epithelial cell line, obtained from a healthy donor and transfected with a SV40 adenovirus [17,18]. Cells were grown in 75 cm2 cell culture flasks (Sarstaedt, Nuembrecht, Germany) in LHC-9 medium (Invitrogen, Karlsruhe, Germany), supplemented with 100 U/mL penicillin and 0.1 mg/mL streptomycin (PAA, Coelbe, Germany). For passaging, they were detached enzymatically with Trypsin/EDTA (PAA, Coelbe, Germany) and seeded in a density of 225,000 cells per flask, which were pretreated with 15 mL LHC-9 medium containing fibronectin (0.01 mg/mL), collagen (0.03 mg/mL, both from Biochrom, Berlin, Germany), and bovine serum albumin (0.01 mg/mL, Biomol, Hamburg, Germany) for at least 2 hours. For the experiments, cells were seeded in 24 or 6 well plates not having been pretreated.
2.3. Flow Cytometry
The presence of the surface antigen CD14 on BEAS-2B cells was investigated by flow cytometry. Cells were grown on 6 well plates, detached with Trypsin/EDTA, centrifuged and resuspended in FACS FlowTM (BD Biosciences, Heidelberg, Germany) at a concentration of 600,000 cells in 100 µl. 4 µl of an APC-conjugated antiCD14 antibody or the matching IgG isotype control were added and incubated for 30 min at 4˚C. After another centrifugation/resuspension step, the fluorescence intensity of the cell population was analyzed by flow cytometry on a FACScalibur (BD Biosciences, Heidelberg, Germany).
2.4. LDH Measurement
LDH is quickly released upon damage of the plasma membrane and its release into the supernatant is an indicator of cell death. LDH was determined by using the reaction from lactate to pyruvate and the photometrical measurement of NADH + H+. Negative and positive controls were included (no stimulation and addition of lysis reagent giving 0 and 100% toxicity effect, respectively). Data are related to 100%, meaning no effect (only background effect) on LDH release.
2.5. MTT Test
The mitochondrial-dependent reduction of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) to formazan was used to measure cell respiration as an indicator of cell viability. 24 h after the test compound MTT solution (final concentration 200 µg/mL) was added and the cells were incubated for another 4 h at 37˚C. After removing the supernatant, 100 µl of DMSO was added to the cells to dissolve formazan. The absorbance was measured by using a microplate reader (570 nm wavelength). The control group consisted of untreated cells and was considered as 100% viable cells. Results are expressed as percentage of viable cells when compared with control group.
2.6. [3H]-Thymidine Incorporation Assay
DNA synthesis, i.e. proliferation, in cells is measured by the incorporation of [3H]-thymidine. Therefore, BEAS- 2B cells were seeded on 24 well plates at a density of 25,000/mL. After 24 hours, cells were incubated for another 48 hours with various concentrations of LPS and 0.1 µCi/mL [3H]-thymidine. Afterwards, cells were fixed with methanol, dried and washed twice with Dulbecco’s PBS (PAA, Coelbe, Germany) and twice with 0.3 M trichloroacetic acid. After lysis with 250 µl 0.3 M NaOH per well (30 min at 37˚C), the lysate was added to 750 µl scintillation cocktail per well and measured in the betacounter (Microbeta 1450, Perkin Elmer, Boston, USA) after shaking overnight. Results are expressed as the relative DNA synthesis; data are normalized to 100%.
2.7. ELISA of NFκB
Cells were grown in 75 cm2 cell culture flasks at a density of 225,000 cells per flask for 4 - 5 days and stimulated with LPS for 6 or 24 hours, respectively. Afterwards, nuclear extracts were prepared following the instructions of the ELISA manufacturer. The protein content of the extracts was measured by a Bradford assay. Extracts were analyzed for p65 NFκB by using a p65 Trans AMTM ELISA kit (Active Motif, Rixensart, Belgium).
2.8. ELISA of Chemokines
Cells were seeded in a density of 100,000/mL and cultured for 48 hours, followed by 24 hours of incubation with the respective substances (LPS in the presence or absence of inhibitors of different intracellular signaling pathways as indicated in the figure legends). Quantification of each sCD14, IL-8, and RANTES in cell supernatants was performed by ELISA kits from R & D Systems, Wiesbaden, Germany (Quantikine® ELISA for sCD14, Duo Set® for IL-8 and RANTES), following the instructions of the manufacturer.
2.9. Proteome ProfilerTM for MAP-Kinases
In R & D Systems’ Proteome ProfilerTM Array, capture antibodies against several intracellular kinases are spotted in duplicate on nitrocellulose membranes. After seeding BEAS-2B cells in 6 well plates at a density of 100,000/mL, culturing them for 48 hours and incubating them with 10 µg/mL LPS for another 24 hours, cells were lysed with the provided lysis buffer and incubated with the membranes. Unbound material was washed away and a cocktail of phosphor-site specific biotinylated secondary antibodies, followed by Streptavidin-HRP incubation, was used to detect phosphorylated kinases. Membranes were then exposed to chemiluminescence reagents (Immobilon Western HRP Substrate, Millipore, Schwalbach, Germany) for 5 minutes and to X-ray film for another 5 minutes. The film was developed (with AGFA Rodinal Developer) and fixed (with Tetenal Superfix Plus, both from Phototec, Norderstedt, Germany). The dark spots, representing the phosphorylated kinases, were identified with a transparent overlay provided by the manufacturer. Arrays were scanned and the pixel density was quantified using GenoSoftTM analysis software.
2.10. Statistical Analysis
The FACS analysis, ELISA and [3H]-thymidine incorporation assays were done by performing at least three independent experiments. Differences between two single values were validated by using an unpaired t-test as a post-hoc test after having used ANOVA-one way analysis. P < 0.05 was regarded as statistically significant.
3. Results
The FACS experiments showed that BEAS-2B cells express the CD14 antigen on their surface. However, the number of receptors was not significantly increased by a 24 hour incubation of the cells with 10 µg/mL LPS (Figure 1), and was hardly greater—though statistically significant—after 48 hour incubation.
Apart from the expression of membrane-bound CD14 on the surface, BEAS-2B cells also secrete the soluble form (sCD14) into the surrounding medium. Figure 2 shows the time-dependent increase of sCD14 in supernatants of unstimulated BEAS-2B cells. It was not yet investigated whether this increase is due to de novo synthesis or additional release. When cells were treated with 1 or 10 µg/mL LPS for the various indicated incubation times, the concentration of sCD14 was not increased (data not shown). Thus other parameters measured in this paper can only be compared at the same indicated time point to avoid indirect effects induced by released sCD- 14.
Since outcome of data shown later (e.g. [3H]thymidine incorporation, NFkB, RANTES and IL-8) may be obscured, the viability (membrane integrity and cell degeneration) was investigated to avoid misinterpretation of data. Table 1 shows that various LPS concentrations have no effect LDH release and formazan test (MTT).
Figure 3 illustrates the increase of [3H]-thymidine incorporation into BEAS-2B cells induced by LPS stimulation. LPS stimulates the proliferation of BEAS-2B cells by maximally 2 fold. This effect is mediated by p38 and JNK MAPK, PI3-kinase, and tyrosine kinases, since inhibitors of these pathways decrease the LPS-stimulated
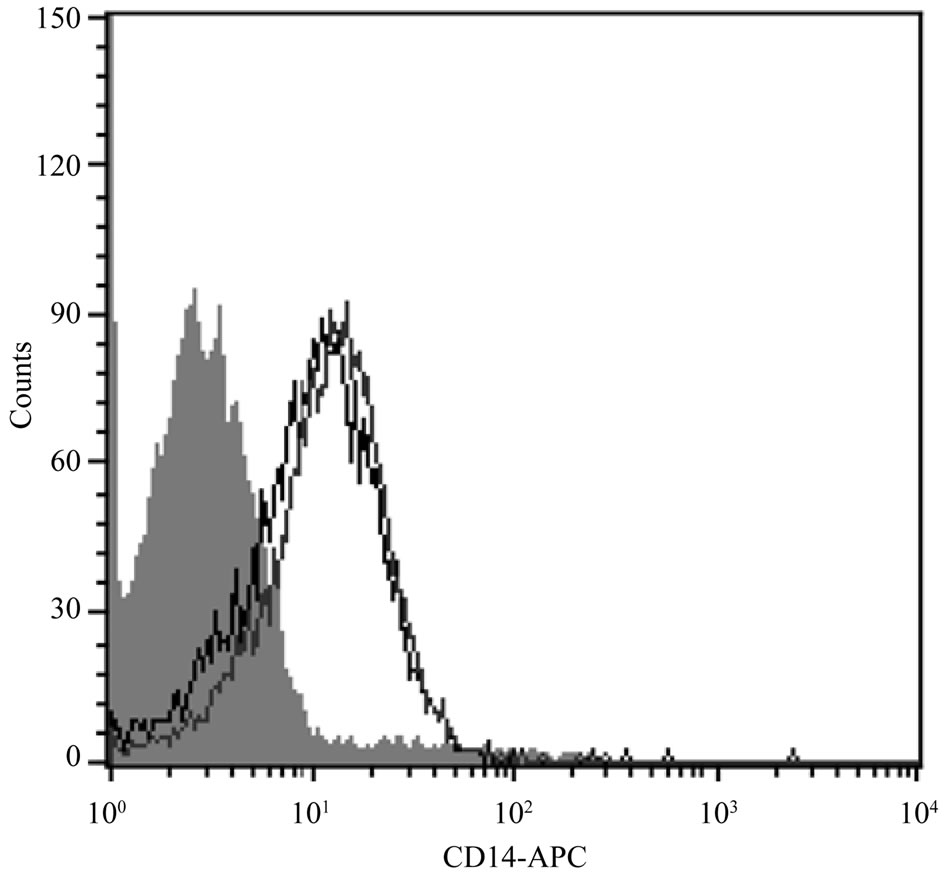
Figure 1. Expression of CD14 on BEAS-2B cells. One out of three similar histograms illustrates the shift of cell populations incubated either with IgG isotype control (grey area) or 18D11 CD14 antibody (black line). The right line represents the cells pretreated with 10 µg/mL LPS for 24 hr; the shift is small (not statistically significant).
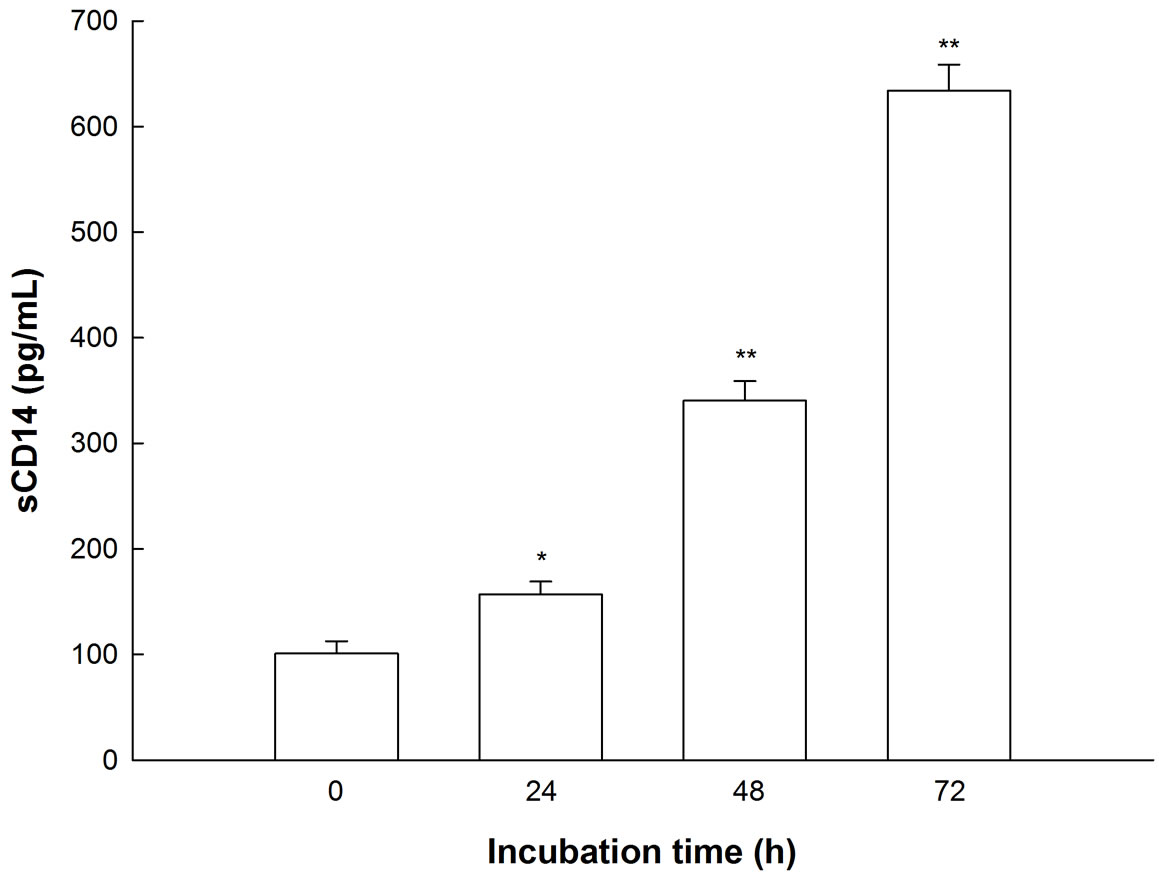
Figure 2. Secretion of sCD14 by BEAS-2B cells. Cells were seeded on 24 well plates and cultivated for the indicated times. Supernatants were collected; sCD14 concentration was measured and compared to fresh medium that had not been in contact with cells. Mean + SEM of three independent experiments. *p < 0.05, **p < 0.01 versus time point 0.
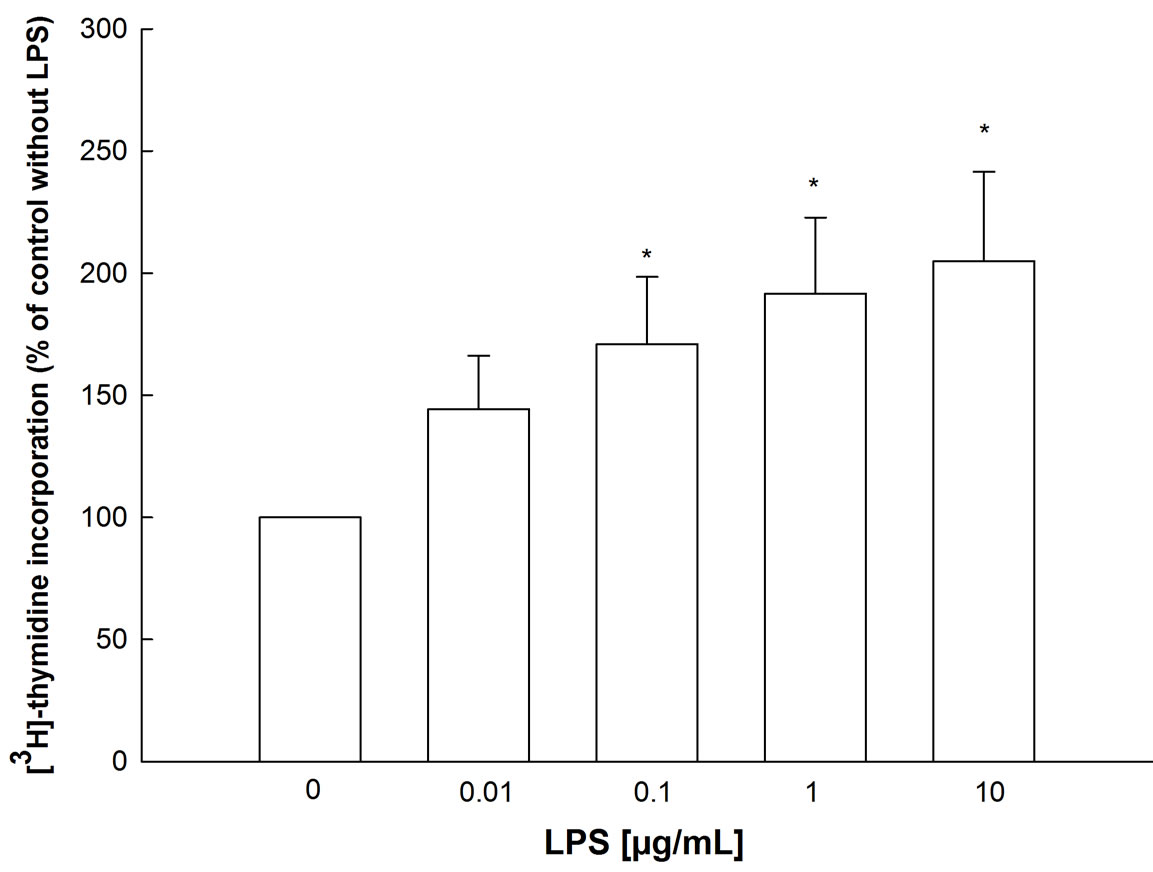
Figure 3. Effects of LPS on [3H]-thymidine incorporation into BEAS-2B cells. Cells were seeded on 24 well plates and grown for 24 hr, then LPS was added together with 0.1 µCi/mL [3H]-thymidine for another 48 hr. After lysis, radioactivity of lysates was measured. Mean + SEM of four independent experiments, performed in triplicates. *p < 0.05 versus control (no LPS).
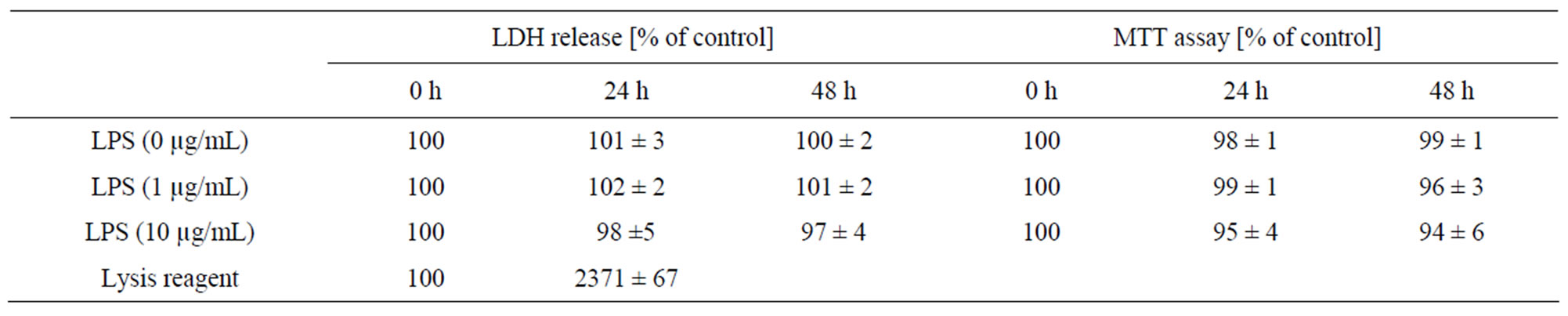
Table 1. Effect of LPS on cell viability/cytotoxicit (MTT) and membrane damage (LDH release test). Various LPS concentrations (0 to 10 µg/mL) and incubation times (0 - 48 h) were used. 100% means no effect (background effect). Mean ± SE; n = 5.
proliferation (Figure 4). The ERK1/2 inhibitor PD 98059 has no impact on proliferation; thus, the ERK1/2 pathway doesn’t seem to be involved.
LPS stimulates the activation of NFκB in a time and dose dependent manner. After 6 hr of incubation, the increase of NFκB is not statistically significant; the effect is stronger after 24 hr (Figure 5) corroborating data of others.
LPS at least at the higher concentration used provokes secretion of the chemotactic cytokine RANTES (Figure 6). The increase of RANTES is modest compared to the positive control consisting of 5 ng/mL TNFα and 5 ng/mL IFNg but at high LPS concentration could be inhibited by polymyxin B. Thus RANTES is involved in contrast to the opinion of others [2] having only used 1 µg/mL LPS (Note: the same others demonstrated an effect of 10, but not 1 µg/mL LPS on IL-4 [2]).
As shown in Figure 7, the expression of IL-8 is increased by LPS in a time and concentration dependent manner. The highest LPS concentration (10 µg/mL) sti-

Figure 4. Influence of specific inhibitors of intracellular signaling on LPS-induced proliferation. Cells were seeded on 24 well plates and grown for 24 hr, then LPS was added together with 0.1 µCi/mL [3H]-thymidine in the presence or absence of SP600125, SB202190, LY294002, or Genistein, for another 48 hr. After lysis, radioactivity of lysates was measured. Mean + SEM of four independent experiments, performed in triplicates. *p < 0.05 versus control (no LPS).
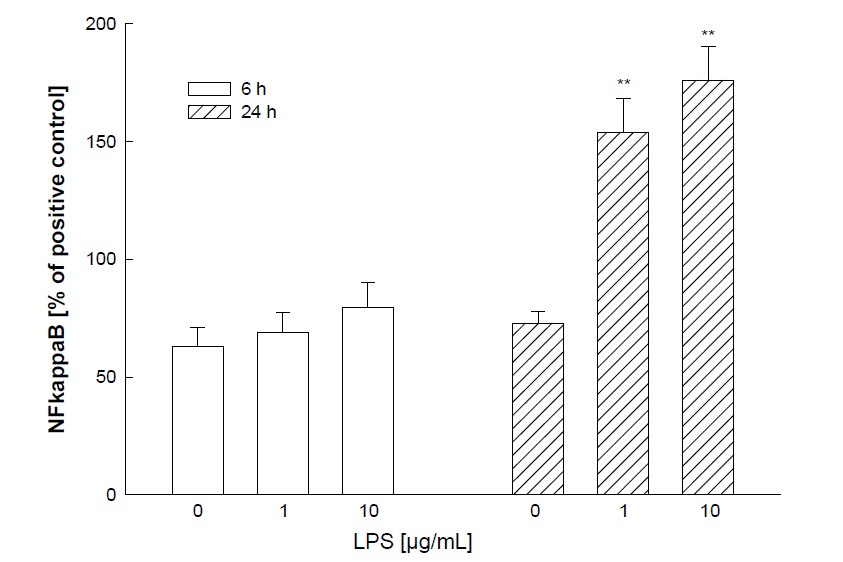
Figure 5. NFκB activation by LPS. BEAS-2B cells were seeded in 75 cm2 cell culture flasks, grown to confluence and stimulated with 1 or 10 µg/mL LPS for 6 or 24 hr. Nuclear extract were prepared and the content of activated p65-NFκB was measured by ELISA. A nuclear extract of Jurkat cells provided by the manufacturer served as positive control. Mean + SEM of four independent experiments, each performed in duplicates. **p < 0.01 versus control (no LPS).
mulates IL-8 release nearly as much as the positive control.
Addition of a functional anti-CD14 antibody (clone 18D11) attenuates the LPS induced IL-8 release (Figure 8). The results suggest that the CD14 receptor is functional and involved in the mechanisms leading to IL-8 secretion. In contrast to our results LPS responses are only modestly enhanced in the same cell line by recombinant sCD14 [19] which is discussed later.
Figure 9 illustrate the results of the Proteome ProfilerTM assay. Each spot in duplicate displays the quantity of a certain phosphorylated (activated) kinase. When

Figure 6. RANTES secretion of LPS stimulated BEAS-2B cells. Cells were seeded on 24 well plates and stimulated with the indicated LPS concentrations for 24 hr. Polym = in the presence of 10 µg polymyxin B. TNFα and IFNg (5 ng/mL each) served as positive controls. Mean + SEM of three independent experiments. **p < 0.01 versus control (no LPS); +p < 0.05 vs absence of polymyxin B.
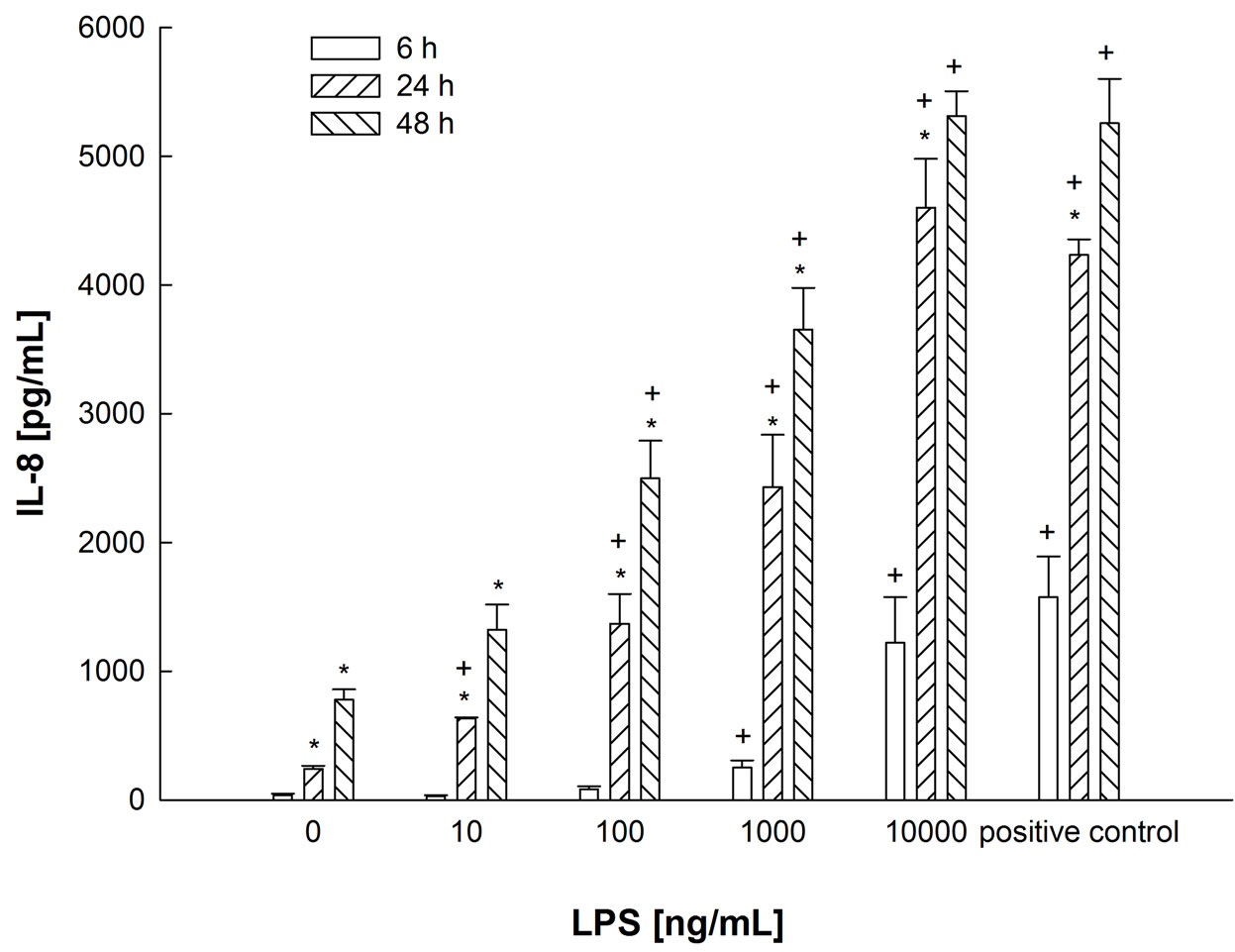
Figure 7. Time and concentration dependent effect of LPS on IL-8 release in BEAS-2B cells. Cells were stimulated with the indicated concentrations of LPS for 6, 24 or 48 hours. TNFα and IFNg (5 ng/mL each) served as positive control. Mean + SEM of three independent experiments. *p < 0.05 for the same LPS concentration at the next shorter time, +p < 0.05 for the value at the same time, but at the next lower concentration.
cells have been stimulated by LPS, the spots are darker, indicating an up-regulation of MAPK (ERK 1/2, JNK and p38). Although the blot concerning the LPS-stimulated cells is a little bit overdone, the outcome nevertheless is clear. A densitometric analysis of the spots confirms the visual impression (Figure 9(b)). Particularly the expression of p38 and JNK MAPK is increased after LPS incubation; the increase of ERK-1/2 was lower than expected.
To investigate the signaling pathways involved in LPS stimulated IL-8 secretion and to see whether the up-regulation of MAPK is linked to the observed effects, specific inhibitors of ERK1/2 (PD98059), p38 (SB202190) and JNK MAPK (SP600125), PI3-kinase (LY294002), tyrosine kinases (Genistein), IRAK 1/4 and NFκB (SN50) were added to the cells 60 minutes prior to the incubation of various concentrations of LPS. Figures 10-13 show the effect of the mentioned inhibitors on LPS-induced IL-8 release. While the ERK 1/2 inhibitor PD98059 does not inhibit IL-8 secretion, the effects of p38 inhibitor SB202190 and PI3 kinase inhibitor LY294002 are evident. For example, LY294002 (25 µM) reduces IL-8 secretion induced by 1 µg/mL LPS from 1311 to 187 pg/mL (Figure 10). The JNK inhibitor SP600125 shows a concentration-dependent effect: 5 µM of the substance attenuate the effect of 100 ng/mL LPS by 25%, whereas

Figure 8. Influence of an anti-CD14 antibody on the LPS-induced IL-8 release in BEAS-2B cells. Cells were incubated with LPS in the presence or absence of an anti-CD14 antibody (clone 18D11) for 24 hr. TNFα and IFNg (5 ng/mL each) served as positive controls: effect of 2104 + 80 pg/mL. Mean + SEM of three independent experiments. The difference between the curves is not statistically significant.
 (a) (b)
(a) (b)
Figure 9. (a) Proteome ProfilerTM for MAPK. BEAS-2B cells were seeded in 6 well plates, grown to confluency and stimulated with 10 μg/mL LPS for 24 hr. Cell lysates were incubated with nitrocellulose membranes on which antibodies against several MAPK were spotted in duplicate. As an example, the spots for p38α-MAPK are marked with an arrow. One of two results from distinct experiments with identical outcome is shown; (b) Densitometric analysis of the Proteome ProfilerTM. Arrays from Figure 9(a) were analysed densitometrically with GeneSoftTM analysis software.
20 µM reduce IL-8 secretion by 70% (Figure 11). At a concentration of 10 µM, the tyrosine kinase inhibitor Genistein only tends to diminish chemokine secretion (not statistically significant). However, at 100 µM, IL-8 concentration is reduced to almost baseline level except when very high LPS concentrations (10 µg/mL) or the positive control is used (Figure 12).
The IRAK-1/4-inhibitor, a specific inhibitor of the Interleukin-1 associated receptor kinase (IRAK) 1 and 4, also strongly inhibits the LPS stimulation (Figure 13). The same is true for the NFκB inhibitor SN50.
These findings suggest that all three MAPK pathways, PI3 kinase, tyrosine kinases, IRAK and NFκB are—in varying degrees—all involved in the LPS-induced IL-8

Figure 10. Influence of the p38 inhibitor SB 202190, the ERK 1/2 inhibitor PD 98059, and the PI3-kinase inhibitor LY 294002 on LPS-induced IL-8 release in BEAS-2B cells. Cells were incubated with LPS in the presence or absence of 10 μM SB 202190, 20 μM PD 98050, or 25 μM LY 294002 for 24 hr. TNFα and IFNg (5 ng/mL each) served as positive controls. DMSO and EtOH at the indicated concentrations served as solvent controls. Mean + SEM of three independent experiments. *p < 0.05, **p < 0.01 versus solvent control at the same LPS concentration.

Figure 11. Influence of the JNK inhibitor SP 600125 on the LPS-induced IL-8 release in BEAS-2B cells. Cells were incubated with LPS in the presence or absence of 5 or 20 μM SP 600125 for 24 hr. TNFα and IFNg (5 ng/mL each) served as positive controls. DMSO served as solvent control. Mean + SEM of three independent experiments. *p < 0.05, **p < 0.01 versus solvent control at the same LPS concentration.

Figure 12. Influence of the tyrosine kinase inhibitor Genistein on the LPSinduced IL-8 release in BEAS-2B cells. Cells were incubated with LPS in the presence or absence of 10 or 100 μM Genistein for 24 hr. TNFα and IFNg (5 ng/mL each) served as positive controls. EtOH served as solvent control. Mean + SEM of three independent experiments. **p < 0.01 versus solvent control at the same LPS concentration.
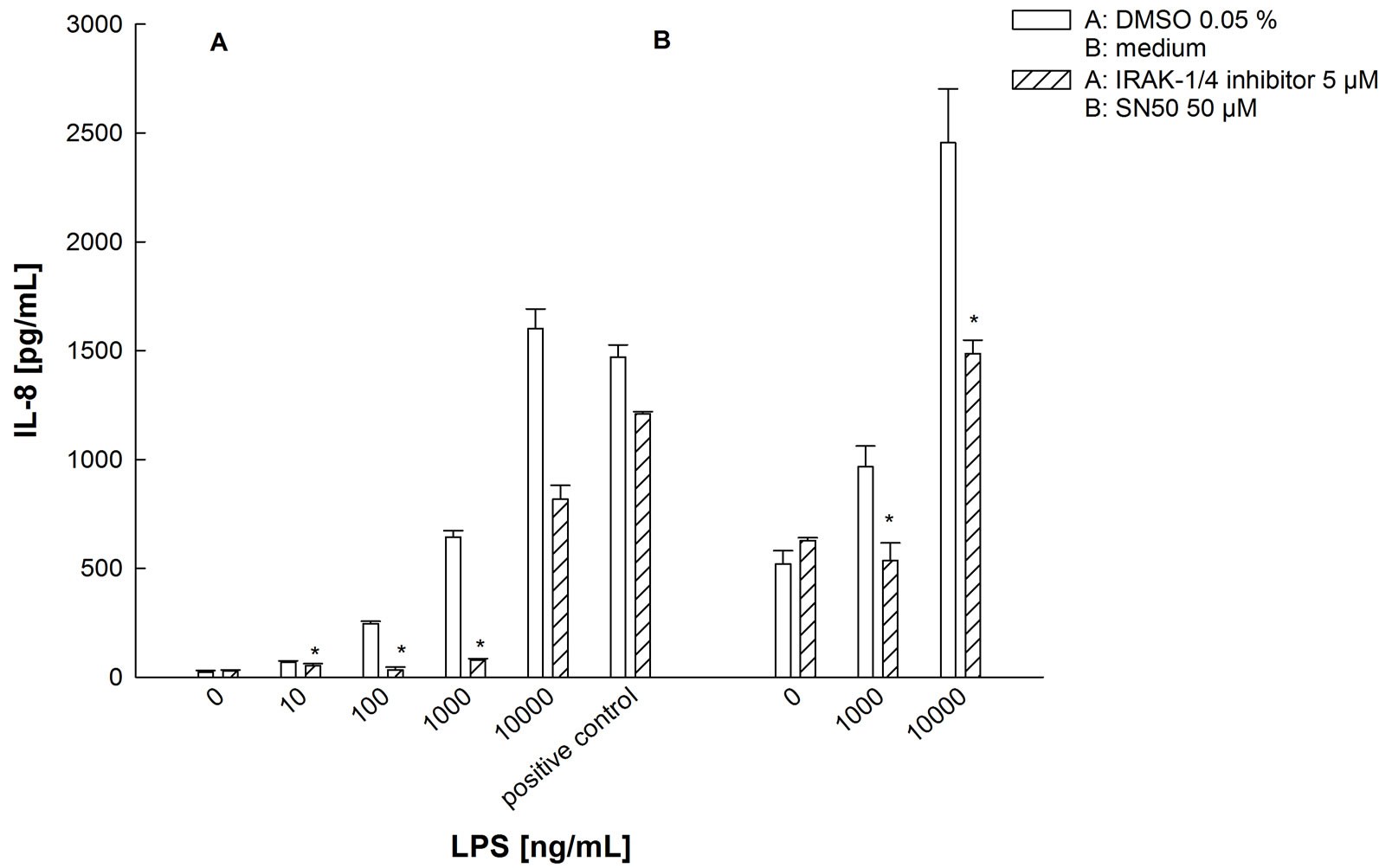
Figure 13. Influence of the IRAK-1/4-Inhibitor and the NFκB-inhibitor SN50 on the LPS-induced IL-8 release in BEAS-2B cells. Cells were incubated with LPS in the presence or absence of 50 μM IRAK-1/4-inhibitor or 50 μg/mL SN50 for 24 hr. TNFα and IFNg (5 ng/mL each) served as positive controls. DMSO or cell medium served as solvent control. Mean + SEM of three independent experimens. *p < 0.05 versus solvent control at the same LPS concentration.
secretion in BEAS-2B cells.
4. Discussion
IL-8 and NFkB are already known to be increased by LPS [2]. We now show the interaction of incubation time and LPS concentration and additionally the interaction with anti-CD14 antibody and various chemical inhibitors to conclude on signaling mediators. The presence of CD14 on the human bronchial epithelial cell line BEAS2B is described for the first time and underlines the crucial role the bronchial epithelial plays in innate immunity. As shown in Figure 2, low levels of sCD14 are detectable in the fresh medium, but this concentration is not in the linear part of the calibration curve and therefore not reliable. For an immune response to LPS, bronchial epithelial cells do not depend on myeloid immune cells, like macrophages, which secrete soluble CD14 and allow surrounding cells to react to LPS; in contrast, BEAS-2B cells even secrete sCD14 by themselves in a time dependent manner. However, it is not clear if this effect is a function of de novo synthesis or of shedding of existing membrane-bound CD14; this could have been examined by using cycloheximide for inhibiting protein synthesis. Since serum-free culture medium was used, this finding cannot be due to carriage of sCD14 by the medium. This increase in sCD14 may overlay results to be shown during all experiments but modify them in parallel during our static incubations and will not influence interpretation of data.
The previous inability to find CD14 on BEAS-2B cells may be due to methodical reasons, i.e. selection of antibody clone and culture conditions, as previously described for HUVECs by Jersmann et al. [12]. Neither the expression of membrane-bound nor of soluble CD14 is increased by LPS stimulation, in contrast to what was described for other epithelial cells by Fearns et al. [20] Differences between species may be one reason for this observation, since those authors used murine epithelial cells for their study, whereas BEAS-2B cells are of human origin. Furthermore, Fearns et al. used living animals so that the increased CD14 expression found by them is not caused by epithelial cells themselves, but the result of adsorption of soluble CD14 from surrounding cells such as macrophages. In addition, although transformed cell lines show many similarities with primary bronchial epithelial cells, there are specific differences such as the expression of antioxidant enzymes [18] and of Toll-like receptors [16].
The functionality of the CD14 receptor is confirmed by the fact that BEAS-2B cells can be stimulated by LPS, resulting in increased proliferation, NFκB activation, upregulation of MAPK and the secretion of RANTES and IL-8. For our experiments, we used LPS. The LPS stimulation may be mediated by other pathways than the CD14/TLR 4 cascade, but the decreased IL-8 secretion in the presence of a functional anti-CD14 antibody shows that the CD14 pathway is at least partly involved. The antibody reduces the LPS effect. This includes that high LPS concentrations (10 µg/mL) are dramatically inhibited.
Since LPS from different species can differ in structure and toxicity, we compared LPS from P. aeruginosa to E. coli-LPS regarding IL-8 release and NFκB activation. Even at a concentration of 10 ng/mL, where effects of P. aeruginosa-LPS were hardly detectable, the latter showed a stronger increase than the former at 10,000 ng/mL (data not shown). Due to the fact that respiratory infections, i.e. in patients with Cystic Fibrosis, are more often caused by P. aeruginosa than by E. coli, we had decided to use continuously P. aeruginosa-LPS, even though higher concentrations than with E. coli-LPS were necessary. The deviation from the Schulz data [19] has to be discussed. Schulz et al. postulate a CD14-independent mechanism for LPS activation of BEAS-2B cells [19]. In contrast these authors used LPS from E. coli instead of P. aeruginosa as we did; different modes of action might explain the differing results. Divergent results may be due to the presence of CD14, since exogenous CD14 not only helped, but can also be antagonized by anti-CD14 antibodies. This indicates that endogenous CD14 is involved.
Many authors investigated the effects of different stimuli on IL-8 and IL-6 secretion, Schulz et al. using BEAS-2B cells [19]. Both IL-6 and IL-8 are up-regulated in the bronchial epithelium of asthmatic patients [21] and the serum levels of both are higher in asthmatic patients compared to healthy subjects [22,23]. Thus, we did not expect to observe major differences between the two cytokines and decided to focus on IL-8.
In BEAS-2B cells, LPS on the one hand up-regulate ERK 1/2, p38 and JNK MAPK, and, on the other hand lead to increased cell proliferation and secretion of IL-8. Here the relationship between these effects was investigated. By adding specific inhibitors of MAPK and other intracellular kinases to LPS stimulated cells, we proved the contribution of ERK 1/2, p38 and JNK MAPK, tyrosine kinases, PI3-kinase, IRAK, and NFκB to increased IL-8 secretion. For the increase in proliferation, it was shown that PI3-kinase, tyrosine kinases, p38 and JNK MAPK, but not ERK1/2 MAPK, are involved. These results do not give information about the way the differrent pathways interact or influence each other. Furthermore, it is not possible to say what kind of tyrosine kinases are involved, since Genistein nonspecifically inhibits various tyrosine kinases [24]. Erk 1/2 may not be involved in proliferation due to our PD 98059 experiments. This was unexpected since in other cell systems they are involved in LPS-induced proliferation, e.g. in smooth muscle cells form trachea [25] or cholangiocytes [26]. With respect to BEAS-2B cells no data were available; however, Erk 1/2 is involved when not LPS but hepatic growth factor is the stimulus [27]. In the Proteome ProfilerTM assay, ERK 1 and ERK 2 are only slightly up-regulated compared to JNK and p38 MAPK. This matches the fact that p38 and JNK inhibitors SB- 202190 and SP600125 show a stronger inhibitory effect on the LPS-induced IL-8 secretion than the ERK 1/2 inhibitor PD98059.
It is always a matter of discussion whether inhibitors are used at too high concentrations questioning their selectivity. The concentrations of the applied inhibitors are the same as used in other studies (e.g. [27] for PD98059 and LY294002, [28] for IRAK-1/4-inhibitor I) or they are within the scope of the supplier’s recommendation. Kinase inhibitors are used in the literature for shorter and longer periods. Since kinases and phosphatases act rather quickly, incubation periods were reduced to 6 hours; the results were the same except the degree of inhibition in some cases was relatively lower (data not shown).
Increased proliferation of airway epithelial cells is closely related to airway remodeling occurring in chronic inflammatory bronchial diseases like asthma. The prolixferative effect of LPS on BEAS-2B cells is contradictory to the findings of Freitag et al., who describe an inhibited proliferation of rat tracheal epithelial cells in the presence of LPS [29]. This may be caused by the use of LPS from different species, differences between rat and human cells and between tracheal and bronchial epithelium. However, cell proliferative effects of LPS were already described for mononuclear cells and keratinocytes [30, 31].
Our results confirm the hypothesis that the bronchial epithelium not only serves as a mechanic barrier, but contributes to innate immunity by reacting to invading pathogens like LPS with increased proliferation and the secretion of chemokines; this was questioned for a long time. We proved the existence and functionality of CD14 receptors on BEAS-2B cells and elucidated signaling pathways involved, which helps to improve the understanding of innate immunity mechanisms in the airways after contact with pathogens.
REFERENCES
- M. Guha, “LPS Induction of Gene Expression in Human Monocytes,” Cell, Vol. 13, No. 2, 2001, pp. 85-94.
- L. Guillot, “Response of Human Pulmonary Epithelial Cells to Lipopolysaccharide Involves Toll-Like Receptor 4 (TLR4)-Dependent Signal Pathways,” Journal of Biological Chemistry, Vol. 279, No. 4, 2003, pp. 2712-2718. doi:10.1074/jbc.M305790200
- J. Pugin, “CD14 Is a Pattern Recognition Receptor,” Immunity, Vol. 1, No. 6, 1994, pp. 509-516. doi:10.1016/1074-7613(94)90093-0
- B. Schmeck, “Pneumococci Induced TLRand Rac1-Dependent NFkB-Recruitement to the IL-8 Promoter in Lung Epithelial Cells,” American Journal of Physiology, Vol. 290, 2006, pp. 730-737.
- A. Haziot, “The Monocyte Differentiation Antigen, CD14, Is Anchored to the Cell Membrane by a Phosphatidylinositol Linkage,” Journal of Immunology, Vol. 141, No. 2, 1988, pp. 547-552.
- V. Bazil, “Biochemical Characterization of a Soluble form of the 53-kDa Monocyte Surface Antigen,” European Journal of Immunology, Vol. 16, No. 12, 1986, pp. 1583-1589. doi:10.1002/eji.1830161218
- V. Bazil, “Structural Relationship between the Soluble and Membrane-Bound Forms of Human Monocyte Surface Glycoprotein CD14,” Molecular Immunology, Vol. 26, No. 7, 1989, pp. 657-662. doi:10.1016/0161-5890(89)90048-5
- J. Pugin, “Lipopolysaccharide Activation of Human Endothelial and Epithelial Cells Is Mediated by Lipopolysaccharide-Binding Protein and Soluble CD14,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, 1993, pp. 2744-2748. doi:10.1073/pnas.90.7.2744
- S. D. Wright, “CD14, a Receptor for Complexes of Lipopolysaccharide (LPS) and LPS Binding Protein,” Science, Vol. 249, No. 4975, 1990, pp. 1431-1433. doi:10.1126/science.1698311
- D. P. Funda, “CD14 Is Expressed and Released as Soluble CD14 by Human Intestinal Epithelial Cells in Vitro: Lipopolysaccharide Activation of Epithelial Cells Revisited,” Infection and Immunity, Vol. 69, No. 6, 2001, pp. 3772-3781. doi:10.1128/IAI.69.6.3772-3781.2001
- H. P. Jersmann, “Time to Abandon Dogma: CD14 Is Expressed by Non-Myeloid Lineage Cells,” Immunology and Cell Biology, Vol. 83, No. 5, 2005, pp. 462-467. doi:10.1111/j.1440-1711.2005.01370.x
- H. P. Jersmann, “Synthesis and Surface Expression of CD14 by Human Endothelial Cells,” Infection and Immunity, Vol. 69, No. 1, 2001, pp. 479-485. doi:10.1128/IAI.69.1.479-485.2001
- J. Nakano, “Endotoxin and Pro-Inflammatory Cytokines Stimulate Endothelin-1 Expression and Release by Airway Epithelial Cells,” Clinical & Experimental Allergy, Vol. 24, No. 6, 1994, pp. 330-336. doi:10.1111/j.1365-2222.1994.tb00243.x
- I. Striz, “The CD14 Molecule Participates in Regulation of IL-8 and IL-6 Release by Bronchial Epithelial Cells,” Immunology Letters, Vol. 62, No. 3, 1998, pp. 177-181. doi:10.1016/S0165-2478(98)00046-7
- M. N. Becker, “CD14-Dependent Lipopolysaccharide-Induced Beta-Defensin-2 Expression in Human Tracheobronchial Epithelium,” Journal of Biological Chemistry, Vol. 275, 2000, pp. 29731-29736. doi:10.1074/jbc.M000184200
- A. K. Mayer, “Differential Recognition of TLR-Dependent Microbial Ligands in Human Bronchial Epithelial Cells,” Journal of Immunology, Vol. 178, No. 5, 2007, pp. 3134-3142.
- R. R. Reddel, “Transformation of Human Bronchial Epithelial Cells by Infection with SV40 or Adenovirus-12 SV40 Hybrid Virus, or Transfection via Strontium Phosphate Coprecipitation with a Plasmid Containing SV40 Early Region Genes,” Cancer Research, Vol. 48, No. 7, 1988, pp. 1904-1909.
- V. L. Kinnula, “Primary and Immortalized (BEAS 2B) Human Bronchial Epithelial Cells Have Significant Antioxidative Capacity in Vitro”, Cell and Molecular Biology, Vol. 11, No. 5, 1994, pp. 568-576.
- C. Schulz, “Differences in LPS-Induced Activation of Bronchial Epithelial Cells (BEAS-2B) and Type II-Like Pneumocytes (A-549),” Scandinavian Journal of Immunology, Vol. 56, No. 3, 2002, pp. 294-302. doi:10.1046/j.1365-3083.2002.01137.x
- C. Fearns, “Murine CD14 Gene Expression in Vivo: Extramyeloid Synthesis and Regulation by Lipopolysaccharide,” Journal of Experimental Medicine, Vol. 181, No. 3, 1995, pp. 857-866. doi:10.1084/jem.181.3.857
- M. Marini, “Expression of the Potent Inflammatory Cytokines, Granulocyte-Macrophage-Colony-Stimulating Factor and Interleukin-6 and Interleukin-8, in Bronchial Epithelial Cells of Patients with Asthma,” Journal of Allergy and Clinical Immunology, Vol. 89, No. 5, 1992, pp. 1001- 1009. doi:10.1016/0091-6749(92)90223-O
- T. L. Wu, “A Panel of Multiple Markers Associated with Chronic Systemic Inflammation and the Risk of Atherogenesis Is Detectable in Asthma and Chronic Obstructive Pulmonary Disease,” Journal of Clinical Laboratory Analysis, Vol. 21, No. 6, 2007, pp. 367-371. doi:10.1002/jcla.20197
- G. Liu, “TNF-Alpha and IL-8 of the Patients with Allergic Asthma,” Journal of Huazhong University of Science and Technology—Medical Sciences, Vol. 25, No. 4, 2005, pp. 274-275, 309.
- T. Akinyama, “Use and Specificity of Genistein as Inhibitor of Protein-Tyrosine Kinases,” Methods in Enzymology, Vol. 201, 1991, pp. 362-370. doi:10.1016/0076-6879(91)01032-W
- T. Pera, “Cigarette Smoke and Lipopolysaccharide Induce a Proliferative Airway Smooth Muscle Phenotype,” Respiratory Research, Vol. 11, 2010, p. 48. doi:10.1186/1465-9921-11-48
- J. Park, “Lipopolysaccharide Induces Cholangiocyte Proliferation via an Interleukin-6-Mediated Activation of p44/p42 Mitogen-Activated Protein Kinase,” Hepatology, Vol. 29, No. 4, 1999, pp. 1037-1043. doi:10.1002/hep.510290423
- K. Takami, “Interferon-Gamma Inhibits Hepatocyte Growth Factor-Stimulated Cell Proliferation of Human Bronchial Epithelial Cells: Upregulation of p27(kip1) Cyclin-Dependent Kinase Inhibitor,” American Journal of Respiratory Cell and Molecular Biology, Vol. 26, No. 2, 2002, pp. 231-238.
- S. Bhattacharyya, “Lipopolysaccharide Activates NF-Kappa B by TLR4-Bcl10-Dependent and Independent Pathways in Colonic Epithelial Cells,” American Journal of Physiology—Gastrointestinal and Liver, Vol. 295, No. 4, 2008, pp. G784-G790.
- A. Freitag, “Effects of Bacterial Lipopolysaccharides (LPS) and Tumour Necrosis Factor-Alpha (TNF Alpha) on Rat Tracheal Epithelial Cells in Culture: Morphology, Proliferation and Induction of Nitric Oxide (NO) Synthase,” Pulmonary Pharmacology, Vol. 9, 1996, pp. 149- 156. doi:10.1006/pulp.1996.0017
- D. Preciado, “Pseudomonas Aeruginosa Lipopolysaccharide Induction of Keratinocyte Proliferation, NF-Kappa B, and Cyclin D1 Is Inhibited by Indomethacin,” Journal of Immunology, Vol. 174, No. 5, 2005, pp. 2964-2973.
- M. E. Dawes, “In Vitro Effects of Lactoferrin on Lipopolysaccharide-Induced Proliferation, Gene Expression, and Prostanoid Production by Bovine Peripheral Blood Mononuclear Cells,” American Journal of Veterinary Research, Vol. 69, No. 9, 2008, pp. 1164-1170. doi:10.2460/ajvr.69.9.1164
Abbreviations
LPS: Lipopolysaccharides;
TLR: Toll-like receptor;
CD: cluster of differentiation;
sCD14: soluble CD14;
IL-6: Interleukin 6;
IL-8: Interleukin 8;
RANTES: regulated upon activation, normal T-cell expressed and secreted;
IFNg: interferon g;
TNFα: tumor necrosis factor α;
mCD14: membrane bound CD14;
MAPK: mitogen-activated protein kinase;
ERK: extracellular regulated kinase;
JNK: c-jun N-terminal kinase;
IRAK: interleukin 1 receptor associated kinase;
ELISA: enzyme linked immunosorbent assay;
FACS: fluorescence activated cell sorting;
MTT: 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide;
LDH: lactate dehydrogenase;
APC: Allophycocyanine;
HRP: horse radish peroxidase.
NOTES
*Corresponding author.