American Journal of Analytical Chemistry
Vol.07 No.01(2016), Article ID:62985,8 pages
10.4236/ajac.2016.71007
The Structures and Properties of Y-Substituted Mg2Ni Alloys and Their Hydrides: A First-Principles Study
Yuanyuan Li, Gaili Sun, Yiming Mi*
School of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, Shanghai, China

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 11 December 2015; accepted 22 January 2016; published 25 January 2016
ABSTRACT
The structures and properties of Y-substituted Mg2Ni alloys and the corresponding hydrides are investigated by a first-principles plane-wave pseudopotential method within density functional theory. Results show that Mg2Ni has the best structural stability when Y atom occupies the Mg(6f) lattice sites. The calculated enthalpies of formation for Mg2Ni, Mg2NiH4 and Mg15YNi8H32 are −51.612, −64.667 and −62.554 kJ/mol, respectively. It is implied that the substitution of Y alloying destabilizes the stability of the hydrides. Moreover, the dissociated energies of H atoms are decreased significantly, indicating that Y alloying benefits the improvement of the dehydrogenating properties of Mg2Ni hydrides. The calculation and analysis of the electronic structures suggest that there is a stronger interaction between H and Ni atoms than the interaction between H and Mg atoms in Mg2NiH4. However, the Ni-H bond is weakened by the substitution of Y. Therefore, the substitution is an effective technique to decrease the structural stability of the hydrides and benefit for hydrogen storage.
Keywords:
Mg2Ni Alloys, Y Substitution, Hydrides, First-Principles

1. Introduction
Due to rich reserves in the earth’s crust, high hydrogen capacity (3.6 wt%), light weight and low cost, Mg2Ni- type alloy hydrides remain as attractive hydrogen storage materials [1] [2] . However, the practical application of the alloy materials has not been achieved because of unfavorable thermodynamics, poor hydrogenation/dehy- drogenation kinetics and releasing undesirable by-products [3] .
Many researches have been devoted to overcoming these drawbacks and improving the properties of hydrogen storage via modifying microstructure by mechanical alloying [4] , alloying with other elements [5] [6] , adding catalysts [7] and composite structures [8] . The effects of transition metals including Cu, Co, Mn, Y, Ti, N band Crelements [9] -[12] on the hydrogen storage properties of Mg-based metal hydrides are investigated and discovered that the properties of hydrogen storage are improved by alloying with a small amount of transition metals in different degrees.
It is believed that alloying of Mg2Ni with transition metals is beneficial to improve the hydrogenating and dehydrogenating kinetics. The electronic structure of element Y is 4d15s2 and it can be incorporated into the metal boride. In addition, its chemical properties and physical performance are similar to La which can be used as an alloy element for hydrogen storage. The density and cohesive energy of Y atom are also relatively small. Therefore, Y has great potential to improve the performance of Mg2Ni alloy and its hydride. Kalinichenka et al. [13] studied that Y can be solved in Mg2Ni and the Mg-Ni-Y alloy exhibits higher dehydrogenation rates comparing with that of the Mg-Ni alloy. Song et al. [14] reported the microstructure and the hydrogenation properties of melt-spun Mg67Ni33−xYx alloys and found that the hydrogen storage capacity and kinetics of Mg2Ni are improved with Y doping. Zhang et al. [15] investigated that the substitution of Y for Mg had an insignificant effect on the activation ability of the Mg2Ni-type alloys, but it dramatically improved the cycle stability of the as- milled alloys. These experiments proved that Y plays an important role in improving the properties of Mg2Ni alloy for hydrogen storage. Thus, my understanding is that, alloying of Mg2Ni with Y can be expected to improve some performances of hydrogen absorption/desorption capacity and kinetics significantly.
In recent years, a number of theoretical investigations about the doped/substituted complex hydrides using first-principles calculations have been reported [16] -[19] . A first-principles study on the structures and properties of hydrogen storage alloy Mg2Ni, of aluminum and silver substituted alloys Mg2−xMxNi (M = Al and Ag), and of their hydrides Mg2NiH4, Mg2−xMxNiH4 was performed by Zeng et al. [20] . Their results show that the hydrogen storage capacity is decreased by the substitution and the substitution destabilizes the hydrides. However, there are no available theoretical reports about the structures and properties of Y substituted Mg2Ni alloys and their respective hydrides to the authors’ knowledge. The models are new for the materials to store hydrogen.
We focus primarily on the stable configuration of Mg2Ni alloys with Y substitution and determine the optimum position of Y. Furthermore, the energies, enthalpies of formation and electronic structures of Y alloying Mg2Ni and its hydrides are also calculated and analyzed using a first-principles plane-wave pseudo potential simulations based on the density functional theory in this paper. These simulations are beneficial to improve our understanding of the effects of substitution on the properties of Mg2Ni, and of the design about advanced magnesium-based hydrogen storage materials.
2. Computational Details
2.1. Computational Model
The crystal structure of Mg2Ni is hexagonal and its space group is P6222 (No.180) [21] , as shown in Figure 1(a). The lattice constants of Mg2Ni are a = b = 5.205 Å, c = 13.236 Å, α = β = 90˚, γ = 120˚. There are 12 Mg and 6 Ni atoms existing in the unit cell of Mg2Ni. The spatial positions Mg and Ni atoms are respectively 6f(0.5, 0, 0.1187), 6i(0.16, 0.324, 0) and 3b(0, 0, 0.5), 3d(0.5, 0, 0.5). Single Y atom substituting for Mg and Ni atoms are investigated respectively. Moreover, it has been shown that Mg2NiH4 forms readily by hydrogenating the alloy Mg2Ni [22] . The space group of Mg2NiH4 is monoclinic C2/c (No.15) and the lattice constants are a = 14.343 Å, b = 6.404 Å, c = 6.483 Å, β = 113.52˚, as shown in Figure 1(b). 16 Mg, 8 Ni and 32 H atoms are in the unit cell of Mg2NiH4 where Mg occupying the 8f, 4e, 4e sites and Ni the 8f site and H the 8f, 8f, 8f, 8f sites [23] [24] . The new systems of Y alloying Mg2NiH4 are studied.
2.2. Computational Method
All the density-functional theory (DFT) calculations are performed using a plane-wave basis set with the projector augmented plane wave (PAW) method as implemented in the Vienna ab initio simulation package (VASP)

Figure 1. Structures of (a) Mg2Ni, (b) Mg2NiH4, (c) Mg15YNi8H32 (where green, purple, red and orange balls denote Mg, Ni, H and Y atoms, respectively).
[25] - [27] . Projector Augmented Wave (PAW) potentials are used to treat the core-valence interaction [28] . The PW91 [29] [30] generalized gradient approximation (GGA) is employed for the exchange-correlation functional. The electronic wave functions are expanded by plane waves with a kinetic energy cutoff of 350 eV to attain the required convergence. All of the self-consistent loops are iterated until the total energy difference of the systems between the adjacent iterating steps is less than 10−7 eV. The Brillouin zone is sampled by 6 × 6 × 2 mesh points in k-space based on Monkhorst-Pack scheme [31] for all systems. The valence electrons of 1s for H, 2p and 3s for Mg, 3p, 3d and 4s for Ni, and 4d and 5s for Y are considered in the calculations.
3. Results and Discussions
3.1. The Structure of Substituted Mg2Ni by Y
In order to check the accuracy of the calculations, we first optimize the structure of Mg2Ni alloy and its hydride and compare the calculated lattice parameters with those determined experimentally. Then we consider the substitution of Mg and Ni by Y in independent spatial positions respectively. To single out a scenario that is most likely responsible for the stabilization of the crystal structure, the lattice parameters and enthalpies of formation ΔH for each case are calculated. The ΔH is calculated by taking the difference in total electronic energy of the products and the reactants [32] :
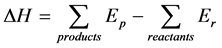 (1)
(1)
In the case of the crystal structure  which including xMg, yY, zNi, the enthalpies of formation are calculated by the following equation:
which including xMg, yY, zNi, the enthalpies of formation are calculated by the following equation:
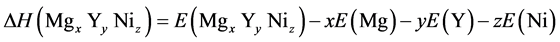 (2)
(2)
where 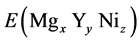 refers to the total energy of substituted Mg2Ni by Y.
refers to the total energy of substituted Mg2Ni by Y.
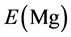 ,
, 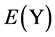 and
and 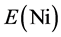 are the energy of every atom in HCP Mg, HCP Y and FCC Ni crystals, respectively. x, y, z are the numbers of Mg, Y and Ni atoms, respectively. Through the calculation, the values of
are the energy of every atom in HCP Mg, HCP Y and FCC Ni crystals, respectively. x, y, z are the numbers of Mg, Y and Ni atoms, respectively. Through the calculation, the values of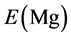 ,
, 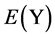 and
and 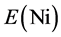 are −1.595, −6.379 and −5.415 eV, respectively.
are −1.595, −6.379 and −5.415 eV, respectively.
Table 1 displays the volume, lattice constant, total energy and enthalpies of formation of all the structures including Mg2Ni, substituted Mg2Ni by Y and their hydrides. The lattice constants of Mg2Ni after geometry optimization are a = b = 5.180 Å, c = 13.232 Å, which agree well with the experimental data a = b = 5.205 Å, c = 13.236 Å [21] . The enthalpy of formation of Mg2Ni is −3.211 eV, which means that the unit cell of Mg2Ni is −51.612 kJ/mol. It is very close to the experimental values −51.9 kJ/mol [33] . When Y atom is added into Mg2Ni, all the volumes of crystal structures will increase compared with the original structures. Moreover, it can be clearly observed that when the position of Mg (6f) is occupied by Y atom in Mg2Ni, the total energy and the enthalpy of formation are the minimum. It indicates that the structure of Mg11Y(6f)Ni6 has the optimal stabilization among all the substituted structures.

Table 1. Volume, lattice constant, total energy, enthalpy of formation of Mg2Ni, Y-substituted Mg2Ni and their hydrides.
3.2. The Properties of Substituted Mg2NiH4 by Y
Based on the stable structure of Mg11Y(6f)Ni6, we study the properties of substituted Mg2NiH4 by Y. Firstly, We have proved that the theoretical lattice constants and internal atomic positions of Mg2NiH4 are in good agreement with experimental results [23] . The states are displayed in Table 1. Various substitutive positions of Mg are considered. We find that the total energy of each new structure is very close. Thereby, a reasonable structure Mg15YNi8H32 is selected to be investigated in detail, as shown in Figure 1(c).
In order to research the effects of Y on the properties of Mg2NiH4, We calculate the enthalpies of formation of Mg2NiH4 and Mg15YNi8H32 respectively. In general, the formation of Mg2NiH4 can be expressed by the following reaction:
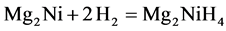 (3)
(3)
The enthalpy of formation of Mg2NiH4 can be expressed in Equation (4):
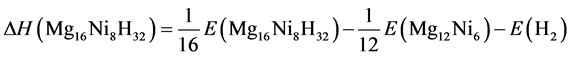 (4)
(4)
In the same way, the reaction of formation and the enthalpy of formation of Mg15YNi8H32 can be respectively written as Equations (5) and (6):
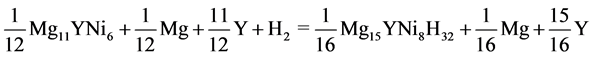 (5)
(5)
 (6)
(6)
where ,
,  ,
,  and
and  are the total energy of Mg2NiH4, Mg2Ni, Mg15YNi8H32 and Mg11Y(6f)Ni6, respectively.
are the total energy of Mg2NiH4, Mg2Ni, Mg15YNi8H32 and Mg11Y(6f)Ni6, respectively.
 is the energy of free H2 molecule. The calculated results are also shown in Table 1. For pure Mg2NiH4, the enthalpy of formation is −64.667 kJ/mol which coincides closely with the experimental result −64.4 ± 4.2 kJ/mol reported by Reilly et al. [22] . Furthermore, the enthalpy of formation of Mg15YNi8H32 is −62.554 kJ/mol which is higher than that of pure Mg2NiH4. It can be clearly seen that the introduction of Y atom has effects on the destabilization of Mg2NiH4 in terms of energy. This is energetically favorable to perform the dehydrogenation reaction of substituted Mg2NiH4 by Y.
is the energy of free H2 molecule. The calculated results are also shown in Table 1. For pure Mg2NiH4, the enthalpy of formation is −64.667 kJ/mol which coincides closely with the experimental result −64.4 ± 4.2 kJ/mol reported by Reilly et al. [22] . Furthermore, the enthalpy of formation of Mg15YNi8H32 is −62.554 kJ/mol which is higher than that of pure Mg2NiH4. It can be clearly seen that the introduction of Y atom has effects on the destabilization of Mg2NiH4 in terms of energy. This is energetically favorable to perform the dehydrogenation reaction of substituted Mg2NiH4 by Y.
To make further investigation about the performance of dehydrogenation, we calculate the energies of Mg2NiH4 and Mg15YNi8H32 which dissociate the nearest 2 H atoms around Ni atoms. The dehydrogenation energy is calculated by Equation (7):
 (7)
(7)
The results are shown in Table 2. From Table 2 we can see that the addition of Y clearly decreases the dehydrogenation energy of Mg2NiH4 by about 47% to 0.983 eV. It suggests that although Y atom has poor effects on the destabilization of Mg2Ni, it breaks down the stability of Mg2NiH4 positively and improve the dehydrogenation kinetics of Mg2NiH4 which as one of the hydrogen storage materials.
3.3. Electronic Structure
In order to further understand the effects of Y atom on the dehydrogenation properties of Mg2NiH4 alloy, the electronic properties of Mg2Ni and Mg15YNi8H32 are studied by calculating total density of states (DOS) and partial density of states (PDOS). Figure 2 displays the DOS and PDOS of Mg2Ni and Mg15YNi8H32 alloys.
Form Figure 2(a) we can see that there are two main peaks in total density of states below Fermi level. The bonding electron of the energy region between −9.2 and −3.7 eV is mainly dominated by Hs, Nis and Nid orbits, partial Mgs orbit. It is implied that H atoms tend to bond with Ni rather than Mg atoms in the structure of Mg2NiH4. The result is in correspondence with the conclusion that the interaction Ni-H is stronger than that of Mg-H which studied by Jasen [34] . There is a major contribution with Ni p, Ni d and Mg s orbits in the region from −2.4 eV to Fermi level. This indicates that Mg and Ni atoms have hybridization which keeps the structure of Mg2NiH4 stable. In addition, Ni d orbit plays the dominating role in the bonding electron.
Table 2. Calculated dehydrogenation energies of Mg15MNi8H32 (M = Mg, Y) (eV).

 (a) (b)
(a) (b)
Figure 2. Total density of states and partial density of states of (a) Mg2NiH4, (b) Mg15YNi8H32.
Compared to pure Mg2NiH4, the enthalpy of formation and dehydrogenation energy change markedly due to the substituted Mg2NiH4 by Y. Figure 2(b) displays that below Fermi level Mg15YNi8H32 has two main bonding peaks from −10.7 to −5.2 eV and −4.1 to −1.6 eV. It is not difficult to find that all the bonding peaks in total density of states move to the energy of deep potential well and the number of bonding electron reduces comparing to Mg2NiH4. It demonstrates that the substitution of Y alloying weakens the interaction of the atoms and destabilizes the structure of the hydride. The effects of Yd orbit on the bonding electron are significant especially for the energy region from −4.1 to −1.6 eV. What is more, Yp and d orbits contribute to the bonding electron and have mutual interaction with Nip and d orbits. It is also worth noting that the overlapping region between Nid and Hs orbits decreases obviously. It means that the interaction between Ni and H atoms become weak.
4. Conclusion
We have investigated the structure and properties of substituted Mg2Ni alloys by Y and the corresponding hydrides. The structure parameter, enthalpy of formation, dehydrogenation energy and electronic structure are calculated by the first-principles method based on density functional theory in this paper. Through analyzing the simulation results, we can draw the conclusions that when Y atom occupies the Mg(6f) lattice site, the structure of Mg2Ni is the optimal stable. The substitution of Y destabilizes the stability of Mg2NiH4 and decreases the dissociated energies of H atoms due to the Ni-H bond weakened by Y. Therefore, the method of substitution is in favor of the dehydrogenation reaction for Mg-based hydrides as hydrogen storage materials. Moreover, we will continue to perfect this respect, for instance, whether the effect of Y elements in the case of different numbers of Y metals and different substituents will change.
Acknowledgements
This work was supported by Innovation Program of Shanghai Municipal Education Commission, China (10YZ172) and Subjects Construction Program of Shanghai University of Engineering Science, China (2012gp43) and Graduated Innovative Research Project of Shanghai University of Engineering Science (E1-0903-14-01107- 14KY0411).
Cite this paper
YuanyuanLi,GailiSun,YimingMi, (2016) The Structures and Properties of Y-Substituted Mg2Ni Alloys and Their Hydrides: A First-Principles Study. American Journal of Analytical Chemistry,07,67-74. doi: 10.4236/ajac.2016.71007
References
- 1. Schlapbach, L. and Züttel, A. (2001) Hydrogen-Storage Materials for Mobile Applications. Nature, 414, 353-358.
http://dx.doi.org/10.1038/35104634 - 2. Jain, I.P., Lal, C. and Jain, A. (2010) Hydrogen Storage in Mg: A Most Promising Material. International Journal of Hydrogen Energy, 35, 5133-5144.
http://dx.doi.org/10.1016/j.ijhydene.2009.08.088 - 3. Liu, T., Wang, C.X. and Wu, Y. (2014) Mg-Based Nano-composites with Improved Hydrogen Storage Performances. International Journal of Hydrogen Energy,39, 14262-14274.
http://dx.doi.org/10.1016/j.ijhydene.2014.03.125 - 4. Shang, C.X. and Guo, Z.X. (2007) Structural and Desorption Characterisations of Milled (MgH2+ Y, Ce) Powder Mixtures for Hydrogen Storage. International Journal of Hydrogen Energy, 32, 2920-2925.
http://dx.doi.org/10.1016/j.ijhydene.2006.11.035 - 5. Shao, H., Asano, K., Enoki, H. and Akiba, E. (2009) Preparation and Hydrogen Storage Properties of Nanostructured Mg-Ni BCC Alloys. Journal of Alloys and Compounds, 477, 301-306.
http://dx.doi.org/10.1016/j.jallcom.2008.11.004 - 6. Kim, H., Nakamura, J., Shao, H., Nakamura, Y., Akiba, E., Chapman, K.W., Chupas, P.J. and Proffen, T. (2011) Insight into the Hydrogenation Properties of Mechanically Alloyed Mg50Co50 from the Local Structure. Journal of Physical Chemistry C, 115, 20335-20341.
http://dx.doi.org/10.1021/jp207197k - 7. Barkhordarian, G., Klassen, T. and Bormann, R. (2003) Fast Hydrogen Sorption Kinetics of Nanocrystalline Mg Using Nb2O5 as Catalyst. Scriptamaterialia, 49, 213-217.
http://dx.doi.org/10.1016/s1359-6462(03)00259-8 - 8. Oelerich, W., Klassen, T. and Bormann, R. (2001) Metal Oxides as Catalysts for Improved Hydrogen Sorption in Nanocrystalline Mg-Based Materials. Journal of Alloys and Compounds, 315, 237-242.
http://dx.doi.org/10.1016/S0925-8388(00)01284-6 - 9. Zhang, Y., Zhao, C., Yang, T., Shang, H.W., Xu, C. and Zhao, D.L. (2013) Comparative Study of Electrochemical Performances of the As-Melt Mg20Ni10-xMx (M = None, Cu, Co, Mn; x = 0, 4) Alloys Applied to Ni/Metal Hydride (MH) Battery. Journal of Alloys and Compounds, 555, 131-137.
http://dx.doi.org/10.1016/j.jallcom.2012.12.016 - 10. Zhang, Q.A., Zhang, L.X. and Wang, Q.Q. (2013) Crystallization Behavior and Hydrogen Storage Kinetics of Amorphous Mg11Y2Ni2 Alloy. Journal of Alloys and Compounds, 551, 376-381.
http://dx.doi.org/10.1016/j.jallcom.2012.11.046 - 11. Cui, J., Liu, J., Wang, H., Ouyang, L.Z., Sun, D.L., Zhu, M. and Yao, X.D. (2014) Mg-TM (TM: Ti, Nb, V, Co, Mo or Ni) Core-Shell Like Nanostructures: Synthesis, Hydrogen Storage Performance and Catalytic Mechanism. Journal of Materials Chemistry A, 2, 9645-9655.
http://dx.doi.org/10.1039/c4ta00221k - 12. Vyas, D., Jain, P., Agarwal, G., Ankur, J. and Jain, I.P. (2012) Hydrogen Storage Properties of Mg2Ni Affected by Cr Catalyst. International Journal of Hydrogen Energy, 37, 16013-16017.
http://dx.doi.org/10.1016/j.ijhydene.2012.08.039 - 13. Kalinichenka, S., Rontzsch, L., Baehtz, C. and Kieback, B. (2010) Hydrogen Desorption Kinetics of Melt-Spun and Hydrogenated Mg90Ni10 and Mg80Ni10Y10 Using in Situ Synchrotron, X-Ray Diffraction and Thermogravimetry. Journal of Alloys and Compounds, 496, 608-613.
http://dx.doi.org/10.1016/j.ijhydene.2012.08.039 - 14. Song, W., Li, J., Zhang, T., Kou, H. and Xue, X. (2014) Microstructure and Tailoring Hydrogenation Performance of Y-Doped Mg2Ni Alloys. Journal of Power Sources, 245, 808-815.
http://dx.doi.org/10.1016/j.jpowsour.2013.07.049 - 15. Zhang, Y., Zhang, P., Yuan, Z., Yang, T., Qi, Y. and Zhao, D.L. (2015) An Investigation on Electrochemical Hydrogen Storage Performances of Mg-Y-Ni Alloys Prepared by Mechanical Milling. Journal of Rare Earths, 33, 874-883.
http://dx.doi.org/10.1016/S1002-0721(14)60499-3 - 16. Haussermann, U., Blomqvist, H. and Noréus, D. (2002) Bonding and Stability of the Hydrogen Storage Material Mg2NiH4. Inorganic Chemistry, 41, 3684-3692.
http://dx.doi.org/10.1021/ic0201046 - 17. Van Setten, M.J., De Wijs, G.A. and Brocks, G. (2007) Ab Initio Study of the Effects of Transition Metal Doping of Mg2NiH4. Physical Review B, 76, Article ID: 075125.
http://dx.doi.org/10.1103/PhysRevB.76.075125 - 18. Jezierski, A., Jurczyk, M. and Szajek, A. (2009) Electronic Structure of Mg2Ni1-xCux. Actaphysicapolonica A, 115, 223-225.
- 19. Huang, L.W., Elkedim, O., Nowak, M., Chassagnond, R. and Jurczyk, M. (2012) Mg2-xTixNi (x = 0, 0.5) Alloys Prepared by Mechanical Alloying for Electrochemical Hydrogen Storage: Experiments and First-Principles Calculations. International Journal of Hydrogen Energy, 37, 14248-14256.
http://dx.doi.org/10.1016/j.ijhydene.2012.07.036 - 20. Zeng, Y., Fan, K., Li, X., Xu, B., Gao, X. and Meng, L. (2010) First-Principles Studies of the Structures and Properties of Al- and Ag-Substituted Mg2Ni Alloys and Their Hydrides. International Journal of Hydrogen Energy, 35, 10349-10358.
http://dx.doi.org/10.1016/j.ijhydene.2010.07.131 - 21. Darriet, B., Soubeyroux, J.L., Pezat, M. and Fruchart, D. (1984) Structural and Hydrogen Diffusion Study in the Mg2Ni-H2 System. Journal of the Less Common Metals, 103, 153-162.
http://dx.doi.org/10.1016/0022-5088(84)90374-6 - 22. Reilly Jr., J.J. and Wiswall Jr., R.H. (1968) Reaction of Hydrogen with Alloys of Magnesium and Nickel and the Formation of Mg2NiH4. Inorganic Chemistry, 7, 2254-2256.
http://dx.doi.org/10.1021/ic50069a016 - 23. Zolliker, P., Yvon, K., Jorgensen, J.D. and Rotella, F.J. (1986) Structural Studies of the Hydrogen Storage Material Magnesium Nickel Hydride (Mg2NiH4). 2. Monoclinic Low-Temperature Structure. Inorganic Chemistry, 25, 3590-3593.
http://dx.doi.org/10.1021/ic00240a012 - 24. Zhang, J., Huang, Y.N., Peng, P., Mao, C., Shao, Y.M. and Zhou, D.W. (2011) First-Principles Study on the Dehydrogenation Properties and Mechanism of Al-Doped Mg2NiH4. International Journal of Hydrogen Energy, 36, 5375-5382.
http://dx.doi.org/10.1021/ic00240a012 - 25. Kresse, G. and Hafner, J. (1993) Ab initio Molecular Dynamics for Open-Shell Transition Metals. Physical Review B, 48, 13115-13118.
http://dx.doi.org/10.1103/PhysRevB.48.13115 - 26. Kresse, G. and Furthmüller, J. (1996) Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Computational Materials Science, 6, 15-50.
http://dx.doi.org/10.1016/0927-0256(96)00008-0 - 27. Kresse, G. and Furthmüller, J. (1996) Efficient Iterative Schemes for Ab initio Total-Energy Calculations Using a Plane-Wave Basis Set. Physical Review B, 54, 11169-11186.
http://dx.doi.org/10.1103/PhysRevB.54.11169 - 28. Perdew, J.P., Burke, K. and Wang, Y. (1996) Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-Electron System. Physical Review B, 54, 16533-16539.
http://dx.doi.org/10.1103/PhysRevB.54.16533 - 29. Perdew, J.P., Chevary, J.A., Vosko, S.H., Jackson, K.A., Pederson, M.R., Singh, D.J. and Fiolhais, C. (1992) Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Physical Review B, 46, 6671-6687.
http://dx.doi.org/10.1103/PhysRevB.46.6671 - 30. Perdew, J.P., Burke, K. and Ernzerhof, M. (1996) Generalized Gradient Approximation Made Simple. Physical Review Letters, 77, 3865-3868.
http://dx.doi.org/10.1103/PhysRevLett.77.3865 - 31. Monkhorst, H.J. and Pack, J.D. (1976) Special Points for Brillouin-Zone Integrations. Physical Review B, 13, 5188-5192.
http://dx.doi.org/10.1103/PhysRevB.13.5188 - 32. Broedersz, C.P., Gremaud, R., Dam, B., Griessen, R. and Lovvik, O.M. (2008) Highlydestabilized Mg-Ti-Ni-H System Investigated by Density Functional Theory and Hydrogenography. Physical Review B, 77, Article ID: 024204.
http://dx.doi.org/10.1103/PhysRevB.77.024204 - 33. Kubaschewski, O. and Alcock, C.B. (1979) International Series on Materials Science and Technology. Pergamon Press, Oxford, 294.
- 34. Jasen, P.V., Gonzalez, E.A., Brizuela, G., Nagel, O.A., González, G.A. and Juan, A. (2007) A Theoretical Study of the Electronic Structure and Bonding of the Monoclinic Phase of Mg2NiH4. International Journal of Hydrogen Energy, 32, 4943-4948.
http://dx.doi.org/10.1016/j.ijhydene.2007.08.011
NOTES

*Corresponding author.