American Journal of Analytical Chemistry
Vol.4 No.10A(2013), Article ID:38571,6 pages DOI:10.4236/ajac.2013.410A1007
Enhanced Electrocatalytic Reduction of Oxygen at Electrodes Coated with a Multi-Metallic Co(II)/Pt(II) Porphyrin
Department of Chemistry, University of Dayton, Dayton, USA
Email: *sswavey1@udayton.edu
Copyright © 2013 Shawn Swavey, David Fresh. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 1, 2013; revised September 1, 2013; accepted September 20, 2013
Keywords: Oxygen Electrocatalysis; Cobalt; Platinum; Porphyrin
ABSTRACT
Edge plane pyrolytic graphite (EPG) electrodes coated with the Co(II)/Pt(II)2 analog of 5,15-bis-(4-pyridyl)-10,20-bis- (3-methoxy-4-hydroxyphenyl)porphyrin undergo an electrochemical-chemical-electrochemical (ECE) reaction when anodically scanned in 1.0 M sulfuric acid. The new redox couple formed from this anodic conditioning of the coated electrode is dependent on the pH of the solution. Roughened EPG electrodes coated with the Co(II)/Pt(II)2 trimetallic porphyrin show a catalytic shift of 400 mV for the reduction of O2 when compared to the reduction of O2 at a bare EPG electrode. An additional catalytic shift of ca. 150 mV is observed for O2 reduction at an EPG electrode coated with the Co(II)/Pt(II)2 porphyrin which has been oxidized in 1.0 M sulfuric acid. In addition to the added electrocatalysis, a significant percentage of O2 reduced at the oxidized Co(II)/Pt(II)2 EPG electrode is converted to H2O as determined by rotating disk electrode measurements.
1. Introduction
Fuel cell technology has gained considerable attention over the past couple of decades in part due to its high efficiency and low pollutant output. Unfortunately the mass production of fuel cells is limited by the costly platinum catalysts which can be as much as 56% of the total cost of the fuel cell [1,2]. This has resulted in extensive research into finding a catalyst capable of replacing platinum in fuel cells. Of the many types of catalysts under investigation, few have received as much attention as cobalt(II) porphyrins [3,4]. Cobalt porphyrins have been shown to reduce oxygen at potentials nearing that of platinum; however, the majority of monometallic cobalt porphyrins reduce oxygen by two electrons to hydrogen peroxide instead of the more desired four electron reduction of oxygen directly to water [5].
Cobalt(II) porphyrins offer an advantage over other transition metal porphyrins in their ability to catalyze the reduction of oxygen; unfortunately, the vast majority of cobalt(II) porphyrins are only capable of reducing oxygen by two electrons to form hydrogen peroxide an unwanted deleterious product for fuel cells. It has been noted that cobalt(II) porphine, the simplest porphyrin, is capable of reducing oxygen to water when coated onto carbon electrodes in acidic media [6,7]. Mass production of cobalt(II) porphine is prohibited, however, by its difficult synthesis and purification. Researchers have found other ways to convert monometallic cobalt(II) porphyrins into catalysts capable of reducing oxygen directly to water. In a series of studies, it was shown that by coordination of 3 or 4 substitutionally inert Ru(II)(NH3)5 moieties to the meso-pyridyl nitrogens of cobalt(II) tetra-(4- pyridyl)porphyrin, adsorbed onto pyrolytic graphite electrodes, the direct four electron reduction of oxygen to water at catalytic potentials could be achieved [8-14]. Subsequent studies revealed that the multi-electron reduction of O2 to H2O with these complexes is the result of increased electron density placed on Co(II) through p-backbonding of the pyridyl groups with the Ru(II) groups. These complexes were found to be unstable in acidic solutions, the medium by which many fuel cells operate.
Stability of the catalyst under the harsh acidic or basic conditions of fuel cells is of paramount importance. To this end researchers have found that conductive polymeric films on glassy carbon electrodes can be produced by continuous anodic cycling in basic solutions of nickel(II) 5,10,15,20-tetra-(3-methoxy-4-hydroxyphenyl) porphyrin [15]. These films have been used to detect trace amounts of nickel. Similar films have also been used to detect nitric oxide from a single cell [16]. Studies of copper(II) 5,10,15,20-tetra-(3-methoxy-4-hydroxyphenyl)porphyrin films as p-type semiconductors have shown promise in photoelectrochemical cells converting light into chemical or electrical energy [17]. Reduction of oxygen to water in buffered pH 7.0 solutions has been accomplished by platinum electrodes modified with Iron(III) 5,10,15,20-tetra-(3-methoxy-4-hydroxyphenyl)porphyrin formed by cycling the electrode in basic solutions of the iron porphyrin [18].
This laboratory has recently shown that by anodically conditioning edge plane pyrolytic graphite (EPG), electrodes coated with cobalt(II) 5,10,15,20-tetra-(3-methoxy-4-hydroxyphenyl) porphyrin in acidic media create a stable electrocatalyst for oxygen reduction [19]. Analysis of rotating disc experiments reveals that approximately 15% of the oxygen is reduced directly to water. In a separate study we incorporated a peripheral transition metal complex to a cobalt(II) porphyrin containing three (3-methoxy-4-hydroxyphenyl) substituents in the hopes of combining the p-backbonding effect with a conductive catalytic film [20]. When EPG electrodes coated with this complex, cobalt(II) 5-(4-pyridyl)-10,15,20-tris-(3-methoxy-4-hydroxyphenyl) porphyrin with PtCl2 (dmso)2 coordinated to the peripheral nitrogen of the pyridyl group was anodically conditioned in acidic media, and a catalytic shift in the reduction of oxygen was observed. Furthermore, rotating disc experiments revealed that this complex reduced almost 65% of the oxygen directly to water. We determined that the peripheral platinum(II) group was acting indirectly in the catalytic process by noting that only the complex containing cobalt(II) showed catalysis of oxygen. From this we presume that the platinum group is acting to strengthen the cobalt(II)-peroxo bond through p-backbonding. To further this study we present herein the synthesis of a new cobalt(II) porphyrin containing two peripheral platinum moieties, complex I (Scheme 1), to determine if further backbonding will facilitate the complete reduction of oxygen to water.
2. Experimental Section
2.1. Materials
All reagents were analytical grade and used without further purification unless stated otherwise. 3-methoxy-4- hydroxybenzaldehyde, 4-pyridinecarboxaldehyde, propionic acid, methanol, N,N’-dimethylformamide (DMF), ethylacetate, methylene chloride, perchloric acid, cobalt(II) acetate, 60 - 200 mesh silica gel (Fisher) were used as received. The buffer solutions and their pH val-
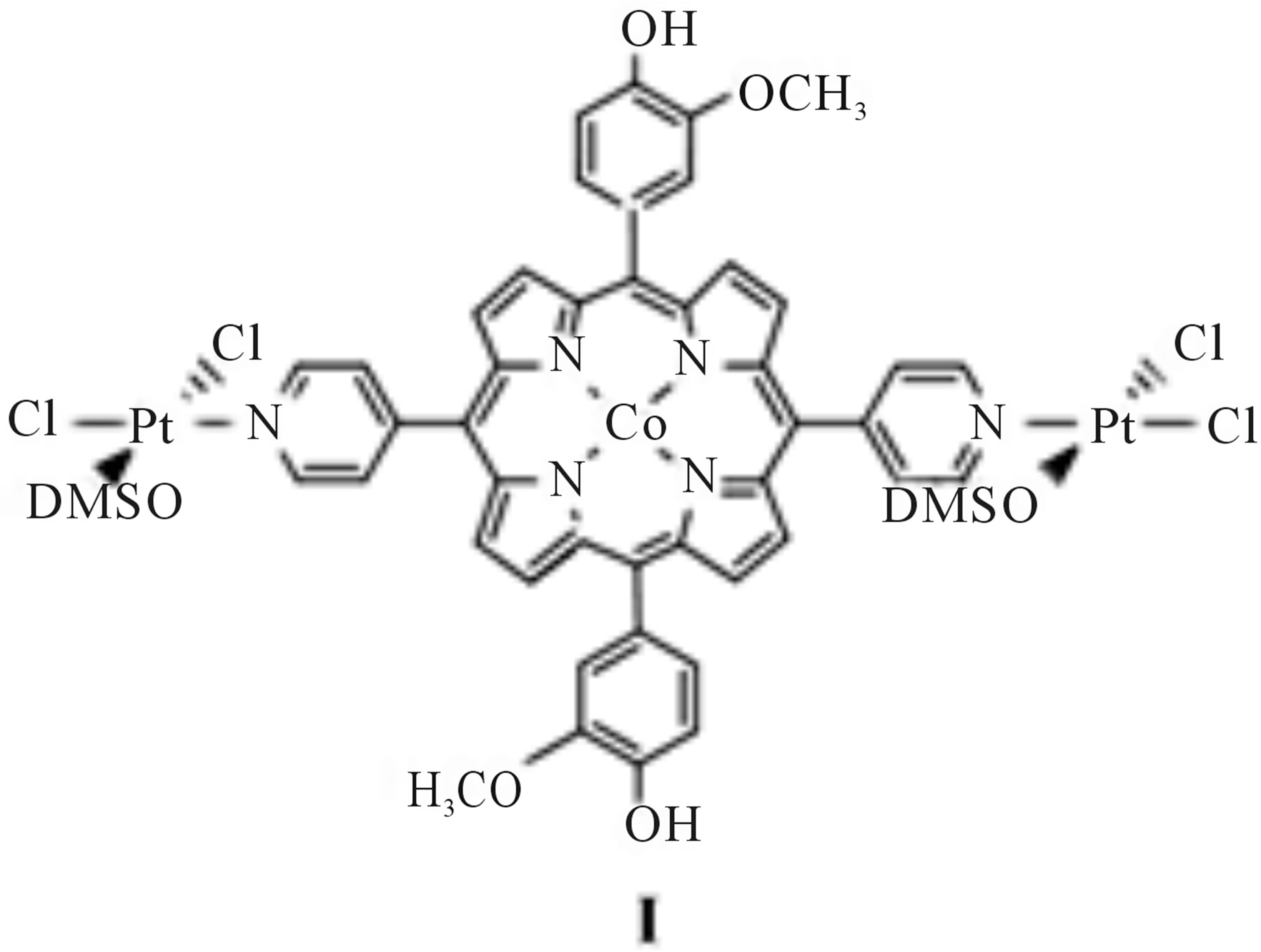
Scheme 1. Structure of diplatinum cobalt porphyrin.
ues measured to ±0.01 at 25˚C using a Denver Instrument UltraBasic pH meter calibrated with standard pH 4.00 and 10.00 buffer solutions (Fisher), were as follows: 0.2 M NaH2PO4, 0.05 M H3PO4 (2.61); 0.24 M CH3COOH, 0.05 M CH3COONa (3.66); 0.2 M CH3COONa, 0.05 M CH3COOH (4.87); 0.2 M NaH2PO4, 0.05 M Na2HPO4 (6.12). Elemental analyses were performed by Atlantic Microlabs, Norcross, GA.
2.2. Preparation of Complexes
5,15-bis-(4-pyridyl)-10,20-bis-(3-methoxy-4-hydro- xyphenyl)porphyrin. A modified procedure to the previous synthetic procedure was used to synthesize this porphyrin. To a solution of 4.9 g (0.032 moles) of 3-methoxy-4-hydroxybenzaldehyde and 3.4 g (0.032 moles) of 4-pyridinecarboxaldehyde in 100 mL of propionic acid was added 4.3 g (0.064 moles) of freshly distilled pyrrole. The solution was refluxed for 1 hr. after which it was cooled to room temperature and cautiously added to 200 mL of a 50:50 methanol:ammonium hydroxide solution cooled in an ice bath. The slurry was refrigerated overnight, filtered and washed (3 × 100 mL) with methanol. The resulting black powder was extracted in a soxhlet with dichloromethane. The volume of the dichloromethane extract was reduced to ca. 50 mL and chromatographed on silica gel using dichloromethane as eluent. The first band was collected using a 1% methanol/dichloromethane mixture, the second band was collected using a 2% methanol/dichloromethane mixture, and the product band was collected with 4% methanol/dichloromethane. Anal. Calc. for C44H32N6O4∙1.5H2O: C 71.82; H, 4.79; N, 11.42. Found: C, 71.81; H, 4.85; N, 11.20.
cis-Pt2(DMSO)2-[5,15-bis-(4-pyridyl)-10,20-bis-(3-methoxy-4-hydroxyphenyl)porphyrin] Cl4. To a solution containing 20 mg (0.028 mmoles) of the porphyrin in 10 mL of CH2Cl2 was added 24 mg (0.056 mmoles) cis-Pt Cl2 (DMSO)2 and the solution was stirred for 24 hrs. at ambient temperature. The reaction was chromatographed on silica gel using a 50:50 ethyl acetate:dichloromethane solution as eluent. The first red band from the column was collected and the solvent removed resulting in 27 mg of a purple powder. Anal. Calc. for C48H52N6O6S2Pt2∙2EtOAc: C, 42.75; H, 3.84; N, 5.34; S, 4.08. Found: C, 43.27; H, 3.85; N, 5.32; S 4.48.
Insertion of the cobalt(II) metal center was performed using a standard procedure using cobalt(II) acetate in slight excess in refluxing chloroform [21]. This gave the desired product I in 38% yield.
2.3. Procedures and Instrumentation
Adsorption of the metalloporphyrin I onto edge plane pyrolytic graphite EPG electrode (Pine Instrument Co.) which had been roughened using 600 grit sandpaper was accomplished by placing 5 - 15 μL aliquots of 1.0 mM acetone solutions of the metalloporphyrin onto the electrode surface and allowing the solvent to evaporate at 20˚C. The electrode was then washed with distilled water and tapped dry with a Kimwipe. Cyclic voltammetry and rotating disc electrode (RDE) experiments of the modified electrodes were performed using a Pine AFCBP1 bipotentiostat and an AFMSRX rotator (Pine Instrument Co.) in 1.0 M sulfuric acid solutions purged with air.
3. Results and Discussion
3.1. Electrode Adsorption Studies
Complex I was adsorbed onto EPG electrodes by taking 5 - 15 μL aliquots of 1.0 mM solutions of I in acetone and dropping them onto the surface of the electrode. The solvent was allowed to evaporate and the modified electrode was rinsed with distilled water and gently tapped dry. Placing the electrode into a 1.0 M sulfuric acid solution with a platinum auxiliary electrode and an SCE reference electrode cyclic voltammetry experiments were performed.
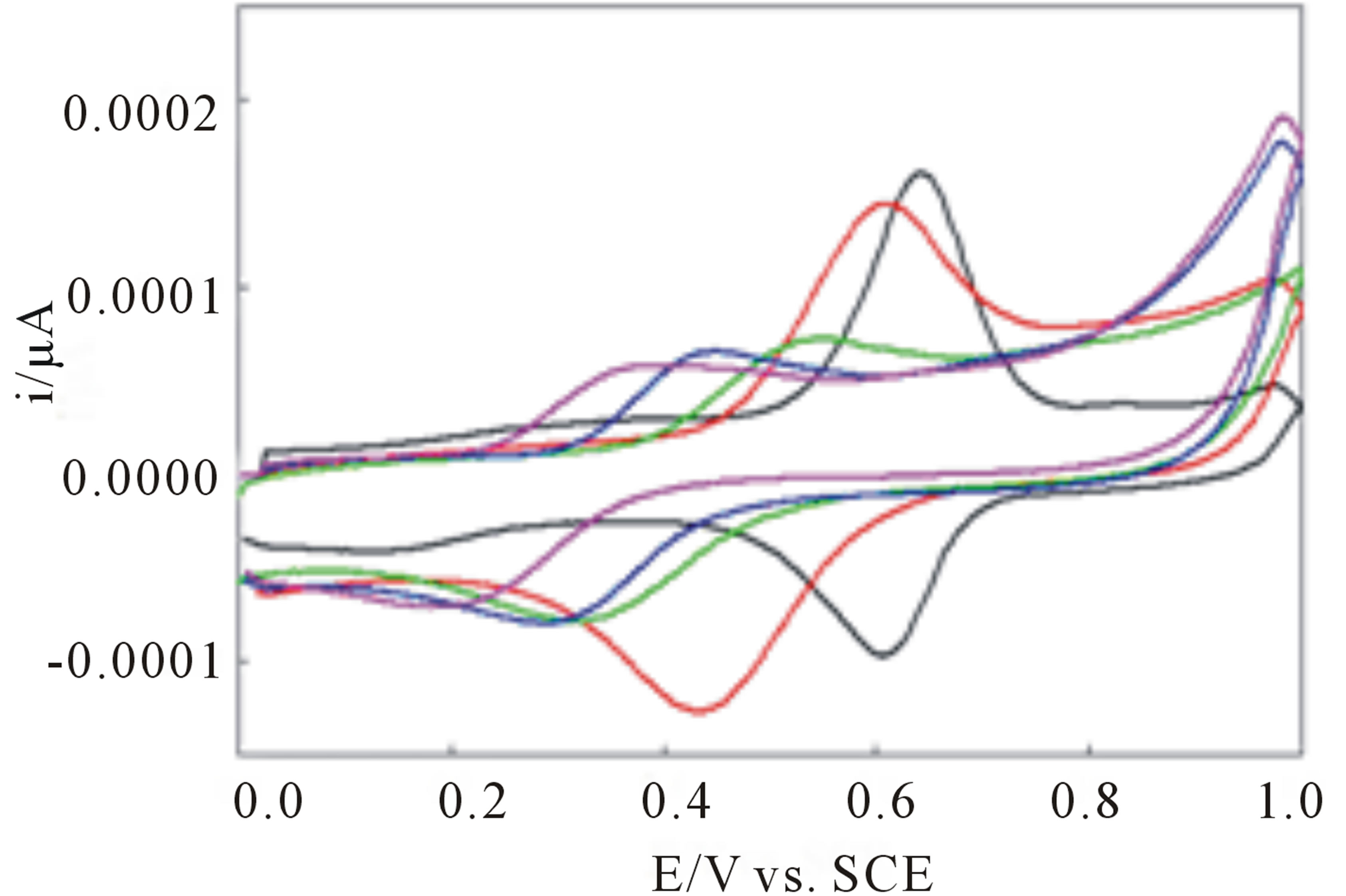
Upon anodic cycling of the modified electrode in 1.0 M sulfuric acid an irreversible oxidation is observed at 0.84 V versus SCE (I, Figure 1). This oxidation is believed to be due to the oxidation of the peripheral 3- methoxy-4-hydroxyphenyl substituents. Reversing the scan reveals a reduction wave at 0.59 V versus SCE (IIa, Figure 1). After oxidation of the peripheral vanillin groups a chemical step, hydrolysis, occurs giving the quinine which is reduced to the hydroquinone electrochemically. On the third cycle a new oxidation wave appears at 0.64 V versus SCE (IIb, Figure 1) coupled to the reduction wave and attributed to the oxidation of the hydroquinone.
The newly formed redox couple (IIa/IIb) is believed to be the quinone/hydroquinone couple illustrated below (Scheme 2). As indicated this chemical reaction depends on the proton concentration.
To give further evidence of the proposed electrochemi-
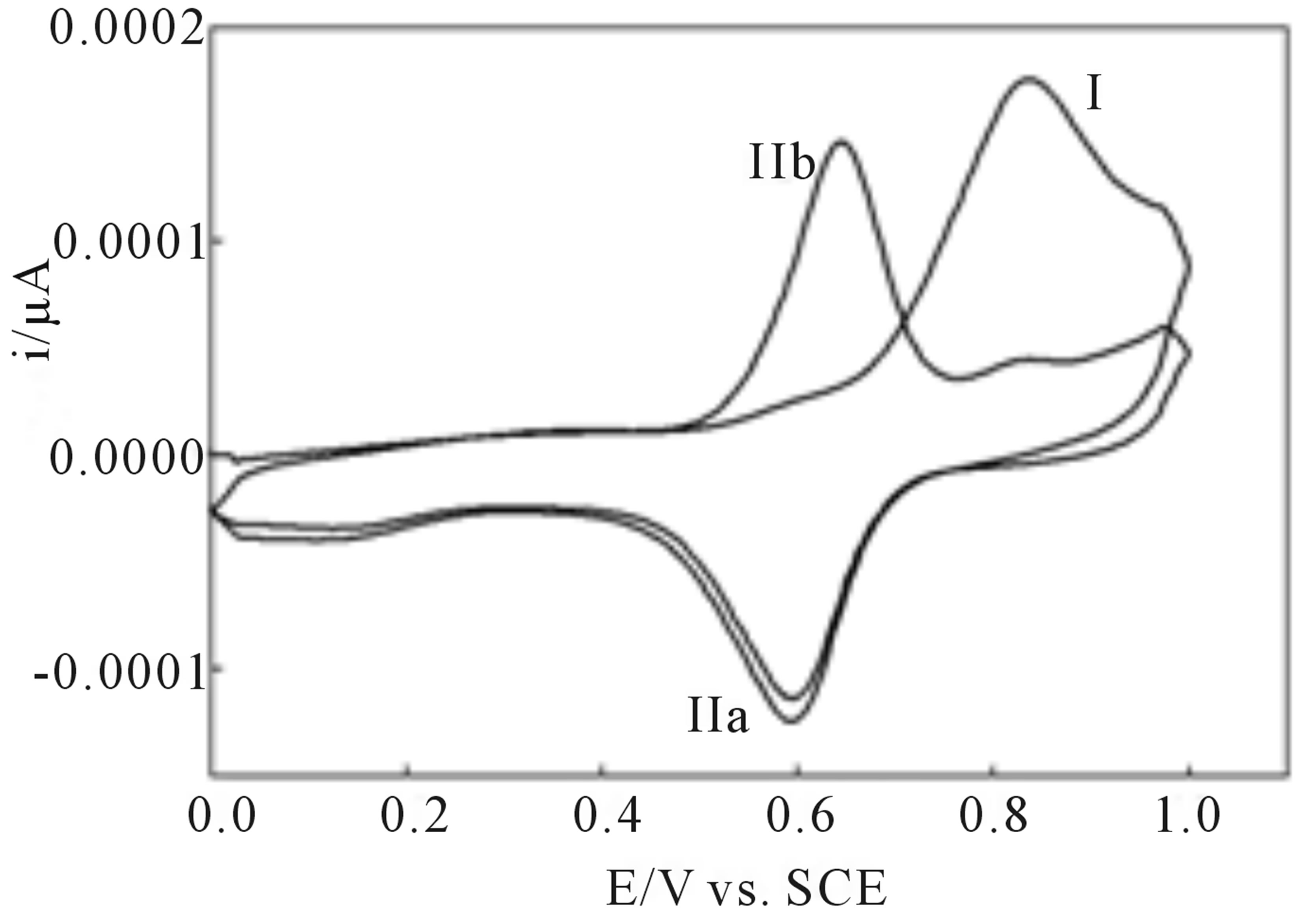
Figure 1. Cyclic voltammograms of complexes I adsorbed on roughened EPG electrodes in 1.0 M H2SO4, scan rate = 50 mV/s, referenced to the saturated calomel electrode (SCE).
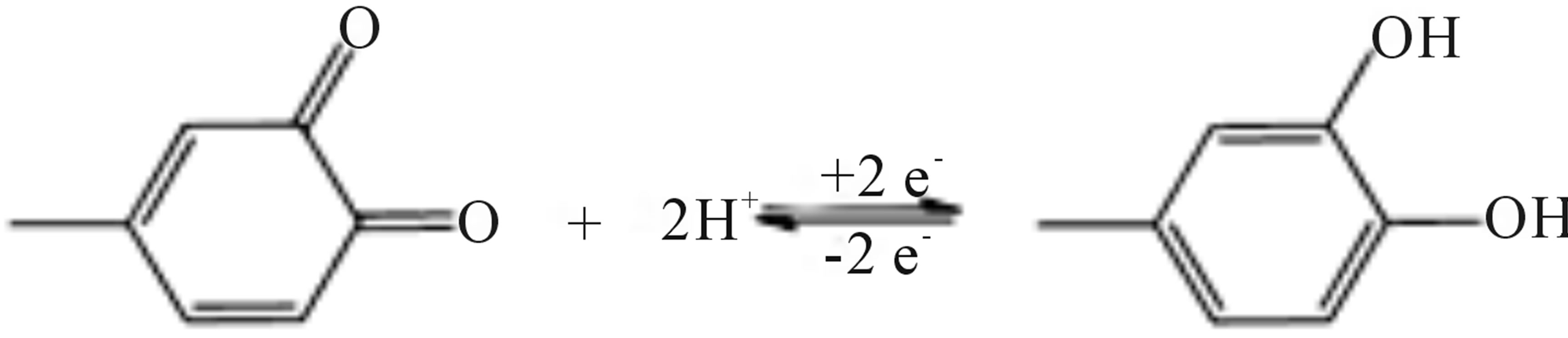
Scheme 2. Redox process for quinone/hydroquinone.
cal process observed in Figure 1 cyclic voltammetry experiments of the anodically conditioned electrode (containing the new redox couple) were run in solutions of varying pH. A plot of the 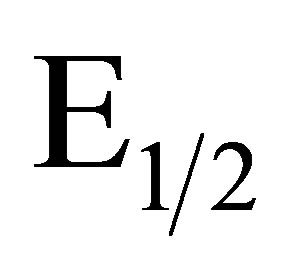 values of the redox couples versus pH should give a negative slope of 59 mV/pH unit for an electron/proton coupled process as shown in Equation (1).
values of the redox couples versus pH should give a negative slope of 59 mV/pH unit for an electron/proton coupled process as shown in Equation (1).
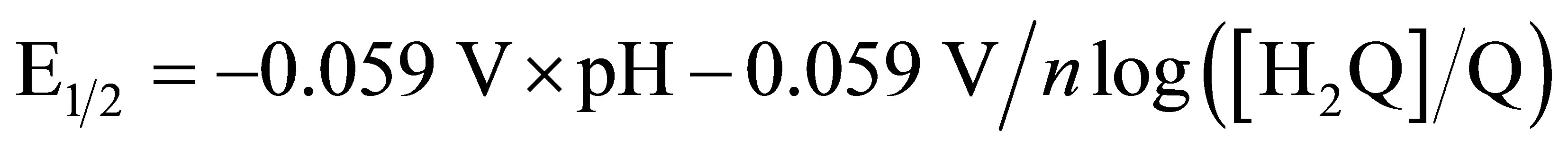 (1)
(1)
A linear relation of 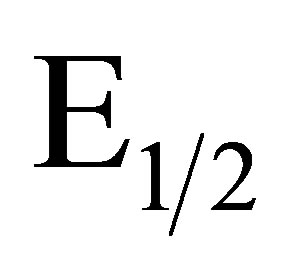 to pH with a slope of −0.059 V ± 0.005 V/pH unit indicates a 1 e−/1 H+ process in agreement with a quinone like surface structure [22]. Figure 2 illustrates the results of a pH study performed on an EPG electrode coated with complex I after it had been oxidized in 1.0 M sulfuric acid. As the [H+] decreases the redox couple shifts to lower potentials. A plot of
to pH with a slope of −0.059 V ± 0.005 V/pH unit indicates a 1 e−/1 H+ process in agreement with a quinone like surface structure [22]. Figure 2 illustrates the results of a pH study performed on an EPG electrode coated with complex I after it had been oxidized in 1.0 M sulfuric acid. As the [H+] decreases the redox couple shifts to lower potentials. A plot of 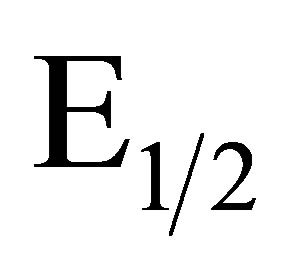 values vs. pH, Figure 2, is linear with a slope of −0.056 V/pH unit consistent with a 2 e−/2 H+ process. Further studies of the electrode surface are needed to allow us to propose an accurate structure of the surface of the electrochemically modified electrodes.
values vs. pH, Figure 2, is linear with a slope of −0.056 V/pH unit consistent with a 2 e−/2 H+ process. Further studies of the electrode surface are needed to allow us to propose an accurate structure of the surface of the electrochemically modified electrodes.
3.2. Oxygen Electrocatalysis Studies
The electrocatalytic reduction of O2 in 1.0 M sulfuric acid was studied at an EPG electrode coated with complex I using rotating disk electrode (RDE) voltammetry. Reduction of O2 at a bare EPG electrode in air saturated 1.0 M sulfuric acid occurs with an 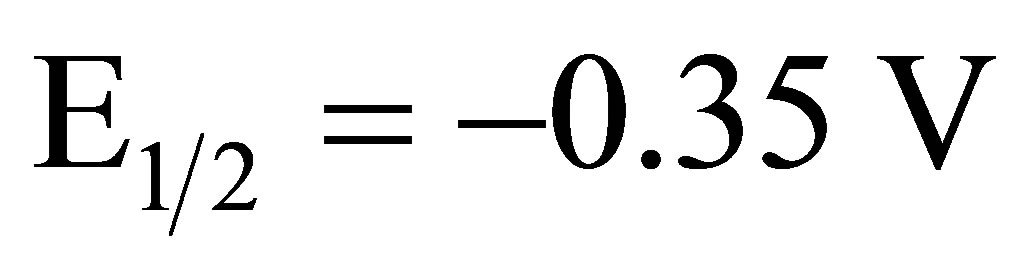 versus SCE. An EPG electrode coated with complex I in air saturated 1.0 M sulfuric acid at 400 rpm reduces O2 with an
versus SCE. An EPG electrode coated with complex I in air saturated 1.0 M sulfuric acid at 400 rpm reduces O2 with an 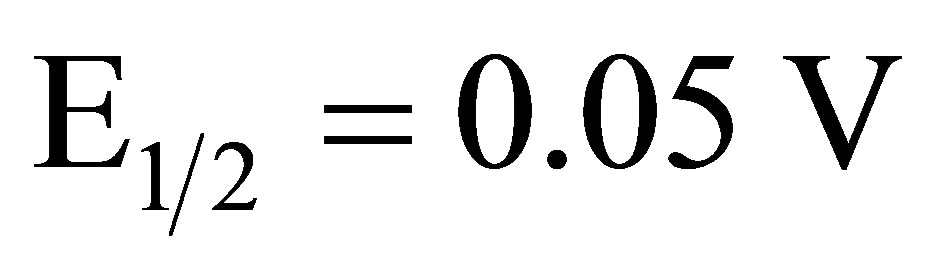 versus SCE, blue line Figure 3, a
versus SCE, blue line Figure 3, a
 (a)
(a)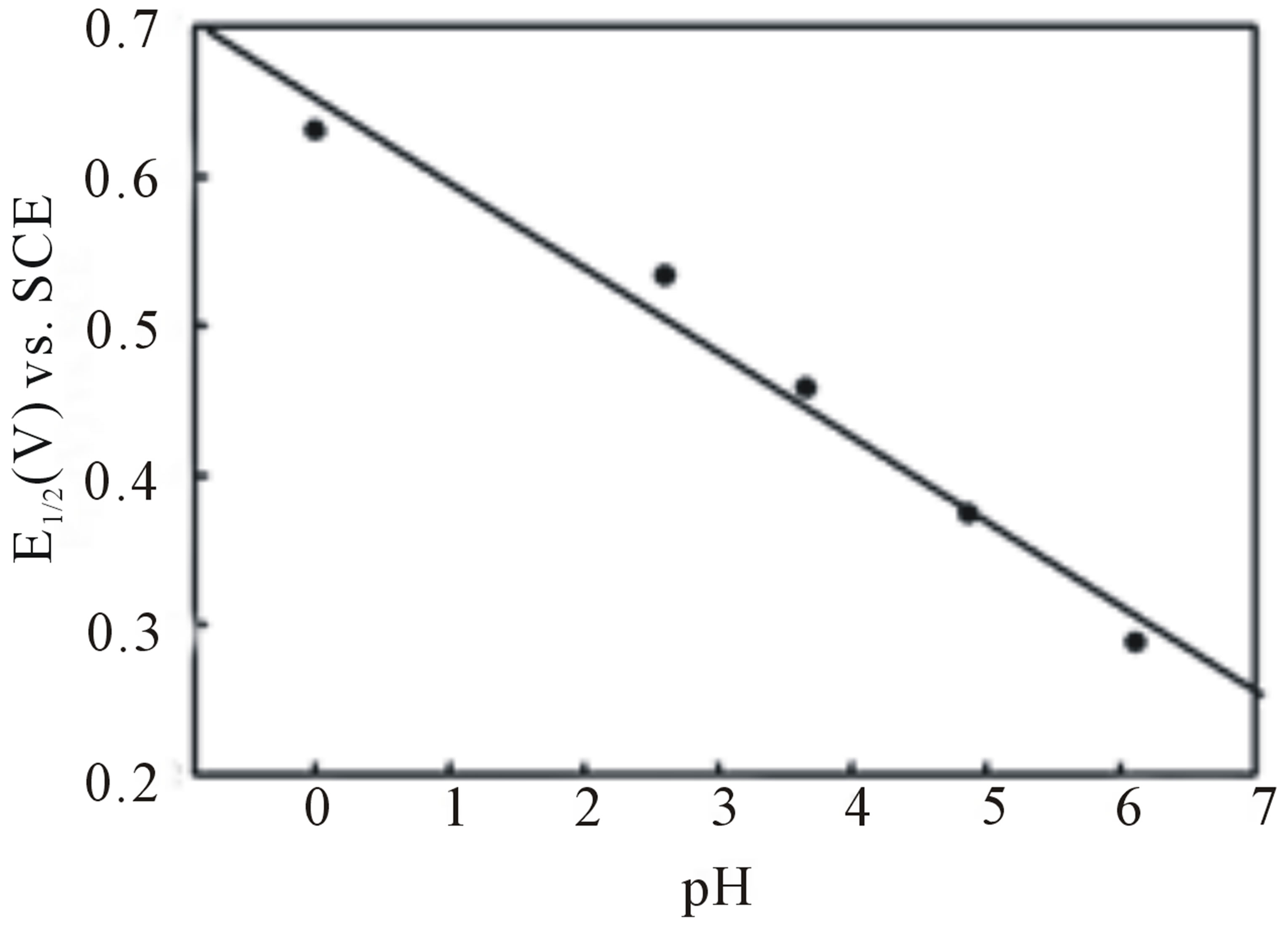 (b)
(b)
Figure 2. Cyclic voltammograms of complex I adsorbed onto an EPG electrode after anodic conditioning in 1.0 M H2SO4, (black line) pH 0.30, (red) pH 2.61, (green) pH 3.66, (blue) pH 4.87, (violet) pH 6.12.
catalytic shift of 400 mV compared to the bare EPG electrode. A catalytic shift of nearly 550 mV when compared to the bare EPG electrode is observed for O2 reduction at an EPG electrode coated with I after oxidation of the surface confined complex, red line Figure 3.
Figure 4 illustrates the results of the RDE experiment performed on an EPG electrode coated with I in air saturated 1.0 M sulfuric acid prior to oxidation of the adsorbed complex. A plot of the inverse of the limiting current density versus the inverse of the square root of the rotation rate, the Koutecky-Levich plot, relates the plateau current density to the rotation rate using Equation (2).
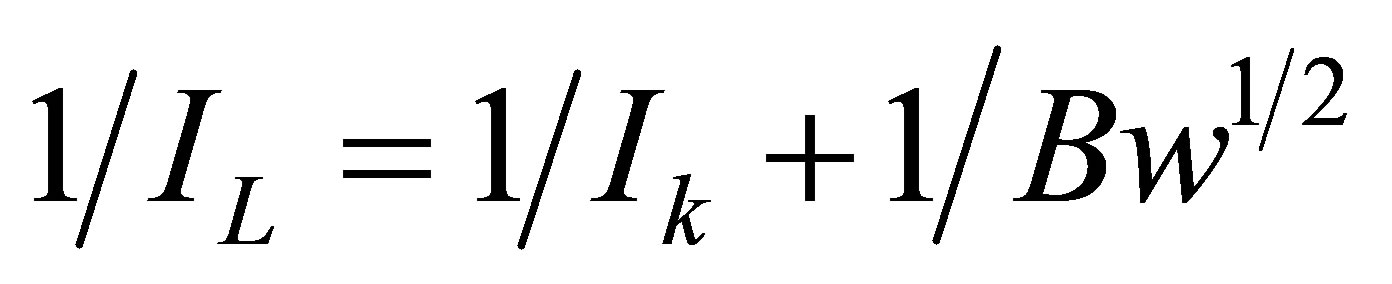 (2)
(2)
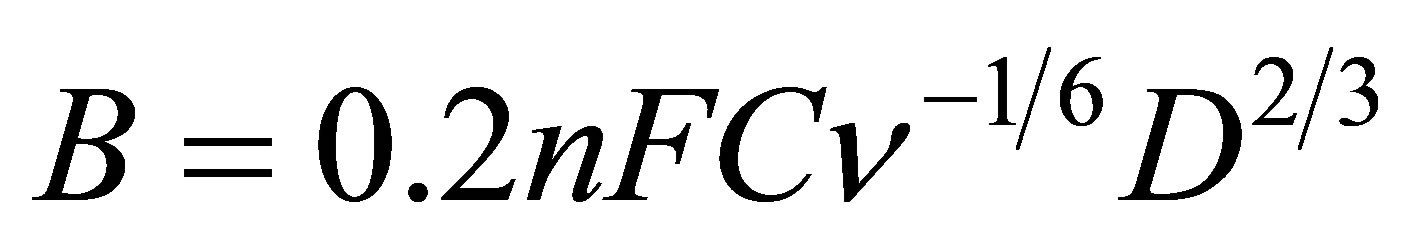
where Ik is the current density (A cm−2), n is the number of electrons for the reaction, F is the Faraday constant (96,500 C mol−1), D is the diffusion coefficient of O2 in the solution (2.0 × 10−5 cm2∙s−1), 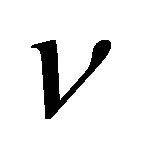 is the kinematic viscosity of the solution (0.01 cm2∙s−1), C is the concentration of O2 in air saturated solution (0.25 mM), and
is the kinematic viscosity of the solution (0.01 cm2∙s−1), C is the concentration of O2 in air saturated solution (0.25 mM), and  is the rotation rate (rpm). Ik is the kinetic current density for O2 reduction and can be calculated from the intercept
is the rotation rate (rpm). Ik is the kinetic current density for O2 reduction and can be calculated from the intercept
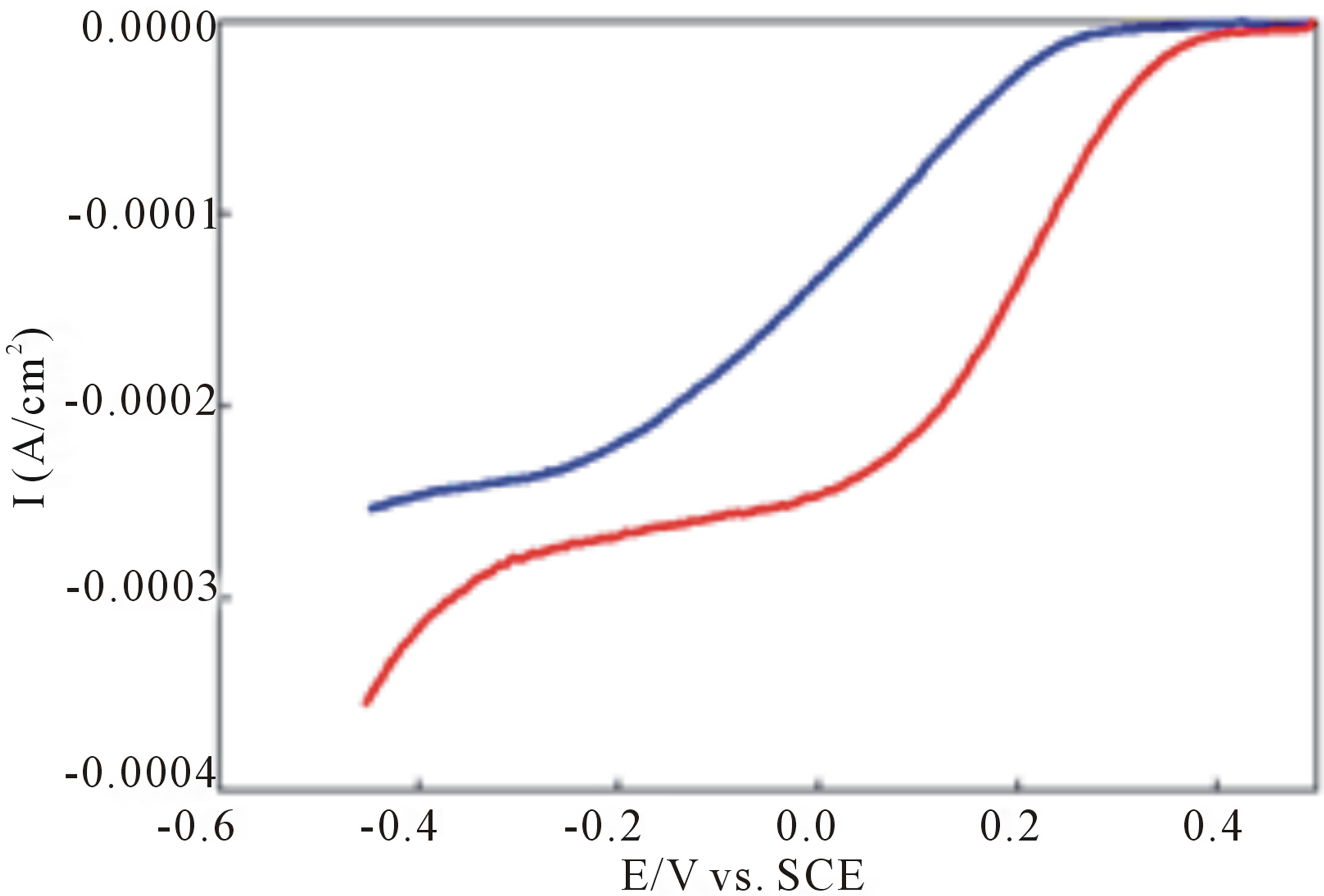
Figure 3. O2 reduction at an EPG electrode coated with complex I in air saturated 1.0 M H2SO4 prior to oxidation of the surface adsorbed complex (blue) and after anodic conditioning (red), rotation rate 400 rpm, scan rate = 5 mV/s, referenced to the saturated calomel electrode (SCE).
 (a)
(a)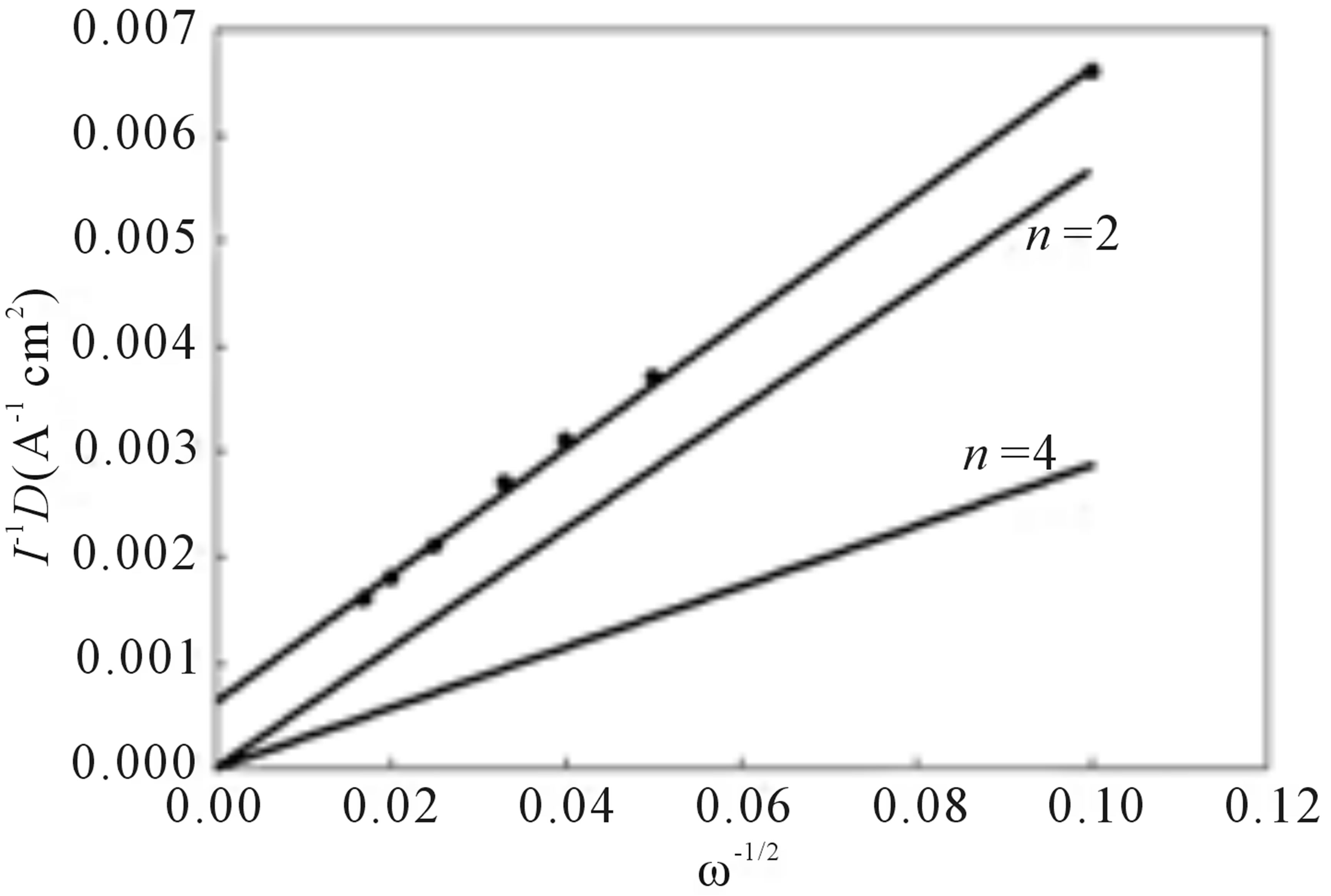 (b)
(b)
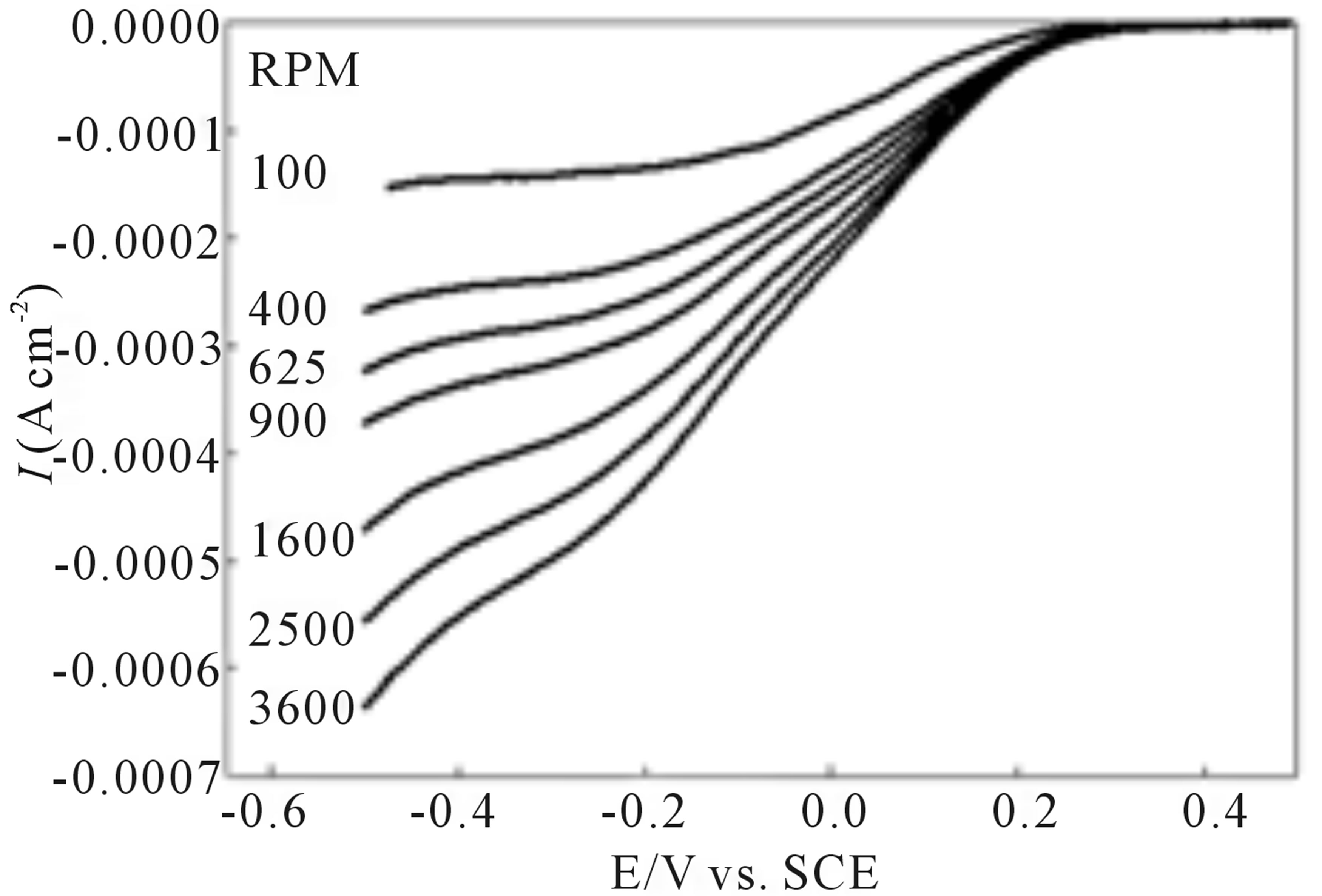
Figure 4. Reduction of O2 at a rotating disk electrode coated with complex I prior to oxidation. Current-potential curves in air saturated 1.0 M H2SO4, scan rate = 10 mV/s; Koutecky-Levich plot of the inverse of the plateau current vs. the inverse of the square root of the rotation rate for the current-potential curves. The theoretical two electron and four electron lines are marked n = 2 and n = 4, respectively.
of the Koutecky-Levich plot.
The slope for the Koutecky-Levich plot for the experimental data is very near the slope for the theoretical n = 2 line giving a value of n = 2.2, Figure 4, indicating that the majority of O2 is electrocatalytically reduced to H2O2, 2 e− process, at the EPG electrode coated with I prior to oxidation of the adsorbed complex.
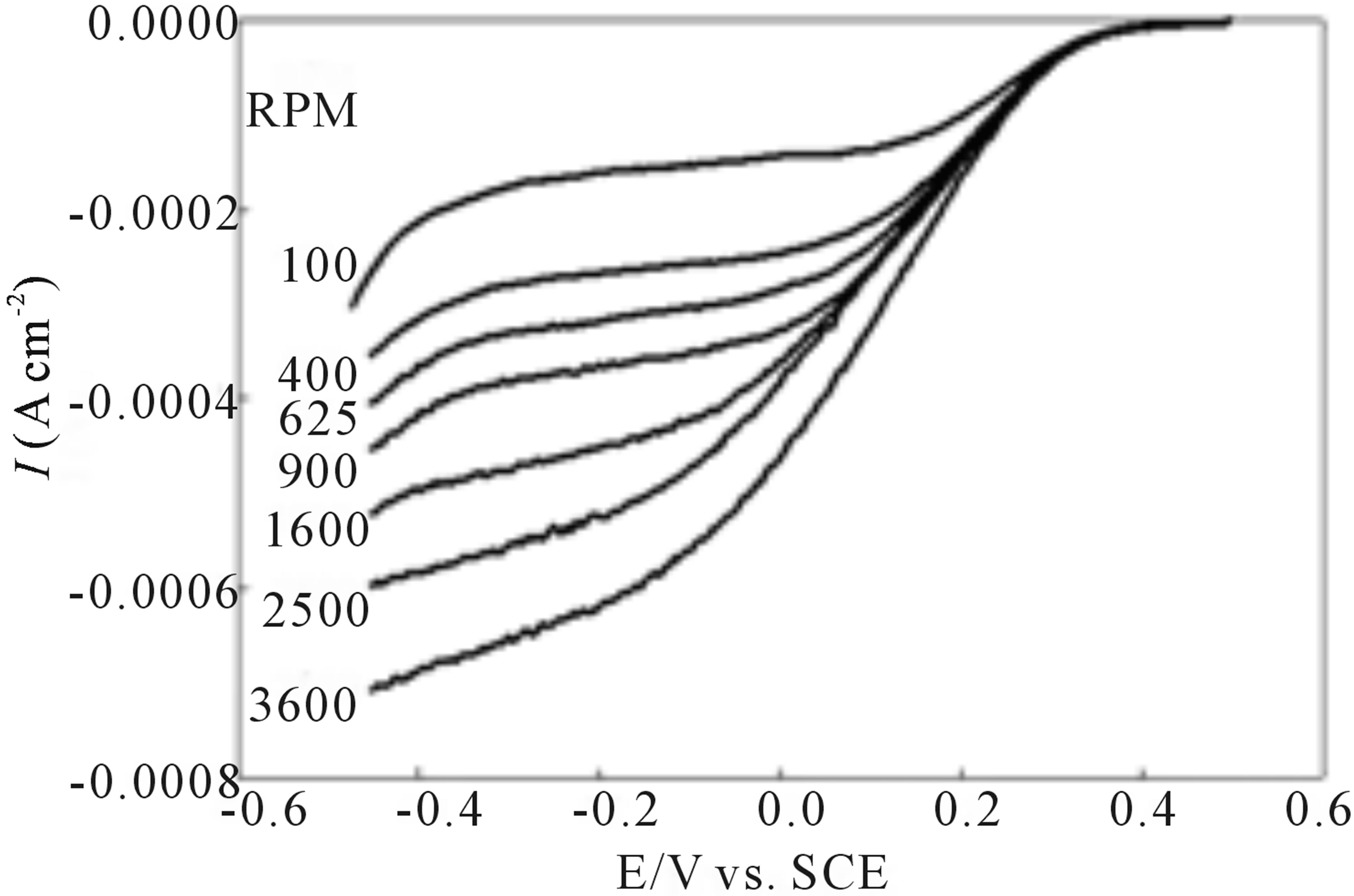
Figure 5 illustrates the results of O2 reduction at an EPG electrode coated with I, after surface oxidation, in an air saturated 1.0 M sulfuric acid solution. The Koutecky-Levich plot of this data, Figure 5, gives an n value of 3.6 suggesting that a significant amount (ca. 80%) of O2 is reduced directly to H2O at this electrochemically modified surface. In addition, the current-potential curves in Figure 5 show enhanced kinetic behavior with steeper increases in current compared to the current-potential curves prior to anodic conditioning of the modified EPG electrode surface.
4. Conclusion
This report describes a new multi-metallic porphyrin complex with catalytic ability toward the reduction of oxygen. RDE data suggest that the majority of the oxygen is
 (a)
(a) (b)
(b)
Figure 5. Reduction of O2 at a rotating disk electrode coated with complex I after anodic conditioning. Current-potential curves in air saturated 1.0 M H2SO4, scan rate = 10 mV/s; Koutecky-Levich plot of the inverse of the plateau current vs. the inverse of the square root of the rotation rate for the current-potential curves. The theoretical two electron and four electron lines are marked n = 2 and n = 4, respectively.
reduced to water, however, only after anodic conditioning of the modified electrode surface. In comparison with the previously studied mono-platinated cobalt(II) porphyrin, this complex (di-platinated cobalt(II) porphyrin) shows less catalytic ability by approximately 50 mV; however, with the di-platinated complex, a greater percentage of oxygen is reduced to water. It is apparent that the addition of more platinum complexes to the periphery of the cobalt(II) porphyrin has both a positive and negative effect on the catalytic reduction of oxygen. On one hand, it appears that the kinetics between the Co(II) metal center and O2 is enhanced with more platinum groups but a small amount of catalytic potential is lost in the process. Further studies are needed to determine if this loss in catalytic potential is truly the result of added platinum(II) or simply an artifact of the modified electrode.
REFERENCES
- G. Farmer and D. J. Hamblin, “First on the Moon,” Little, Brown, and Co., 1970.
- S. Wasmus and A. Küver, “Methanol Oxidation and Direct Methanol Fuel Cells: A Selective Review,” Journal of Electroanalytical Chemistry, Vol. 461, No. 1-2, 1999, pp. 14-31.
- H. Jahnke, M. Schönborn and G. Zimmermann, “Organic Dyestuffs as Catalysts for Fuel Cells,” Topics in Current Chemistry, Vol. 61, 1976, pp. 133-181. http://dx.doi.org/10.1007/BFb0046059
- Y. Kiros, “Metal Porphyrins for Oxygen Reduction in PEMFC,” International Journal of Electrochemical Science, Vol. 2, 2007, pp. 285-300.
- J. E. Newton and M. B. Hall, “Generalized Molecular Orbital Calculations on Transition-Metal Dioxygen Complexes: Models for Iron and Cobalt Porphyrins,” Inorganic Chemistry, Vol. 23, No. 26, 1984, pp. 4627-4632. http://dx.doi.org/10.1021/ic00194a043
- C. Shi, B. Steiger, M. Yuasa and F. C. Anson, “Electroreduction of O2 to H2O at Unusually Positive Potentials Catalyzed by the Simplest of the Cobalt Porphyrins,” Inorganic Chemistry, Vol. 36, No. 20, 1997, pp. 4294-4295. http://dx.doi.org/10.1021/ic970516s
- E. Song, C. Shi and F. C. Anson, “Comparison of the Behavior of Several Cobalt Porphyrins as Electrocatalysts for the Reduction of O2 at Graphite Electrodes,” Langmuir, Vol. 14, No. 15, 1998, pp. 4315-4321. http://dx.doi.org/10.1021/la980084d
- C. Shi and F. C. Anson, “Multiple Intramolecular Electron Transfer in the Catalysis of the Reduction of Dioxygen by Cobalt meso-Tetrakis(4-pyridyl)porphyrin to Which Four Ru(NH3)5 Groups are Coordinated,” Journal of the American Chemical Society, Vol. 113, No. 25, 1991, pp. 9564-9570. http://dx.doi.org/10.1021/ja00025a022
- C. Shi and F. C. Anson, “Electrocatalysis of the Reduction of O2 to H2O by Tetraruthenated Cobalt meso-Tetrakis(4-pyridyl)porphyrin Adsorbed on Graphite Electrodes,” Inorganic Chemistry, Vol. 31, No. 24, 1992, pp. 5078-5083. http://dx.doi.org/10.1021/ic00050a029
- B. Steiger, C. Shi and F. C. Anson, “Electrocatalysis of the Reduction of Dioxygen by Adsorbed Cobalt 5,10,15, 20-Tetraarylporphyrins to Which One, Two, or Three Ru (NH3)52+ Centers are Coordinated,” Inorganic Chemistry, Vol. 32, No. 10, 1993, pp. 2107-2113. http://dx.doi.org/10.1021/ic00062a038
- C. Shi and F. C. Anson, “Comparison of the Catalytic Reduction of Dioxygen by [5,10,15, 20-tetrakis((pentaamineruthenium(II))4-pyridyl)porphyrinato]cobalt(II) in Solution and on Graphite Electrode Surfaces,” Inorganic Chemistry, Vol. 34, No. 18, 1995, pp. 4554-4561. http://dx.doi.org/10.1021/ic00122a010
- C. Shi and F. C. Anson, “Cobalt meso-Tetrakis(N-methyl-4-pyridiniumyl)porphyrin Becomes a Catalyst for the Electroreduction of O2 by Four Electrons When [(NH3)5Os]n+ (n = 2,3) Groups are Coordinated to the Porphyrin Ring,” Inorganic Chemistry, Vol. 35, No. 26, 1996, pp. 7928-7931. http://dx.doi.org/10.1021/ic9607189
- F. C. Anson, C. Shi and B. Steiger, “Novel Multinuclear Catalysts for the Electroreduction of Dioxygen Directly to Water,” Accounts of Chemical Research, Vol. 30, No. 11, 1997, pp. 437-444. http://dx.doi.org/10.1021/ar960264j
- K. Araki and H. E. Toma, “Synthesis and Electrochemical Behavior of a Tetrametallated Cobalt Porphyrin,” Inorganica Chimica Acta, Vol. 179, No. 2, 1991, pp. 293-296.
- T. Malinski, A. Ciszewski, J. Bennet, J. R. Fish and L. Czuchajowski, “Characterization of Conductive Polymeric Nickel(II) Tetrakis(3-methoxy-4-hydroxyphenyl) Porphyrin as an Anodic Material for Electrocatalysis,” Journal of The Electrochemical Society, Vol. 138, No. 7, 1991, pp. 2008-2015. http://dx.doi.org/10.1149/1.2085915
- T. Malinski and Z. Taha, “Nitric Oxide Release from a Single Cell Measured in situ by a Porphyrinic-Based Microsensor,” Nature, Vol. 358, 1992, pp. 676-677. http://dx.doi.org/10.1038/358676a0
- T. Malinski, A. Ciszewski, J. Fish, E. Kubaszewski and L. Czuchajowski, “Concuctive Polymeric Cu(II) Tetrakis (3-methoxy-4-hydroxyphenyl) Porphyrin as a Photosensitizer in a Photoelectrochemical Cell,” Advanced Materials, Vol. 4, No. 5, 1992, pp. 354-357. http://dx.doi.org/10.1002/adma.19920040508
- X. Wu, Y. Li, B. Grundig, N.-T. Yu and R. Rennenberg, “A Novel Iron-Porphyrin Derived Oxygen Sensor Working Near 0 V (vs. Ag/AgCl) in Neutral Solution,” Electroanalysis, Vol. 9, No. 16, 1997, pp. 1288-1290. http://dx.doi.org/10.1002/elan.1140091614
- G. Richards and S. Swavey, “Electrooxidation of Fe, Co, Ni, and Co Metalloporphyrins on Edge-Plane Pyrolytic Graphite Electrodes and Their Electrocatalytic Ability towards the Reduction of Molecular Oxygen in Acidic Media,” European Journal of Inorganic Chemistry, Vol. 2009, No. 35, 2009, pp. 5367-5376. http://dx.doi.org/10.1002/ejic.200900651
- K. Gadamsetti and S. Swavey, “Electrocatalytic Reduction of Oxygen at Electrodes Coated with a Bimetallic Cobalt(II)/Platinum(II) Porphyrin,” Dalton Transactions, No. 46, 2006, pp. 5530-5535.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, “Palladium(II) and Platinum(II) Alkyl Sulfoxide Complexes. Examples of Sulfur-Bonded, Mixed Sulfur and Oxygen-Bonded, and Totally Oxygen-Bonded Complexes,” Inorganic Chemistry, Vol. 11, No. 6, 1972, pp. 1280-1284. http://dx.doi.org/10.1021/ic50112a025
- F. Pariente, E. Lorenzo and H. D. Abruna, “Electrocatalysis of NADH Oxidation with Electropolymerized Films of 3,4-Dihydroxybenzaldehyde,” Analytical Chemistry, Vol. 66, No. 23, 1994, pp. 4337-4344. http://dx.doi.org/10.1021/ac00095a034
NOTES
*Corresponding author.