American Journal of Analytical Chemistry
Vol.4 No.5(2013), Article ID:31707,8 pages DOI:10.4236/ajac.2013.45033
Simultaneous Quantification of Carbohydrates, Alcohols, and Toxic Components in a Bio-Based Medium Using Dual-Detection HPLC Analysis
Department of Chemical and Biological Engineering, University of Saskatchewan, Saskatoon, Canada
Email: *mas233@mail.usask.ca
Copyright © 2013 Majid Soleimani, Lope Tabil. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 18, 2013; revised April 25, 2013; accepted May 1, 2013
Keywords: Dual-Detection Analysis; Retention Time; Carbohydrates; Alcohols; Furans
ABSTRACT
Bio-based media, derived from cellulosic biomass depolymerisation or bioconversion processes, are composed of several chemicals and biochemicals. In this study, the main components of a hemicellulosic-based medium were analyzed using a dual detection HPLC method to separate and determine concentrations of the major monosaccharides (glucose, xylose, and arabinose), alcoholic and acidic components (ethanol, xylitol, and acetic acid) and furanic compounds (furfural and hydroxymethylfurfural (HMF)). The analyses, which employed a single stationary phase (Aminex HPX-87H) and two detection methods, were carried out under isocratic conditions and involved mobile phases consisting of 5 mM sulfuric acid and acetonitrile in different mix ratios from 0 to 0.061 mole fraction of acetonitrile. Based on the analysis run time and the chromatogram quality, the optimum condition was determined for the simultaneous quantification of the components.
1. Introduction
Lignocellulosic resources provide a renewable source of carbon mainly (75%) as carbohydrates for the biotransformation-based processes that produce fuels and chemicals for a wide variety of applications [1]. During depolymerization of the natural polymers in the biomass, a complicated conversion of the components to the simpler chemicals occurs. The mechanism of conversion of monosaccharides including hexoses and pentoses, as well as acetyl groups, respectively, to hydroxymethylfurfural (HMF), furfural, and acetic acid and also the mechanisms of their inhibitive impact on cell metabolic activities and regeneration have been studied in detail by Palmqvist and Hahn-Hagerdal [2].
The analytical methods for xylitol determination and quantification could be classified as: 1) methods based on high performance liquid chromatography (HPLC); 2) methods based on gas chromatography (GC); 3) liquid chromatography-mass spectroscopy (LC-MS) methods; and 4) capillary electrophoresis (CE). Since derivatization steps are not required to generate volatile derivatives applicable to the final analytical study in the HPLC analytical methods [3], HPLC is the preferred method compared with GC.
Different types of columns such as HPX-87H, TSK amide—80 columns, amino-based carbohydrate column, and ion exclusion column have been used for xylitol analysis. Several kinds of detectors based on various detection methods and chromatographic systems have been used to quantify carbohydrates; these approaches include refractive index (RI) detection, mass spectroscopy (MS), pulsed amperometric detection (PAD), evaporative light scattering (ELS), and ultraviolet (UV) detection. Less sensitive methods such as RI detection and ELS have more applications for xylitol analysis [3,4].
The biomass hydrolysis byproducts (furans) derived from the degradation of monosaccharides (hexose and pentose) are detected by UV, GC-mass spectrometry and GC-FID methods [4]. The UV spectral method, as suggested by Martinez and co-workers [5], is a rapid and convenient means to estimate and monitor total furans (furfural and HMF) in the biomass hydrolysate. Individual determination of methyl furfural (MF), furfural, and HMF was developed using a spectrophotometric method based on the reaction of the components with 2-thiobarbituric acid (TBA) [6]. Scarlata and Hyman [7] reported of a study wherein a fast acid method using an HPLC ion exclusion column based on cross-linked sulfonated styrene-divinyl benzene (SDVB) and RI detection method, successfully analyzed organic acids, alcohols, and furans; however in some cases, carbohydrates were eluted together. Even levulinic acid co-eluted with acetic acid, and glycerol co-eluted with formic acid using the fast acid system. In another study reported by Xu and coworkers [8], a combination of RI and UV detection methods was used for the simultaneous analysis of dextrose and 5-HMF in an aqueous system, attaining a high sensitivity (30 - 50 ppb) for 5-HMF detection. The use of the anion exchange chromatography with the PAD method showed that the simultaneous quantification of dextrose and 5-HMF could be impractical because of the low loading capacity of the anion exchange column as well as the mismatch of the concentrations of these two components. The analytical study of the catalytic dehydration of xylose for furfural production was carried out using an HPLC system coupled with an ion exchange stationary phase equipped with RI and UV detection systems by Dias et al. [9]. The application of HPLC in combination with ELS and photodiode array (PDA) detection systems resulted in a reproducible quantification of carbohydrates (monosaccharides and cellobiose) and furans, respectively [4].
The application of a single method of detection coupled with liquid chromatography for the simultaneous analysis of furans, carbohydrates, alcohols, and organic acids is a challenge because of the great difference in their concentrations at a single sample concentration. Fast eluting methods using fast acid columns result in the co-elution of some of the important components during the analytical work. The objective of this work, therefore, is to apply a dual detection system using UV and RI detection methods to simultaneously quantify the hydrolysis products (monosaccharides) and hydrolysis byproducts, such as the inhibitors (toxic components), and the bioconversion products (alcohols: ethanol and xylitol) in an aqueous system. For this purpose, eluent components are to be optimized to achieve a high resolution chromatography with less elution time.
2. Materials and Methods
2.1. Chemicals
Acetonitrile and sulfuric acid were used as the components of the mobile phase in the HPLC analysis, and other materials including glucose, xylose, arabinose, acetic acid, HMF and furfural were used as the standards for the hydrolysate or bioconversion medium analysis. Xylitol and ethanol were used as standards for the dual-detection analysis using HPLC in experiments related to the bioconversion process. All monosaccharides and xylitol were purchased from Alfa Aeser (Ward Hill, MA); ethanol, HMF, and acetic acid were supplied by Sigma Aldrich Canada (Oakville, ON), and furfural was obtained from J. I. Baker Chemical Co. (Phillipsburg, NJ).
2.2. Samples and Instrument
The oat hull feedstock was supplied by Can-Oat Milling Inc. (Martensville, SK), and the impurities, including endosperm grits and dust, were separated using a sieving machine (Link Aero, Fargo, ND). To prepare the biobased medium, the biomass was hydrolyzed in a 4-liter reactor (Hoppes Inc., Springfield, OH) under acidic conditions using dilute sulfuric acid solution (as catalyst) in a concentration of up to 0.55 N and at a temperature of up to 130˚C within a period of 150 minutes and a 10% (w/w) solid concentration. To simulate the hydrolysate to a xylitol bioconversion medium, xylitol and ethanol were added to the hydrolysate with concentrations of up to 25 and 5 g/l, respectively. These samples were then diluted with water, centrifuged to separate suspended materials and, finally, filtered through 0.2 µm pore-sized syringe filters. A dual-detection HPLC analytical approach was employed using the Agilent 1100 series HPLC (Agilent 1100; Hewlett-Packard, Waldbronn, Germany) on an anion exchange column (HPX-87H) packed with sulfonated polystyrene-divinylbenzene. A mobile phase flow rate of 0.5 ml/min at 30˚C, was used to determine the concentrations of the major and minor components. This system was equipped with an RI detector and a DAD (diode array detector) in a series. The density and viscosity data of the acetonitrile/water mixtures was obtained from the literature [10].
3. Results and Discussion
The linear relationship between the density and concentration (mole fraction) of acetonitrile in its mixture with water is shown in Figure 1. Because of the lower density of acetonitrile (0.782 g/ml at 20˚C) compared to water, any fractional increase of the former substance in the mixture results in a linear reduction of the mixture density. Another important point with the mobile phase is the back pressure (DP) in the liquid chromatography system, which could be affected by the mobile phase viscosity, as included in the following equation for a packed bed:
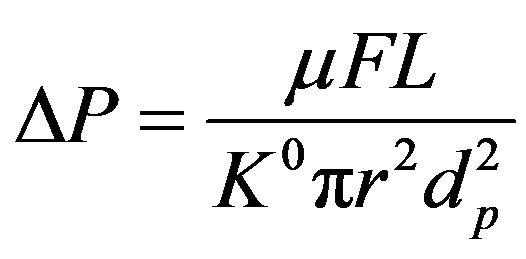 (1)
(1)
where µ is the eluent viscosity (Pa·s), F is flow rate (m3/s), L is length of the column (m), and K0, r and dp are specific permeability (m2), column radius (m) and particle diameter (m), respectively. The results obtained by observations indicated that the back pressure in the system increased parallel to the viscosity of the eluent. Just at the end of the curves (Figure 2), the slopes were slightly dissimilar, and this difference is probably due to the interaction of other factors with the flow of the mobile phase in the system. In a study on temperature and pressure behaviours of the mobile phase in LC systems using reversed phase columns, Aburjai et al. [11] concluded that changing the mobile phase fractions could be effective on the back pressure of the system; these variations are possibly dependent on the type of the organic modifier as well as the stationary phase.
3.1. Analysis of Carbohydrates and Alcohols
The results of some HPLC analyses of the samples on carbohydrates consisting of glucose, xylose, arabinose, and of alcohols, which include xylitol and ethanol, are shown in Figure 3. From the peaks and the results in Table 1, it could be concluded that loading more acetonitrile into the system in the mobile phase resulted in the reduction of the retention times of the components. However, this reduction is more significant for some of the components such as ethanol and less significant for others such as all monosaccharides and xylitol. This agrees with the result reported by Yuan and Chen [12], who did not observe any significant change in the retention times of the carbohydrates by acetonitrile concentration in the mobile phase. Table 1 indicates that increasing the acetonitrile concentration (mole fraction) from 0 to 0.061 for ethanol results in a reduction (from 25.79 to 23.61) of 2.18 min (8.5% of the retention time at initial condition where no organic solvent was used). This
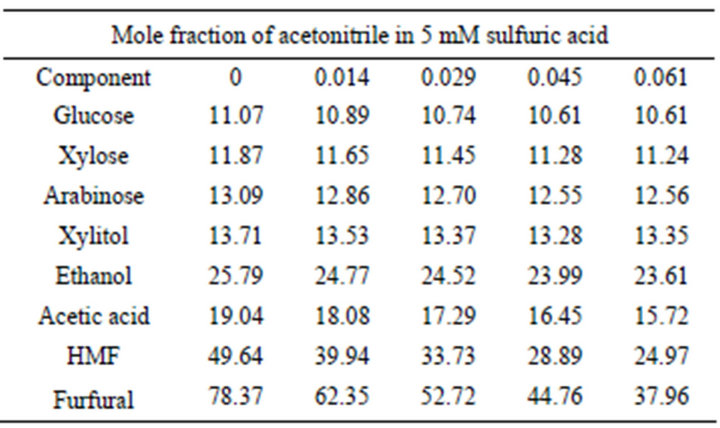
Table 1. Retention times (min) of the components at different acetonitrile concentrations (mole fraction) in dilute sulfuric acid.
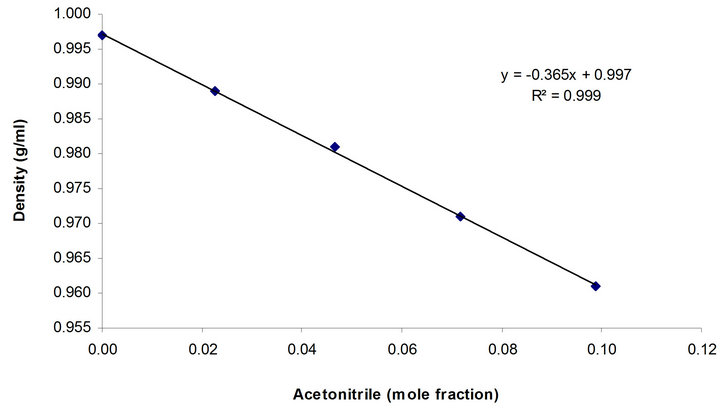
Figure 1. Relationship of acetonitrile density to its concentration (mole fraction) in water.
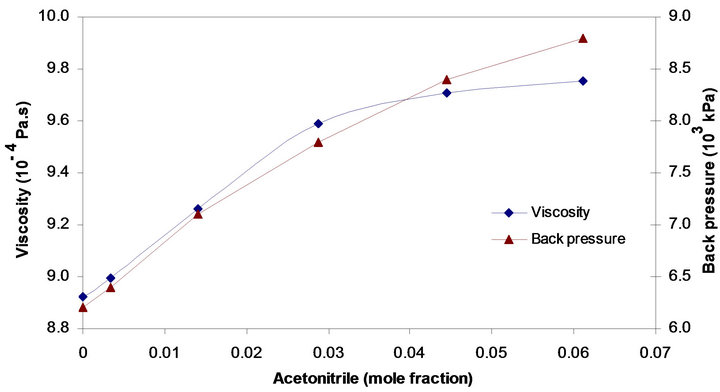
Figure 2. Effect of acetonitrile concentration in acetonitrile-water mixture on its viscosity and back pressure in the system.
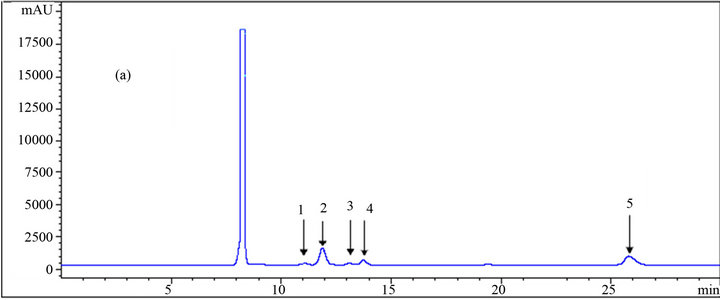
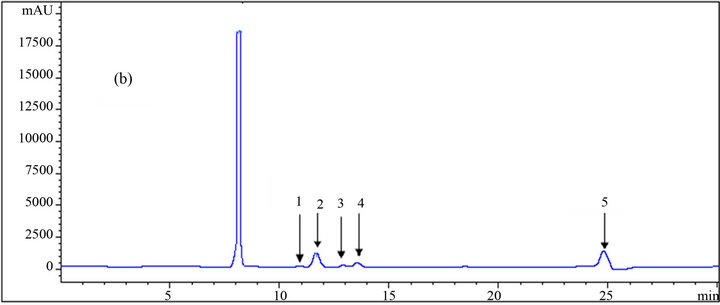

Figure 3. HPLC chromatograms of (1) glucose, (2) xylose, (3) arabinose, (4) xylitol, and (5) ethanol using an RI detector; (a), (b) and (c) are the results of the analysis at different mole fractions of acetonitrile: (a) 0; (b) 0.029; (c) 0.061 as the mobile phase.
number for glucose, xylose, arabinose, and xylitol are 4.15%, 5.31%, 4.05% and 2.63%, respectively.
On the other hand, the negative effect of the organic solvent loading into the mobile phase was the appearance at certain points of a negative peak in the baseline which could be critical in the determination of peak areas and the concentration measurement. This occurred for mole fractions of 0.014 and 0.029 on the right side and left side of the peaks, respectively, resulting in large errors in reading the areas under such conditions. At the other two mole fractions (0.045 and 0.061) of the organic solvent in the mobile phase, void volumes appeared between xylitol and ethanol far from the peaks. Also, the baselines of the chromatograms were wavy compared to the pure 5 mM sulfuric acid of the mobile phase, which gave a very straight and stable baseline. In a study of the determination of carbohydrates and organic acids, a very high baseline noise, which was not acceptable, resulted from the application of acetonitrile as the organic modifier in the mobile phase [13]; this caused the lower sensitivity of the system to the analytes, especially the ones in lower concentrations.
However, in the present study, the height of the noises was not too high to develop errors in such conditions. Altogether, pure dilute (5 mM) sulfuric acid gave the best result and the two higher mole fractions (0.045 and 0.061) of the organic solvent gave better results in terms of the chromatogram quality than the other ones. The detailed results of the HPLC analysis relating to the retention times of the components are listed in Table 1.
3.2. Analysis of Toxic Components
Toxic components (microbial growth inhibitors) including acetic acid, furfural, and HMF were analyzed, and the chromatogram results using different eluents are presented in Table 1, as well as in Figures 4 and 5. The effect of the eluent components on the retention time and separation process is more significant for these materials, especially furfural and HMF, than for carbohydrates.
Yuan and Chen [12], who did the analytical work on six furanic compounds, achieved a similar result. By increasing the mole fraction of acetonitrile in the mobile phase from 0 to 0.61, the retention times of acetic acid, HMF, and furfural were reduced from 19.04 to 15.72 min, 49.64 to 24.97 min and from 78.37 to 37.96 min, respectively (Table 1 and Figure 6). If furfural was assumed to be the final chemical being analyzed, there would be over 51% reduction in its retention time, resulting in the shortening of the duration of analysis. Application of the mobile phase containing 0.045, 0.029, and 0.014 acetonitrile mole fractions resulted in reduced furfural retention times by 43%, 33%, and 20%, respectively, compared to
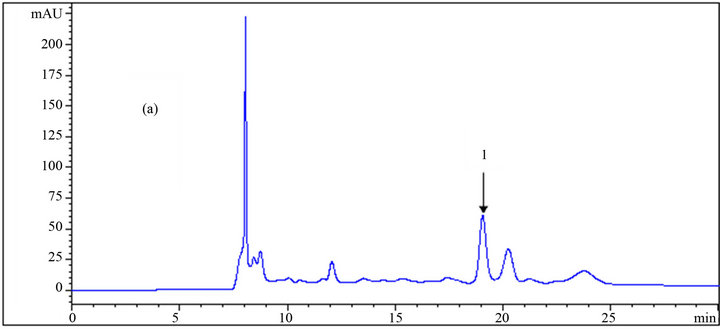
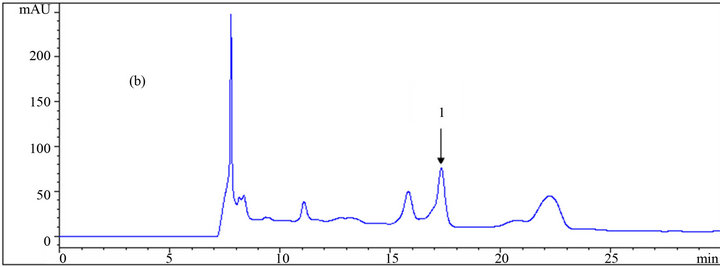

Figure 4. HPLC chromatograms of acetic acid (1) using a DAD detector at 205 nm wavelength; (a), (b) and (c) are the results of the analysis at different mole fractions of acetonitrile: (a) 0; (b) 0.029; (c) 0.061 as the mobile phase.
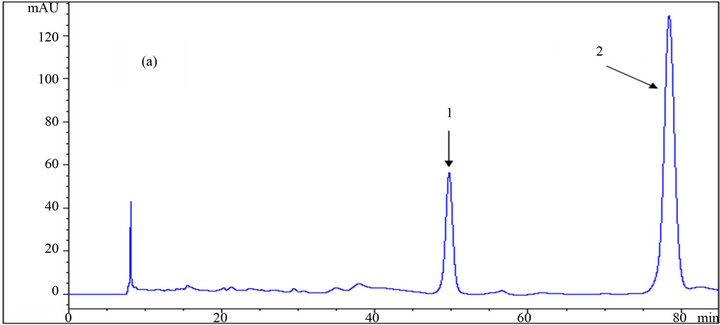

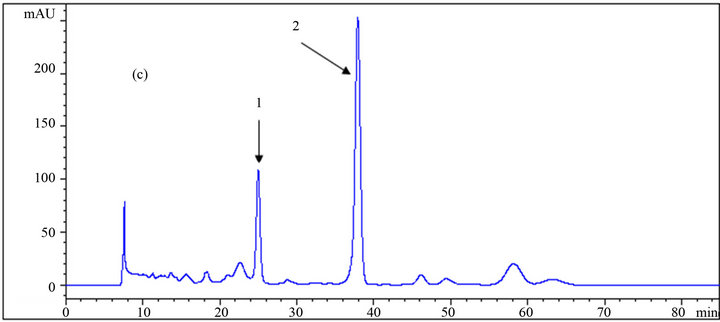
Figure 5. HPLC chromatograms of (1) HMF, and (2) furfural using a DAD detector at 280 nm wavelength; (a), (b) and (c) are the results of the analysis at different mole fractions of acetonitrile: (a) 0; (b) 0.029; (c) 0.061 as the mobile phase.
when sole dilute sulfuric acid was used as the mobile phase. In addition to the time savings, appropriate application of the organic fraction in the mobile phase results in less depreciation of the analytical device. However, the resolution and quality of the chromatogram is an important factor in making decisions. As shown in the chromatograms, good resolutions were achieved in almost all conditions for HMF and furfural. However, in some cases of acetic acid, a shoulder appeared on the left side of the peaks using mobile phases with 0.014 and 0.061 acetonitrile mole fractions resulting in an error in the final concentration measurement. This did not happen in the other cases. Altogether, regarding the short analysis period with the mobile phase containing 0.045 acetonitrile mole fraction among others, this ratio is recommended over the other ones.
4. Conclusion
Access to a reliable method in quantifying the components of a complex medium such as a hemicellulose hydrolysate or a biobased medium could be an important factor in monitoring and controlling the depolymerization and bioconversion processes which lead to the final product (xylitol). In this study, the objective parameters of the chromatogram resolution and analysis time for the
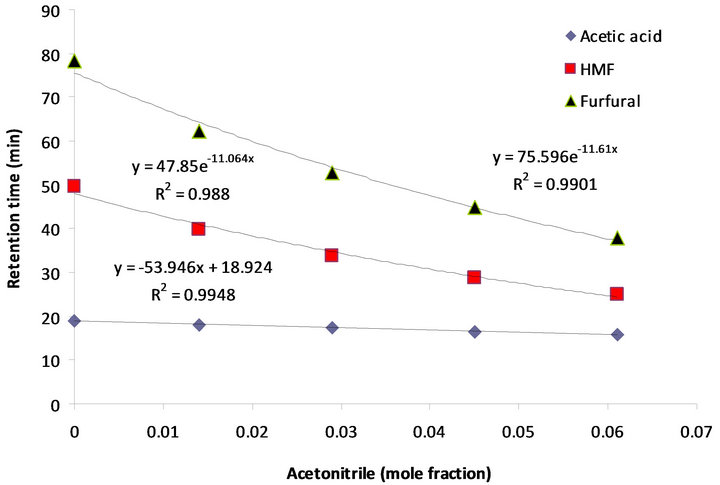
Figure 6. Dependence of the retention times of acetic acid, HMF and furfural to acetonitrile concentration (mole fraction) in the mobile phase.
quantitative determination of the intended components (monosaccharides, alcohols and toxic components) were investigated. HPLC system properties indicated that the back pressure on an ion exchange stationary phase could be altered by changing the ratio of the organic modifier. The retention times of the compounds, mainly furans and to a lesser extent ethanol and acetic acid, were shortened by increasing the organic part of the mobile phase mixture; however, those of the monosaccharides were not affected by this change in the eluent. To optimize the ratio of the organic solvent, it was important to consider the quality of the peaks and the resolution of the chromatogram in addition to the retention times of the components. Overall, in the range studied here, the mole fraction of 0.045 for the organic solvent (acetonitrile) resulted not only in a much shorter analysis run time (43% shorter) than the pure inorganic solvent, but also in a high quality of the chromatogram comparable to the one obtained by pure dilute sulfuric acid as the mobile phase.
REFERENCES
- A. Corma, S. Iborra and A. Velty, “Chemical Routes for the Transformation of Biomass into Chemicals,” Chemical Reviews, Vol. 107, No. 6, 2007, pp. 2411-2502. doi:10.1021/cr050989d
- E. Palmqvist and B. Hagerdal, “Fermentation of Lignocellulosic Hydrolysates. II: INHIBITORS and Mechanisms of Inhibition,” Bioresource Technology, Vol. 74, No. 1, 2000, pp. 25-33. doi:10.1016/S0960-8524(99)00161-3
- X. Chen, Z. H. Jiang, S. Chen and W. Qin, “Microbial and Bioconversion Production of Xylitol and Its Detection and Application,” International Journal of Biological Sciences, Vol. 6, No. 7, 2010, pp. 834-844. doi:10.7150/ijbs.6.834
- X. Liu, N. Ai, H. Zhang, M. Lu, D. Ji, F. Yu and J. Ji, “Quantification of Glucose, Xylose, Arabinose, Furfural and HMF in Corncob Hydrolysate by HPLC-PDA-ELSD,” Carbohydrate Research, Vol. 353, 2012, pp. 111-114. doi:10.1016/j.carres.2012.03.029
- A. Martinez, M. E. Rodriguez, S. W. York, J. F. Preston and L. O. Ingram, “Use of UV Absorbance to Monitor Furans in Dilute Acid Hydrolysates of Biomass,” Biotechnology Progress, Vol. 16, No. 4, 2000, pp. 637-641. doi:10.1021/bp0000508
- S. M. Rocha, M. A. Coimbra and I. Delgadillo, “Occurrence of Furfuraldehydes during the Processing of Quercus suber L. cork. Simultaneous Determination of Furfural, 5-Hydroxymethylfurfural and 5-Methylfurfural and Their Relation with Cork Polysaccharides,” Carbohydrate Polymers, Vol. 56, No. 3, 2004, pp. 287-293. doi:10.1016/j.carbpol.2004.03.002
- C. J. Scarlata and D. A. Hyman, “Development and Validation of a Fast High Pressure Liquid Chromatography for the Analysis of Lignocellulosic Biomass Hydrolysis and Fermentation Products,” Journal of Chromatograpghy A, Vol. 1217, No. 14, 2010, pp. 2082-2087. doi:10.1016/j.chroma.2010.01.061
- H. Xu, A. C. Templeton and R. A. Reed, “Quantification of 5-HMF and Dextrose in Commercial Aqueous Dextrose Solutions,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 32, No. 3, 2003, pp. 451-459. doi:10.1016/S0731-7085(03)00163-8
- A. S. Dias, S. Lima, M. Pillinger and A. A. Valente, “Acidic Cesium Salts of 12-Tungstophosphoric Acid as Catalysts for the Dehydration of Xylose into Furfural,” Carbohydrate Research, Vol. 341, No. 18, 2006, pp. 2946-2953. doi:10.1016/j.carres.2006.10.013
- K. Hickey and W. E. Waghorne, “Viscosities and Volumes of Dilute Solutions of Formamide in Water + Acetonitrile and for Formamide and N,N-Dimethylformamide in Methanol + Acetonitrile Mixed Solvents: Viscosity BCoefficients, Activation Free Energies for Viscous Flow, and Partial Molar Volumes,” Journal of Chemical and Engineering Data, Vol. 46, No. 4, 2001, pp. 851-857. doi:10.1021/je0003647
- T. Aburjai, M. Alzweiri and Y. M. Al-Hiari, “Temperature and Pressure Behaviours of Methanol, Acetonitrile/ Water Mixtures on Chromatographic Systems,” American Journal of Analytical Chemistry, Vol. 2, No. 8, 2011, pp. 934-937. doi:10.4236/ajac.2011.28108
- J. P. Yuan and F. Chen, “Simultaneous Separation and Determination of Sugars, Ascorbic Acid and Furanic Compounds by HPLC-Dual Detection,” Food Chemistry, Vol. 64, No. 3, 1999, pp. 423-427. doi:10.1016/S0308-8146(98)00091-0
- H. A. Eyeghe-Bickong, E. O. Alexandersson, L. M. Gouws, P. R. Young and M. A. Vivier, “Optimisation of an HPLC Method for the Simultaneous Quantification of the Major Sugars and Organic Acids in Grapevine Berries,” Journal of Chromatography B, Vol. 885-886, 2012, pp. 43-49. doi:10.1016/j.jchromb.2011.12.011
NOTES
*Corresponding author.