American Journal of Analytical Chemistry
Vol. 3 No. 4 (2012) , Article ID: 18437 , 8 pages DOI:10.4236/ajac.2012.34044
Determination of 8 Diuretics and Probenecid in Human Urine by Gas Chromatography-Mass Spectrometry: Confirmation Procedure
Taras Shevchenko National University of Kiev, Kiev, Ukraine
Email: iuna_tsyrulneva@yahoo.com
Received February 16, 2012; revised March 15, 2012; accepted March 25, 2012
Keywords: Diuretics; Urine; Gas Chromatography; Tandem Mass Spectrometry; Confirmation
ABSTRACT
A fast and sensitive method for determination of 8 diuretics (acetazolamide, bendroflu-methiazide, bumetanide, chlorthalidone, furosemide, hydrochlorothiazide, metolazone, triamterene) and masking agent (probenecid) in human urine using gas-chromatography with mass spectrometric detection is described. The extraction of the substances as function of the nature of organic solvent, mixing time and pH of aqueous phase was studied. The tandem mass spectrometry was used to increase selectivity of diuretics determination due to elimination of background interferences. Fragmentation reactions were studied for each compound and their collision energies were optimized to obtain the best selectivity. The results of method’s validation demonstrate its suitability in routine analysis for confirmation purposes.
1. Introduction
It is known [1] that athletes misuse diuretics for several reasons: to reduce body weight in order to be qualified for a lower weight category, to decrease fluid retention in body caused by the application of steroids and to reduce the concentration of other prohibited substances in urine to avoid positive doping results. International Olympic Committee and World Anti-Doping Agency (WADA) prohibited the use of diuretics in sport. WADA established the requirement for anti-doping laboratories to control the presence of these compounds in urine at the level less or equal to minimum required performance limit (MRPL), which is 0.25 µg·ml–1 [2]. Most of the banned substances should not be present at any concentration in the specimens (mainly urine) collected for the analysis. Therefore, they are often detected only on a qualitative basis.
Diuretics belong to non-threshold substances, which means the absence of specific threshold concentration in urine. Table 1 shows that diuretics include compounds which have differences in molecular structures and chemical properties [3], and this fact complicates the elaboration of the only method of their extracting from biological matrix and their detection at the necessary selectivity level.
According to WADA requirements the only methods of analysis that should be applied at anti-doping laboratories are liquid or gas chromatography with mass spectrometric detection. Though liquid chromatography with mass spectrometric detection (LC-MS) causes the decrease in sensitivity due to the suppression of ionization by biological matrix [4,5] and shift in retention times in case of direct injection of sample [6] it also allows to achieve low detection limits and short time of analysis [7]. Because of relatively high cost of LC-MS this method is more preferable for quantative determination of diuretics in biological objects and for screening purpose. Gas chromatography with mass detection (GC-MS) is suitable for multicomponent, selective and sensitive diuretics determination in human urine, as well as liquid chromatography. GC-MS method which includes two-step liquid-liquid extraction and derivatization was proposed for quantative determination of 16 diuretics [8]. Long preliminary sample preparation of urine limits the use of the method in routine analysis. Moreover, this technique does not provide high recovery of chlorthalidone and triamterene. At the same time GC-MS demonstrates the best results for qualitative determination and for confirmation.
The GC-MS method was proposed for confirmation in [9]. The recovery of diuretics and impeding effect of urine matrix and other diuretics were not studied in this work as well as in [10]. Besides, both methods were not validated. Moreover, the technique proposed in [11] is long lasting which is caused by long retention time.
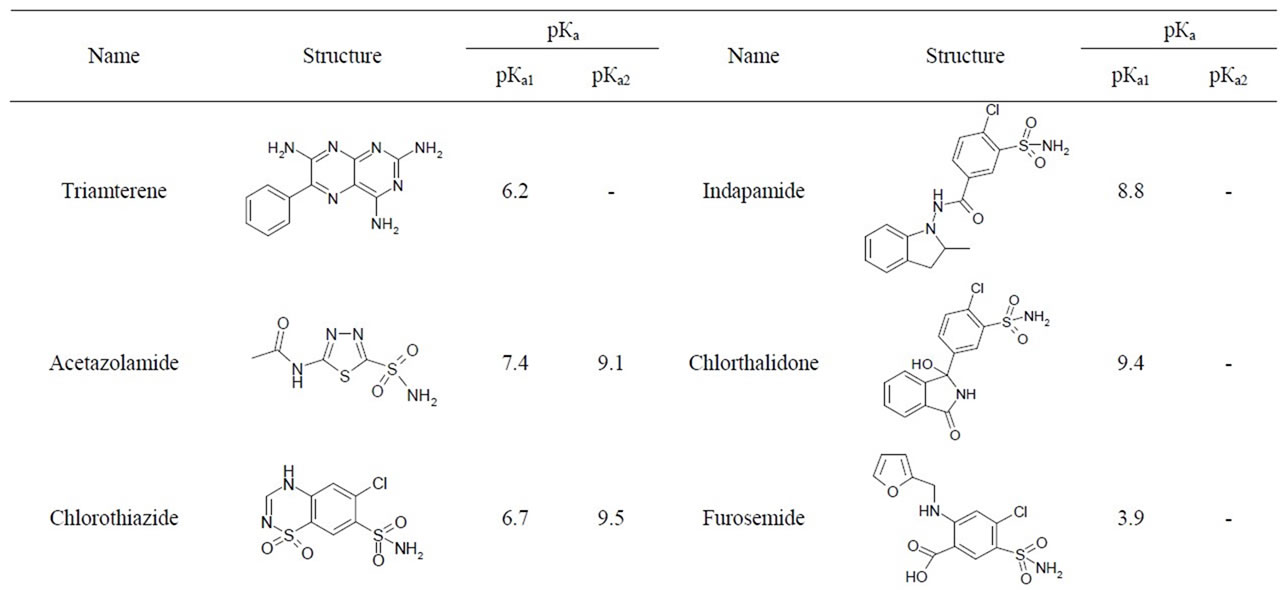
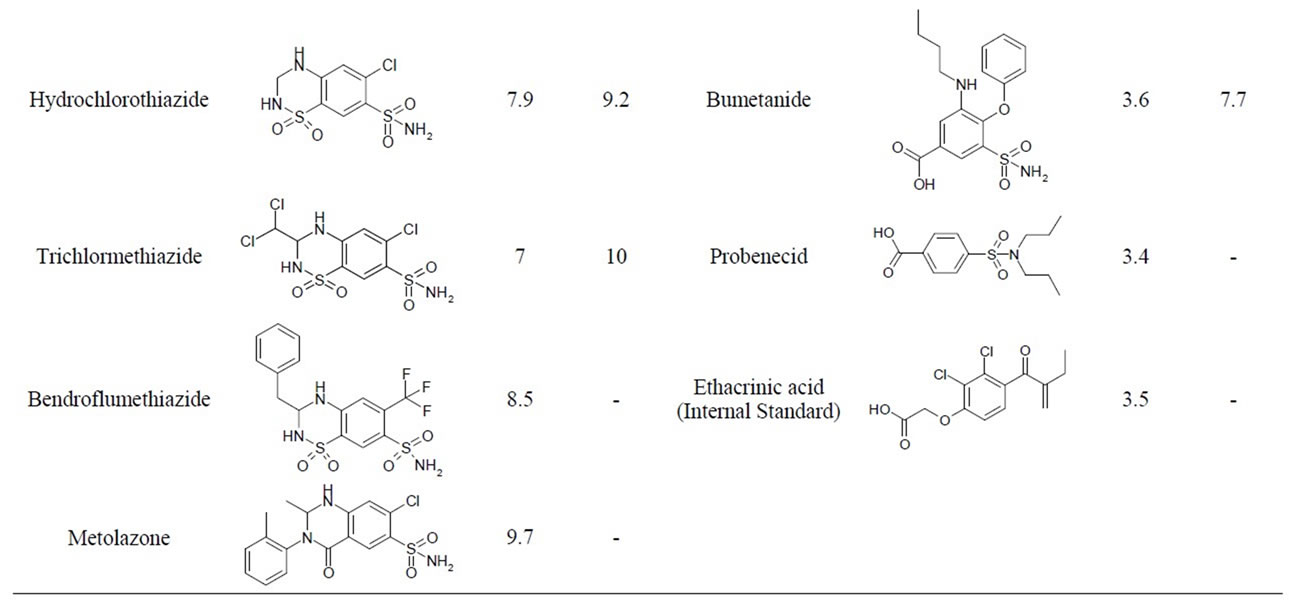
Table 1. General characteristics of diuretics.
Despite the great number of methods proposed, gas or liquid chromatography with mass spectrometric detection do not always provide the necessary selectivity towards the urine components [10,12]. In case of liquid chromatography, the use of tandem mass-spectrometry allowed to pass over these problems and to ensure the satisfactory result [13-15].
Despite liquid chromatography is more preferable for screening purposes, gas chromatography with mass detection demonstrates the best results for confirmation. According to WADA requirements the optimal method for confirmation of presence of diuretics is the method different from the screening method. Therefore, the goal of the present work was to develop a method of confirmation of presence of diuretics and probenecid in urine by gas chromatography with tandem mass spectrometric detection.
2. Experimental Procedure
2.1. Preparation of Reagent Solutions
Stock standard solutions were prepared for 12 diuretic drugs (acetazolamide, bendroflumethiazide, bumetanide, chlorthalidone, chlorothiazide, ethacrinic acid, furosemide, hydrochlorothiazide, indapamide, metolazone, triamterene, trichlormethiazide) and a masking agent (probenecid) (all by SIGMA, St. Louis, MO, USA) by dissolving in methanol at concentration of 1.0 mg·ml–1.
Diethyl ether, ethyl acetate, isopropanol, acetone (Merck, Darmstadt, Germany) and methanol (SIGMA, St. Louis, MO, USA) had a qualification “for chromatography”. Methyl iodide with purity 99% (SIGMA, St. Louis, MO, USA), sodium sulfate (Merck, Darmstadt, Germany) were used without further purification, potassium carbonate (Merck, Darmstadt, Germany) was used after calcination in oven. Hydrochloric acid and sodium hydroxide were prepared by dilution of the concentrated initial solution (Merck, Darmstadt, Germany).
2.2. Instruments and Materials
Gas chromatograph Varian 3800 (Australia) equipped with an automatic sample injector system Combipal and FactorFour Capillary Column VF-DA 12 m, 0.20 mm I.D. for mass spectrometric determination of narcotic drugs and pharmaceuticals. Mass spectrometer Varian 1200 (Australia), directed by computer system of data processing Varian MsWorkstation 6.9, with a system of three quadruples. Auxiliary equipment: thermostat VLM EC1 (Germany), evaporator in nitrogen stream Liebisch (Germany), centrifuge OPN-3Y4.2 (Russia), shaker Heidolph Vibramax 110 (Germany), oven Binder (Germany), pHmeter inoLab pH level 1 (Germany), analytical balance KERN ABS (Germany), pipettes of adjustable volume Eppendorf (Germany) 0 - 20 µl, 20 - 200 µl, 100 - 1000 µl, 500 - 5000 µl.
2.3. Instrumental Parameters
Instrumental parameters of chromatographic determination: carrier gas—helium, flow rate—1 ml·min–1, injector temperature—290˚C, injector mode—split 20; volume of sample injected—2 µl; flushing liquid—acetone, source temperature—225˚C, interface temperature—250˚C; SIM Width—0.7; Scan Time—0.2 s; start of the ion current registration—5.3 min. Instrumental parameters of tandem mass spectrometry: auxiliary gas pressure (argon) on the second quadruple—2.10 - 1.65 mTorr, electron energy at the source—20 eV. Registration mode: selected ion monitoring (SIM) or multiple reaction monitoring (MRM). In experiment mass spectrometry detection was carried out in SIM mode using ions listed in Table 2 [12].
2.4. Sample Preparation
Negative urine samples were taken from volunteers who did not consume banned substances; the positive (spiked) urine samples were prepared by adding mixtures of standard diuretics solutions to the negative urine samples prior to the sample preparation. The urine samples were stored in polypropylene bottles at a temperature of –20˚C. Before conducting the experiment samples were defreezed, centrifuged for 10 min at 1500 rpm, and the pH value was measured. Ethacrinic acid (internal standard, IS), mixture of standard diuretics, 200 mg of Na2SO4 and 5.0 ml of urine were transferred in a 12 ml glass tube. The pH value was adjusted by adding NaOH or buffer solution and 5.0 ml of organic solvent. The tubes were shaken in a shaker for 5 - 20 min and then centrifuged. The top layer was totally transferred to a 10 ml glass tubes. Solutions were evaporated in nitrogen stream at 40˚C (for solvents based on diethyl ether) or 80˚C (for solvents based on ethyl acetate) to obtain solid residue, the tubes were cooled to room temperature. The derivatization (methylation by methyl iodide in acetone in an alkaline environment) was performed according to [16]. Afterwards the samples were immediately sent to gaschromatographic analysis. The degree of substances recovery from matrix achieved by liquid-liquid extraction was investigated at a concentration of 0.9 MRPL (0.225 µg·ml–1). The mixture of standard diuretics and IS were added to the urine samples and sample preparation was performed by the method described. Simultaneously, the derivatization of mixture of diuretics standards with the same concentration was conducted. All samples were analyzed by chromatographic-mass spectrometric method. The recovery degree was calculated by taking the ratio of peak height obtained at the retention time for the analyte of interest compared to that obtained from an IS in spiked urine and mixture of standards.
2.5. Method Validation
To determine all parameters (capability to identification, selectivity, detection limit), that are necessary for suitability of verification in accordance with ISO/IEC 17025: 2006 [17], the investigation was performed. Linearity, accuracy and precision were not examined since the method is intended for confirmation and is not intended for quantitative determination of diuretics, as established in [17,18]. Therefore more consideration was given to definite identification of the analyte in urine.
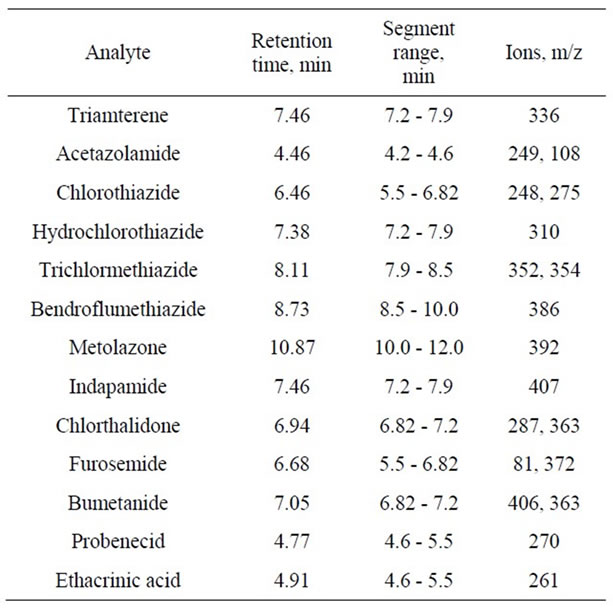
Table 2. Characteristic ions of diuretics in Selected Ion Monitoring mode.
Identification by the retention time. A series of spiked urine samples and mixture of standards were simultaneously analyzed to check the capability of identification. Absolute and relative differences in retention times were calculated.
Mass spectrometry identification in tandem mass spectrometry (MS-MS) mode. A series of extracts of spiked urine samples and mixture of standards at concentrations 0.5, 1.0 and 5.0 MRPL were simultaneously analyzed to prove the possibility of mass spectrometric identification of the analytes. The relative intensity of the diagnostic ion was calculated on the base of the chromatographic peaks’ area or height, integrated by corresponding ion.
The selectivity of method was tested by analyzing six different urine samples after sample preparation without analytes.
The limit of detection was evaluated according to [19]. The samples were prepared by addition of the mixture of standards to urine to achieve the concentrations 0.2, 0.5, 1.0, 2.0 MRPL, and analyzed by marking the number of positive determinations regarding to total number of determinations. That concentration level, which corresponded to an unambiguous detection in all the samples, was considered as a limit of detection.
2.6. Quality Control
While carrying out the experiment, the mixture of diuretics standards at 0.9 MRPL (standard sample), the urine sample obtained from a volunteer who did not consume prohibited substances (negative sample), spiked urine sample with concentration equal to 0.9 MRPL (positive sample) and reagent blank (distilled water) were also analyzed using the same method of sample preparation for each analytical run. All samples, except for the reagent blank, were prepared with the addition of IS.
3. Results and Discussion
3.1. Optimization of Gas-Chromatographic Method
The method of diuretics determination includes their extraction by organic solvents, subsequent derivatization and following chromatographic separation and detection. In this work the recovery degree as the function of the nature of organic solvent, mixing time and pH of aqueous phase were investigated to optimize the conditions of diuretics determination.
The liquid-liquid extraction of diuretics from urine was carried out with the use of diethyl ether, ethyl acetate and mixtures of organic solvents (ratio): diethyl ether and isopropanol (19:1), diethyl ether and methanol (19:1), ethyl acetate and isopropanol with different ratio of components (19:1, 9:1, 17:3). The obtained results show (Figure 1) that the higher recovery degree for most of
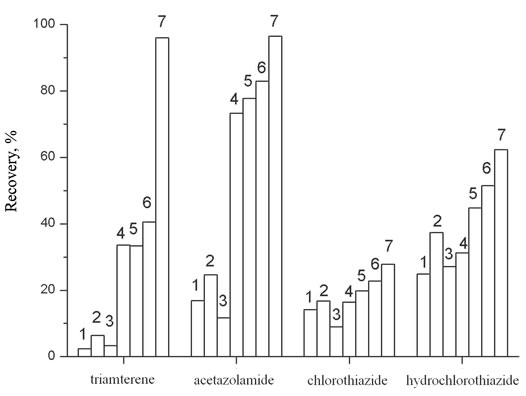
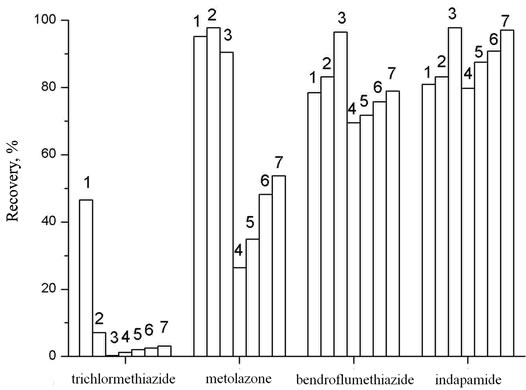
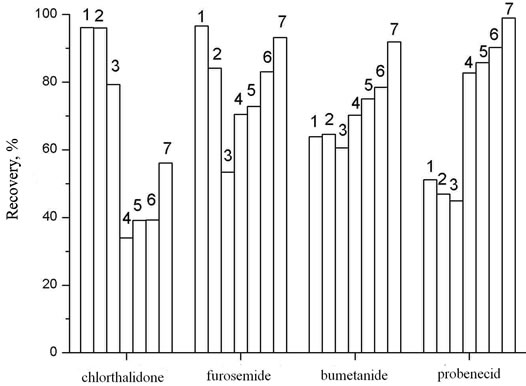
Figure 1. The recovery of diuretics as function of solvents. Vsolv = 5 ml, Vaq = 5 ml, рН = 7.0, Т = 295 ± 1 K. 1: Diethyl ether; 2: Diethyl ether:Isopropanol = 19:1; 3: Diethyl ether: Methanol = 19:1; 4: Ethylacetate; 5: Ethylacetate:Isopropanol = 19:1; 6: Ethylacetate:Isopropanol = 9:1; 7: Ethylacetate:Isopropanol = 17:3.
diuretics has been achieved at their extraction from mixture ethyl acetate:isopropanol = 17:3. The extraction degree of all substances is satisfactory, though the recovery of metolazone and trichlormethiazide under these conditions is 53.7% and 3.1%, respectively, but the signal intensity is sufficient enough to detect these compounds at the MRPL level. The extraction equilibrium in the systems is reached for 5 minutes at given conditions.
The diuretics can be classified as weak acids (carboanhydrase inhibitors, such as acetazolamide; thiazide derivatives and those that belong to them, for example, chlorthalidone), strong acids (loop diuretics such as furosemide) and base compounds (triamterene). Considering this fact extraction of selected diuretics as the function of pH value of the aqueous phase was investigated. It was found experimentally that weakly acidic diuretics are effectively extracted in molecular form in the range of pH 6 - 8 (pH of urine is ~5.5 - 7.0). The degree of their extraction exceeds 50%. Extraction degree of triamterene also increases. Extraction degree of more acidic diuretics such as furosemide, bumetanide and probenecid is lower under these conditions, but high chromatographic yield of characteristic ions of these compounds allows to detect them at the MRPL level. Further the acidity of aqueous phase was adjusted by NaOH addition to achieve pH 6 - 8.
Considering the demands made for chromatographic column, such as providing maximum separation of analytes in a short time and compatibility with mass detector, capillary polymethacrilate column FactorFour VF-DA 12 m, 0.20 mm I.D., which was successfully used for determination of narcotic drugs and pharmaceuticals, was chosen. To increase the efficiency of chromatographic separation of diuretics and to provide high rate of the analysis the optimization of temperature program was carried out. The following parameters were varied: initial and final temperature, heating rate and held. The optimal temperature program that meets all the requirements has following parameters: initial and final temperatures 120 and 300˚C, respectively; first held for 0.5 min, followed by heating rate 30˚C·min–1 and subsequent held for 8.5 min. The chromatogram of diuretics standards mixture under optimal conditions is shown in Figure 2. It is seen that complete chromatographic separation of hydrochlorothiazide, triamterene and indapamide at this temperature program is not achieved. However, these compounds can be easily discriminated due to different mass spectral fea-
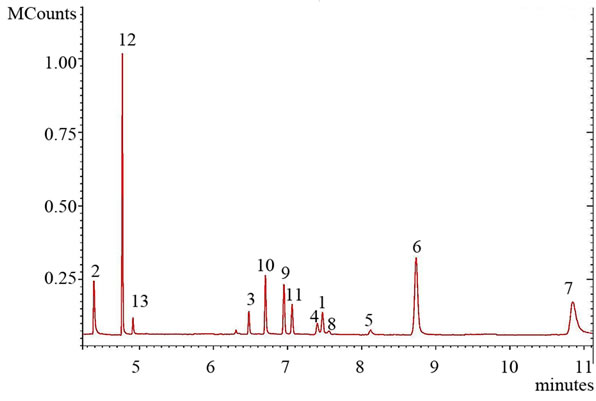
Figure 2. The chromatogram of the diuretics standard mixture, analyzed in SIM acquisition mode. С = 0.225 µg·ml–1. 1: Triamterene; 2: Acetazolamide; 3: Chlorothiazide; 4: Hydrochlorothiazide; 5: Trichlormethiazide; 6: Bendroflumethiazide; 7: Metolazone; 8: Indapamide; 9: Chlorthalidone, 10: Furosemide; 11: Bumetanide; 12: Probenecid; 13: Ethacrinic acid.
tures of diuretics. Besides, the presence of various diuretics in the same sample is unlikely. Time of chromatography is 12 min under the optimal conditions.
3.2. Optimization of MS-MS Conditions
One of the major disadvantages of mass spectrometric methods is low selectivity in the analysis of biological objects. The identification of analytes in biological matrices is complicated due to misrepresentation of mass spectrum caused by concomitant substances. It can lead to appearance of false-positive results. In this case tandem mass spectrometry is more preferable method [20]. The combination of mass selection of the precursor followed by potentially unique collision-induced dissociation and mass selection or scanning of the product ion provides higher specificity, since coincidence of spectrum of dissociated ion and ion with known structure is a conclusive proof of ions structure identity. The tandem mass spectrometry method can be used either in full scan acquisition mode or multiply reaction monitoring mode. The second technique is more sensitive in comparison with the first one. Typically, two transitions precursorproduct must be monitored, but in some cases the combination of a single precursor-product ion pair is sufficiently unique to be definitive.
For each diuretic molecular ions, product ions and collision energy (E) were chosen. Collision energy should ensure maximum yield of product ions [21]. The data for transition reaction molecular ion—product ion are presented in Table 3.
Chromatograms of spiked with bendroflumethiazide and triamterene chosen as examples urine samples and negative urine samples at these conditions are presented in Figure 3.
It is shown that the compounds may be detected in each of the reactions. Besides, peaks with signal-to-noise ratio ≥ 3 at the time which corresponds to analyte retention time, are absent in chromatogram of negative sample. This result was observed for other diuretics and probenecid as well. This fact proves sufficient selectivity of method.
3.3. Method Validation
According to the requirements of WADA [21] the relative intensities of any of the ions in mass shall not differ by more than the amount in Table 4 from the relative intensities of the same ions acquired from spiked urine.
The retention times of all diuretics (except metolazone) do not differ by more than 1% from that of the same substance in a spiked urine sample. However, there are no peaks with signal-to-noise ratio ≥ 3 in chromatograms of six analyzed blanks for the following compounds: acetazolamide, furosemide, bumetanide, indapamide, chlorothiazide, hydrochlorothiazide, metolazone, bendroflumethiazide, trichlormethiazide, chlorthalidone, triamterene.
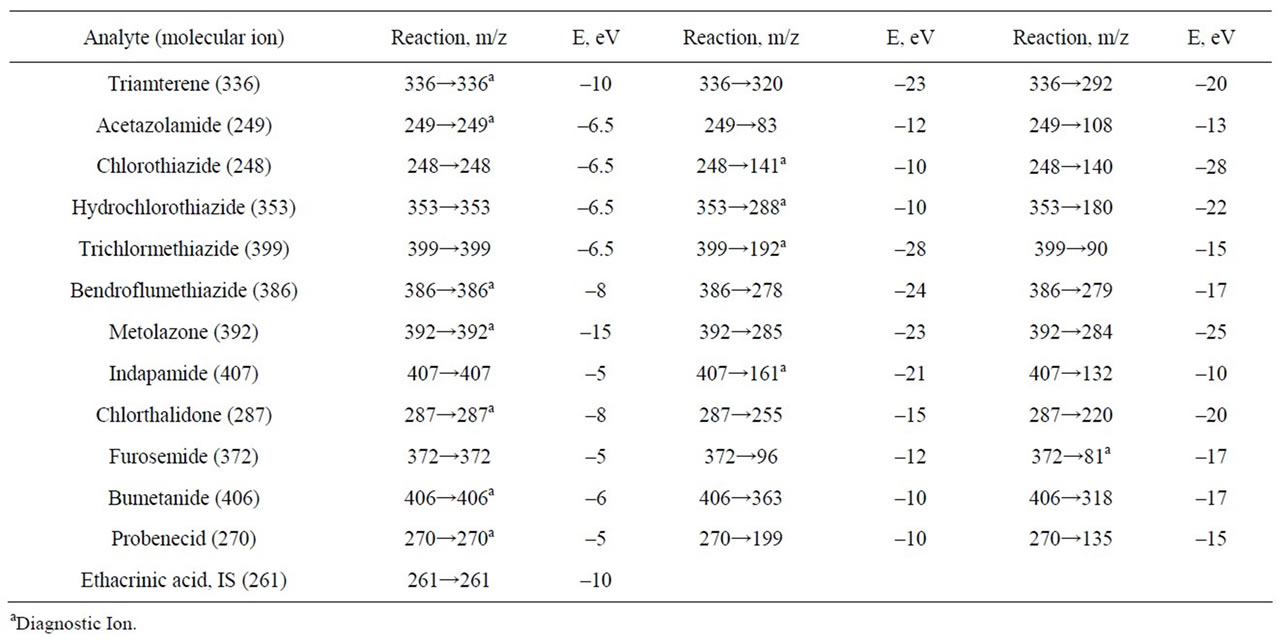
Table 3. The diagnostic reactions and optimal collision energies for diuretics determination in Multiple Reaction Monitoring mode.
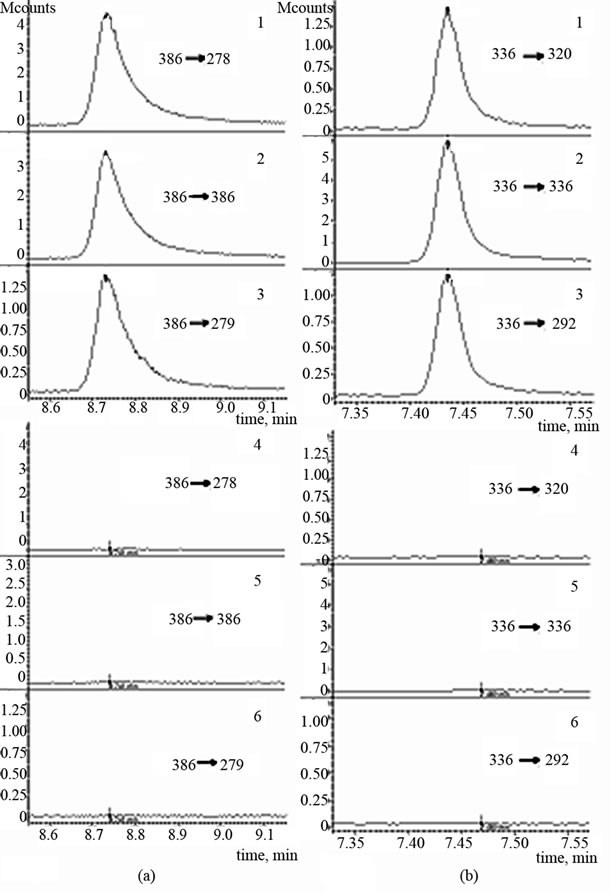
Figure 3. The chromatograms of spiked (1-3) and negative urine sample (4-6), (a) Bendroflumethiazide; (b) Triamterene. С = 0.25 µg·ml–1.
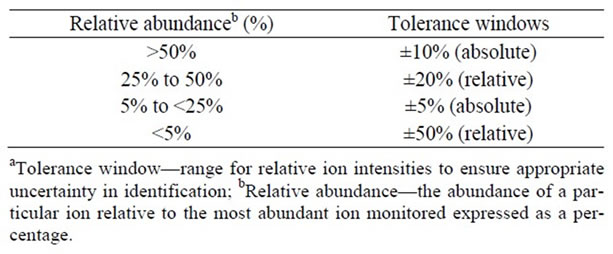
Table 4. The maximum tolerance windowsa for relative ion intensities.
Peak of small intensity with signal-to-noise ratio ≥ 3 is observed for probenecid in chromatogram of negative urine sample (m/z 270→135, Table 3). This fact is possibly caused by the influence of matrix. But the integrated intensity of this peak is negligible which according to WADA recommendations [2] can not be a cause of false identification. Besides, there are no peaks with signal-to-noise ratio ≥ 3 in the chromatograms of negative urine sample obtained using other diagnostic ions.
The ratio of intensities of diagnostic ions of acetazolamide, chlorothiazide, furosemide, chlortha-lidone, bumetanide, hydrochlorothiazide, triamterene and probenecid in the standard solutions and spiked urine samples meets WADA requirements (Tables 3 and 5). In case of indapamide and trichlormethiazide this parameter does not fit the tolerance window. Moreover, significant change of relative intensities dependent on concentration is observed which obviously can be explained by matrix influence at relative intensity. Thereby, this method can not be recommended for confirmation of indapamide and trichlormethiazide in urine.
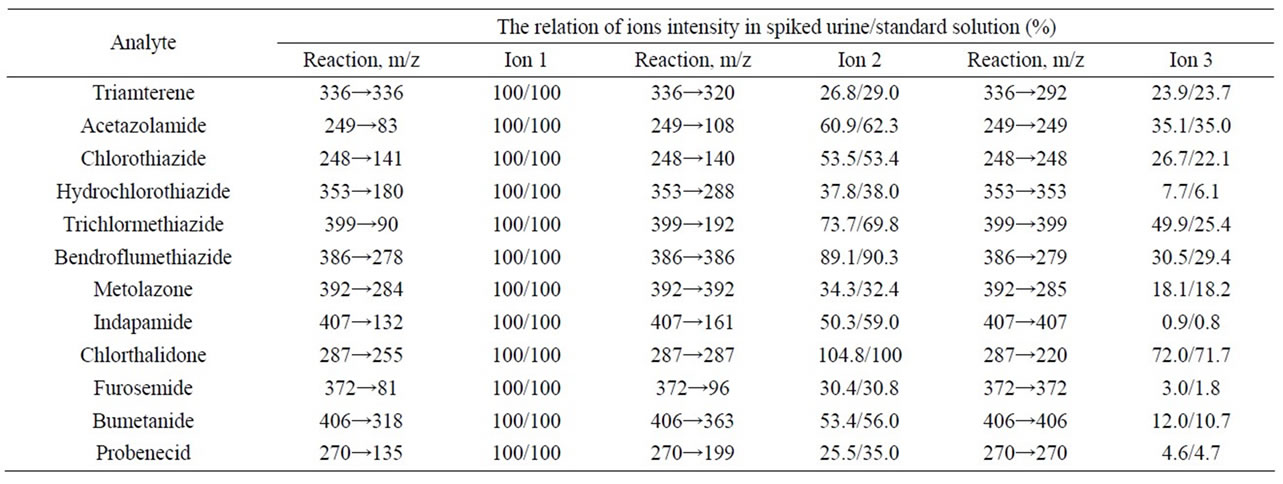
Table 5. Relative abundance of diagnostic ions in Multiple Reaction Monitoring mode, established during the method validation.
Limits of detection of acetazolamide, probenecid, furosemide, chlorthalidone, bumetanide, hydrochlorothiazide, triamterene, bendroflumethiazide, metolazone by tandem mass spectrometry are 0.050 µg·ml–1. Limit of detection of chlorothiazide which is 0.500 µg·ml–1 does not meet WADA requirements. High limit of detection may be explained by its partial dissociation at the column.
4. Conclusions
The developed method proves the suitability of gas chromatography and tandem mass spectrometry for confirmation of presence of 8 diuretics and probenecid in urine. It is selective towards acetazolamide, furosemide, bumetanide, hydrochlorothiazide, bendroflumethiazide, metolazone, probenecid, chlorthalidone and triamterene, since the components of urine do not interfere with the detection of diuretics. The method can not be recommended for determination of indapamide, trichlormethiazide and chlorothiazide.
The reduction of time for sample preparation and sufficient recovery of all diuretics merit the method proposed. Optimization of temperature program for diuretics determination at chromatographic column which wasn’t previously used for this purpose allowed to achieve chromatographic separation of all diuretics in short time. Selection of diagnostic fragmentation reactions and optimal collision energies allowed to achieve high selectivity.
Limits of detection of diuretics and probenecid by MS-MS are lower than that regulated by WADA and are 0.050 µg·ml–1.
The method was validated that proves the possibility of its application in anti-doping laboratories.
REFERENCES
- WADA, “The world Anti-Doping Code—The 2009 Prohibited List: International Standard,” World Anti-Doping Agency, Montreal, 2009.
- WADA, “Minimum Required Performance Limits for Detection of Prohibited Substances (Technical Document TD2009MRPL),” World Anti-Doping Agency, Montreal, 2009.
- R. Ventura and J. Segura, “Detection of Diuretic Agents in Doping Control,” Journal of Chromatography B, Vol. 687, No. 1, 1996, pp. 127-144. doi:10.1016/S0378-4347(96)00279-4
- M. Mazzarino, X. de la Torre and F. Botrè, “A Screening Method for the Simultaneous Detection of Glucocorticoids, Diuretics, Stimulants, Anti-Estrogens, Beta-Adrenergic Drugs and Anabolic Steroids in Human Urine by LC-ESI-MS/MS,” Analytical and Bioanalytical Chemistry, Vol. 392, No. 4, 2008, pp. 681-698. doi:10.1007/s00216-008-2292-5
- O. J. Pozo, P. Van Eenoo, K. Deventer and F. T. Delbeke, “Development and Validation of a Qualitative Screening Method for the Detection of Exogenous Anabolic Steroids in Urine by liquid Chromatography-Tandem Mass Spectrometry,” Analytical and Bioanalytical Chemistry, Vol. 389, No. 4, 2007, pp. 1209-1224. doi:10.1007/s00216-007-1530-6
- K. Deventer, O. J. Pozo, P. Van Eenoo and F. T. Delbeke, “Qualitative Detection of Diuretics and Acidic Metabolites of Other Doping Agents in Human Urine by HighPerformance Liquid Chromatography-Tandem Mass Spectrometry. Comparison between Liquid-Liquid Extraction and Direct Injection,” Journal of Chromatography A, Vol. 1216, No. 31, 2009, pp. 5819-5827. doi:10.1016/j.chroma.2009.06.003
- R. Ventura, M. Roig, N. Monfort, P. Saez, R. Berges and J. Segura, “High-Throughput and Sensitive Screening by Ultra-Performance Liquid Chromatography Tandem Mass Spectrometry of Diuretics and Other Doping Agents,” European Journal of Mass Spectrometry, Vol. 14, No. 3, 2008, pp. 191-200. doi:10.1255/ejms.920
- V. Morra, P. Davit and P. Capra, “Fast Gas Chromatographic/Mass Spectrometric Determination of Diuretics and Masking Agents in Human Urine. Development and Validation of a Productive Screening Protocol for Antidoping Analysis,” Journal of Chromatography A, Vol. 1135, No. 2, 2006, pp. 219-229. doi:10.1016/j.chroma.2006.09.034
- L. Amendola, C. Colamonici, M. Mazzarino and F. Botrè, “Rapid Determination of Diuretics in Human Urine by Gas Chromatography-Mass Spectrometry Following Microwave Assisted Derivatization,” Analytica Chimica Acta, Vol. 475, No. 1-2, 2003, pp. 125-136. doi:10.1016/S0003-2670(02)01223-0
- C. Brunelli, C. Bicchi, A. Di Stilo, A. Salomone and M. Vincenti, “High-Speed Gas Chromatography in Doping Control: Fast-GC and Fast-GC/MS Determination of β- Adrenoceptor Ligands and Diuretics,” Journal of Separation Science, Vol. 29, No. 18, 2006, pp. 2765-2771. doi:10.1002/jssc.200500387
- Y.-L. Tseng, M.-H. Shieh, C.-T. Lin and F.-H. Kuo, “Detection of Diuretics in Urine during Sports Events in Taiwan,” Tzu Chi Medical Journal, Vol. 16, No. 2, 2004, pp. 69-77.
- P. Van Eenoo and F. T. Delbeke, “Criteria in Chromatography and Mass-Spectrometry—A Comparison between Regulations in the Field of Residue and Doping Analysis,” Chromatographia, Vol. 59, Suppl. 1, 2004, pp. 39- 44. doi:10.1365/s10337-004-0198-8
- L. Politi, L. Morini and A. Polettini, “A Direct Screening Procedure for Diuretics in Human Urine by Liquid Chromatography-Tandem Mass Spectrometry with Information Dependent Acquisition,” Clinica Chimica Acta, Vol. 386, No. 1-2, 2007, pp. 46-52. doi:10.1016/j.cca.2007.07.018
- C. Goebel, G. Trout and R. Kazlauskas, “Rapid Screening Method for Diuretics in Doping Control Using Automated Solid Phase Extraction and Liqud Chromatography-Electrospray Tandem Mass Spectrometry,” Analytica Chimica Acta, Vol. 502, No. 1, 2004, pp. 65-74. doi:10.1016/j.aca.2003.09.062
- D. Thieme, J. Grosse, R. Lang, R. K. Mueller and A. Wahl, “Screening, Confirmation an Quantification of Diuretics in Urine for Doping Control Analysis by High-Performance Liquid Chromatography-Atmospheric Pressure Ionization Tandem Mass Spectrometry,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 757, No. 1, 2001, pp. 49-57. doi:10.1016/S0378-4347(01)00058-5
- S. J. Park, H. S. Pyo, Y. J. Kim, M. S. Kim and J. Park, “Systematic Analysis of Diuretic Doping Agents by HPLC Screening and GC/MS Confirmation,” Journal of Analytical Toxicology, Vol. 14, No. 2, 1990, pp. 84-90.
- ISO/IEC 17025, “General Requirements for the Competence of Testing and Calibration Laboratories,” 2006.
- Eurachem Working Group, “A Laboratory Guide to Method Validation and Related Topics,” Eurachem, Teddington, 1998.
- WADA, “The International Standard for Laboratories, Version 6.0,” World Anti-Doping Agency, Montreal, 2009.
- E. Hoffmann and V. Stroobant, “Mass Spectrometry. Principles and Applications,” 3rd Edition, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007.
- WADA, “Identification Criteria for Qualitative Assays Incorporating Column Chromatography and Mass Spectrometry (Number TD2010IDCR),” World Anti-Doping Agency, Montreal, 2010.