American Journal of Analytical Chemistry
Vol. 3 No. 3 (2012) , Article ID: 18267 , 8 pages DOI:10.4236/ajac.2012.33032
Quantification of Thiamethoxam in Rhizomes and Leaves of the Hedychium coronarium and Water and Soil by High-Pressure Liquid Chromatography
1Course of Chemistry, State University of Mato Grosso do Sul, Dourados, Brazil
2Department of Mechanical Engineering, Federal University of Rio Grande do Sul, Rio Grande do Sul, Brazil
Email: *claudia@uems.br
Received December 30, 2011; revised February 10, 2012; accepted February 24, 2012
Keywords: LC-UV; Thiamethoxam; Hedychium coronarium; Zingiberaceae
ABSTRACT
The linearity, stability, accuracy and inter-day precisions of the assay method were evaluated in water, soil and rhizomes and leaves of Hedychium coronarium of the Zingiberaceae family. Mato Grosso do Sul is a predominantly agricultural state in Brazil and has many rivers and permanent and seasonal lakes, in which the Hedychium coronarium, an aquatic plant, is found the swampy environments. The prepared samples were analyzed quantitatively by High Performance Liquid Chromatography with PDA and UV detection for the presence of thiamethoxam. The thiamethoxam was recovered from these samples at rates ranging from 81.16% - 99.93%. The coefficient of variation in the quantitative analysis of the thiamethoxam was under 5%. The linearity of the method was determined by linear regression. The analysis of the samples spiked with known amounts of analyte demonstrated that the response was proportional to the concentrations of the samples with determination coefficients of r2 = 0.9992 (water and soil) and r2 = 0.9990 (leaves and rhizomes) for the linear range of the analytical calibration curves of the samples. The detection limit was 0.36 µg∙L–1 and quantification limit was 1.2 µg∙L–1 for thiamethoxam. The method was considered sensitive for quantification of the thiamethoxam in water, soil and rhizomes and leaves of Hedychium coronarium.
1. Introduction
Pesticides are usually studied in terms of their effectiveness in controlling pests and diseases, but recently synthesized molecules have caused physiological effects on plants, capable of modifying their metabolism and morphology, thereby influencing their growth and production [1,2].
The introduction of Thiamethoxam® (Novartis, Switzerland) took place in 1998. Thiamethoxam, 3-[(2- chloro-5-thiazoly)methyl]tetrahydro-5-methyl-N-nitro4H-1,3,5-oxadiozin-4-imine, belongs to a relatively new class of insecticides known as neonicotinoids, which act as agonists of the post-synaptic nicotinic acetylcholine receptors [1].
Their physicochemical properties make them useful for a wide range of application techniques, including foliar, seed treatment, soil drench, and stem application. Neonicotinoids show good activity against pest insects resistant to other classes of insecticides such as organophosphates, carbamates, pyrethroids, chlorinated hydrocarbons, and several other classes of compounds [3].
Thiamethoxam is marketed in Brazil under the brand names of ActaraR (Syngenta Crop Protection Ltd., São Paulo, SP) for foliar and soil application and of Cruiser (Syngenta Crop Protection Inc., São Paulo, SP) for seed treatment.
High performance liquid chromatography (HPLC) with UV detection or HPLC-mass spectrometry (HPLC-MS) are currently preferred for the determination of neonicotinoids in environmental samples [4-10]. Although these methods have provided reliable and accurate quantification of neonicotinoids, they involve tedious sample extraction and cleanup procedures. The thermolability and high polarity of neonicotinoids make them difficult to analyze by gas chromatography (GC) or GC-MS, requiring derivatization of the analytes prior to analysis [11-14].
The extensive use of neonicotinoids has led to concern about contamination of surface and ground water [1], since these compounds show high toxicity for some aquatic organisms, such as Hedychium coronarium Koenig, an aquatic plant of the Zingiberaceae family, which is found in swampy environments of the several rivers and permanent and seasonal lakes of Mato Grosso do Sul, a predominantly agricultural state in Brazil.
The rhizome of H. coronarium is used in Chinese natural medicine, and has been prescribed for treatment of headaches, lancinating pain and contusion inflammatory [15].
In pharmacological studies of this natural medicine, it was reported that sesquiterpenes of H. coronarium showed inhibitory effects on the release of beta-hexosaminidase. Diterpenes of H. coronarium showed anti-inflammatory and cytotoxic activities [16-22].
One study showed that the presence of H. coronarium in wastewater for a period of four months led to a percentage reduction of Biochemical Oxygen Demand in 90.7%, of Chemical Oxygen Demand in 81.1% and of Thermotolerant Coliforms in 99.99998%. The system was therefore considered efficient in treating the wastewater, meeting the standards established by current Brazilian laws [23].
The present study developed and validated a methodology to analyze thiametohxam in samples of soil, water and rhizomes and leaves of H. coronarium.
2. Materials and Methods
2.1. Reagents
Spectroscopy grade methanol was purchased from Merck (Darmstadt). Stock mixture of thiamethoxam was constituted from individual solution, dissolved in methanol and then used as external standard. Standard of thiamethoxam was purchased from Sigma Aldrich (99.7%).
2.2. Plant Material
Rhizomes and leaves of H. coronarium and soil were collected, respectively, in october 2010 in Dourados city (latitude 22˚09'24.2''S e longitude 54˚57'08.5''W), Mato Grosso do Sul state, Brazil, and authenticated by Msc. Vali Joana Pott of the Federal University of Mato Grosso do Sul (UFMS), Mato Grosso do Sul state, Brazil. Voucher specime (CGMS/31466) was deposited at the Herbarium of the UFMS.
2.3. Experiment in a Greenhouse
Nine samples of 1 kg collected soil were placed in plastic cylindrical containers having radius and heights of 9 cm 25 cm, respectively. Of the samples, three were noncontaminated, three were contaminated each with 4.0 mg of insecticide Actara 250 WG (corresponding to 1.0 mg of thiamethoxam) and three were contaminated with 9.6 mg of the insecticide (corresponding to 2.4 mg of thiamethoxam). After contamination of the soil samples, five H. coronarium plants were transplanted to each container, and water was added to maintain the natural environment of the plant. The samples were collected after twenty one days of culture to apply the method of extraction that was developed and validated in this study.
2.4. Extraction
2.4.1. Soil
Soil was dried in an oven at a temperature of 40˚C, and then sieved with a mesh of 1 mm. For the extraction of thiamethoxam, 1 g of soil was put in contact with 2 mL of ultra-purified water for 12 hours, after which it was placed in the ultrasound (Ultrasonic Cleaner 1440 D Odontobrás) for 40 minutes and filtered through quantitative paper (porosity: 8 μm). After filtering, to the solid material was added another 1 mL of ultra-purified water for 40 minutes, and the ultrasound was applied prior to filtering through quantitative paper (porosity: 8 μm). The latter extraction process was performed in duplicate. Water was added to the extracts to complete the 10 mL volumetric flask, after which it was filtered through ultra-membrane filter of 0.45 µm and 0.20 µm, and subsequently analyzed with HPLC. Samples that exceeded the analytical curve were diluted and those samples that showed negative response were concentrated in down to 100 µL.
2.4.2. Water
One gram of water was placed in contact with 2 mL of ultra-purified water for 1 hour and after 40 minutes the mixture was subjected to ultrasound and completed with ultra-purified water to a 5 mL volumetric flask, filtered through ultra-membrane filter of 0.20 µm and analyzed with HPLC. Samples that exceeded the analytical curve were diluted and those samples that showed negative response were concentrated to 100 µL.
2.4.3. Leaves and Rhizomes
Samples in nature and powdered of leaves having 0.2 g were placed in contact with 2 mL of ultra-purified water for 40 minutes in ultrasound. Each sample was filtered in quantitative paper (porosity: 8 μm). The solid material was subjected to the same steps above, using first 2 mL and finally 1 mL of ultra-purified water. The samples were completed with water to a 5 mL volumetric flask, and filtered in an ultra-0.20 µm membrane filter prior to analysis with HPLC. The rhizomes (0.2 g) had the same extraction procedure that was applied to the leaves. Samples that exceeded the analytical curve were diluted and those samples that showed negative response were concentrated to 100 µL.
2.5. Preparation of Thiamethoxam for Analysis by LC
A sample of 0.2393 g thiamethoxam was dissolved in 5 mL of ultra-purified water, filtered through a 0.45 mm Millex filter. The resulting solution was diluted in water in a volumetric flask of 10 mL (stock solution). The stock solution was dissolved ultra-purified water in order to obtain solutions for analysis with LC.
2.6. LC Analysis of Standards and Samples
The aqueous extracts obtained from the samples were analyzed in an analytical LC (Varian 210) system with a ternary solvent delivery system equipped with an autosampler, a photodiode array detector (PDA) monitored at λ = 200 - 800 nm, and in an analytical LC (Shimadzu LC-6AD) system with a binary solvent delivery system equipped with an manual injection, a UV-Vis detector (UV) monitored in 254 nm. In both analytical LC equipment, the LC column was a C-18 (25 cm ´ 4.6 mm; particle size, 5 mm; Luna, Phenomenex, Torrance, CA, USA), with a small pre-column (2.5 cm ´ 3 mm) containing the same packing, used to protect the analytical column. In each analysis, the flow rate and the injected volume were set as 1.0 mL/min and 20 μL, respectively. All chromatographic analyses were performed at 22˚C. Elution was carried out using the following solvent-gradient program for thiamethoxam: water: methanol (60:40, v/v) by 7 minutes, taking 23 minutes to reach 100% metanol, then returning in exactly 5 minutes to the initial conditions in water and soil (Figures 1 and 2) and then returning in exactly 10 minutes to the initial conditions in leaves and rhizomes H. coronarium (Figures 3 and 4).
2.7. Validation Method
The validation procedure was performed according to the International Conference on Harmoni-zation Guidelines (Validation of Analytical Procedures, Text on Validation of Analytical Procedures Q2A and Q2B) [24]. The samples were spiked during a period of 12 hours, after which the extraction was performed as described in Section 2.4.
2.7.1. Determination of the Detection and Quantification Limits
The detection limit was determined by injecting (n = 5) solutions of thiamethoxam of known concentration (20 µL each) and then decreasing the concentrations of the samples until detection of a peak with a signal/noise ratio of 3. The corresponding concentration was considered the minimal detectable concentration. The quantification limit was determined by performing the same methodology and, thus, the quantification limit was defined as the chromatographic peak having a signal/noise ratio of 10.
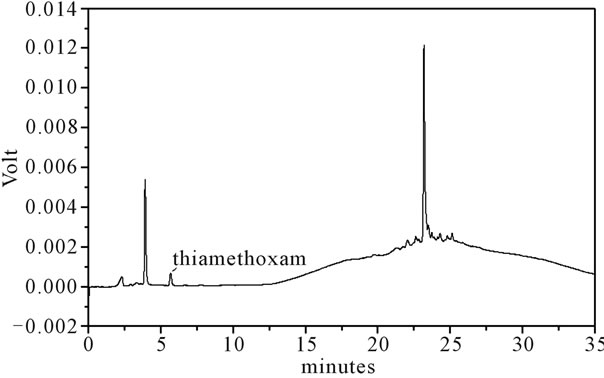
Figure 1. LC-UV of extract from water. For chromatographic conditions, see the experimental section.
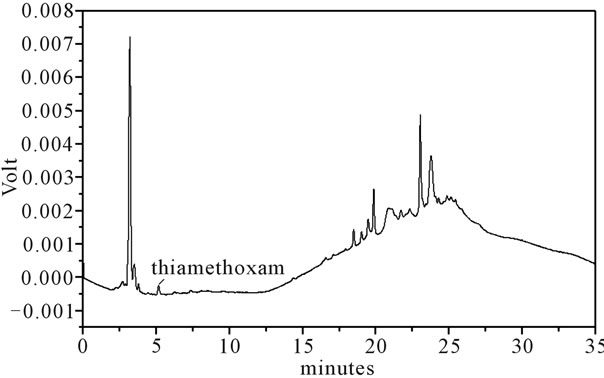
Figure 2. LC-UV of extract from soil. For chromatographic conditions, see the experimental section.
2.7.2. Extraction Recovery
The extraction efficiency (recovery) was determined by analyzing aliquots of each sample (water, rhizomes, soil and leaves) spiked with standards corresponding to low, medium and high concentrations. The spiked samples were submitted to the same procedure as described in Section 2.4.
2.7.3. Linearity
The content estimation of the thiamethoxam (1.2 - 192.0 µg∙L–1) in the samples was performed by external calibration. Aliquots of 20 µL of the dilutions was analyzed via LC with each determination being carried out five times. For the standard, the corresponding chromatogram was obtained and a graph was constructed from the mean of the chromatogram areas plotted against the thiamethoxam concentration. A linear least-square regression of the peak areas as a function of the concentrations was performed to determine the correlation coefficients. The equation parameters (slope and intercept) of the standard curve were used to obtain the concentration values for the samples. Specimens with an analytic concentration exceeding the analytical curve were re-assayed upon appropriate dilution of the samples.
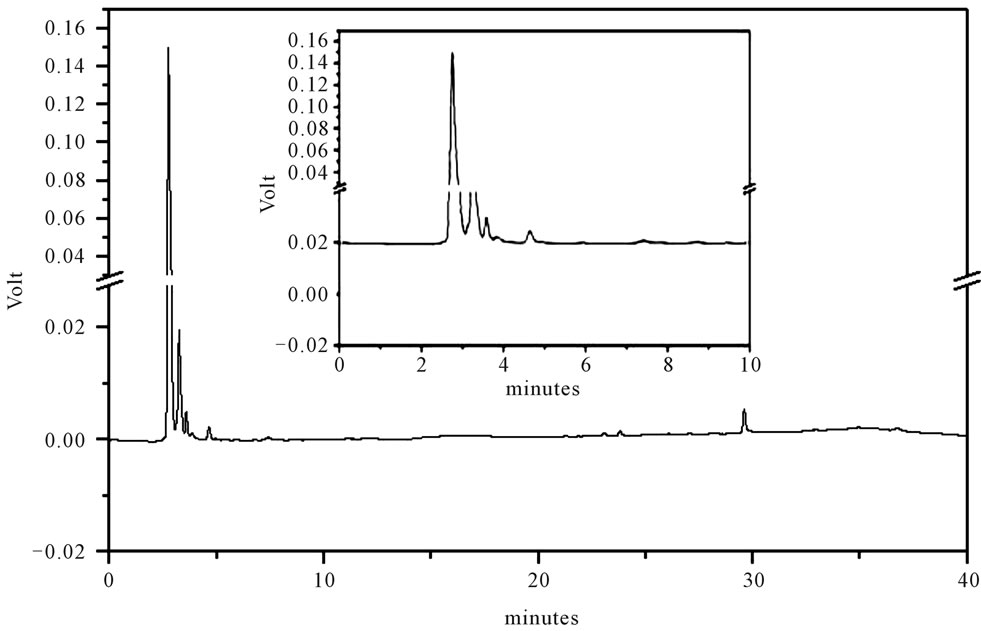
Figure 3. LC-UV of extract from rhizomes of the H. coronarium. For chromatographic conditions, see the experimental section.
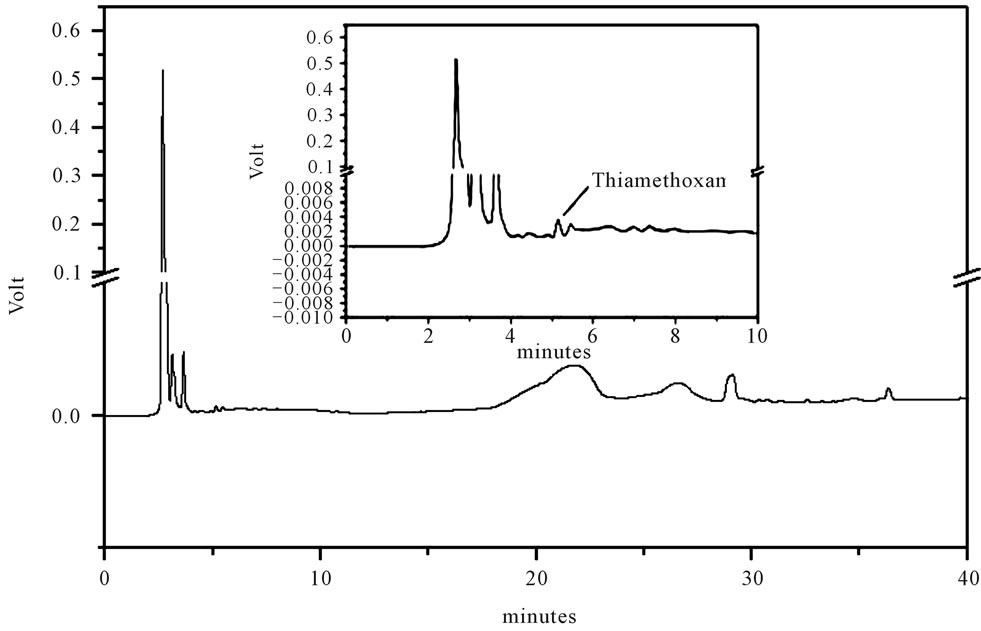
Figure 4. LC-UV of extract from leaves of the H. coronarium. For chromatographic conditions, see the experimental section.
2.7.4. Accuracy and Precision
The accuracy of the method was evaluated by performing replicate analyses against an analytical calibration curve and calculating the mean percentage differences between the theoretical values and the measured values. The accuracy values in the inter-day variation studies using LC at low, medium and high concentrations of thiamethoxam were evaluated in samples (water, rhizomes, soil and leaves). The precision of the method, expressed as the percentage relative standard deviation (RSD) of the replicate measurements, was tested for inter-day repeatability via LC. The inter-day variability of the method was determined from three different analyses (n = 5) of each sample with an addition of known amounts of analyte at low, medium and high concentrations.
2.7.5. Stability Study
The stability of the working standard solutions was tested at 22˚C (working temperature), 4˚C and –20˚C (storage temperatures). The stability of thiamethoxam in the samples was evaluated during all the storage steps (i.e., at room temperature, at 4˚C and at –20˚C). Spiked samples were analyzed against the analytical calibration curves immediately after preparation (reference values) and after storage. Stability was defined as being less than 2% loss of the initial drug concentration in the stated time.
3. Results and Discussion
3.1. Sample Preparation and LC-UV Analysis
The main objective of this work was developing a rapid, selective and sensitive analytical method including an efficient and reproducible sample for quantitative analysis of thiamethoxam in water, soil, rhizomes and leaves (Figures 1-4).
Identification of the thiamethoxam with the aid of PDA detector scanning in the spectral range of 200 - 800 nm did not reveal interferences in retention time of the extracts of water, soil rhizomes and leaves of H. coronarium in LC by the developed elution method. Thiamethoxam was easily identified based on their absorption spectra in the UV region and in retention time.
Thiamethoxam found in extracts of water, rhizomes, soil and leaves were unambiguously identified by performing co-injection experiments in which aliquots of the extracts and standard were mixed and diluted to a known volume, and analyzed through LC by more than one elution method. Thus, the peak of the chromatogram in Figures 1-4 with retention times tr of 5.25 ± 0.23 min were identified as thiamethoxam. The relative standard deviation for the retention times of replicated injections (n = 5) were less than 2%, thereby demonstrating good repeatability.
3.2. Method Validation
3.2.1. Linearity
The calibration curve was determined by linear regression using LC (Table 1). The linearity for thiamethoxam (1.2 - 192.0 µg∙L–1) was assessed for 11 concentration ranges. The average standard errors for the peak areas of replicated injections (n = 5) were less than 1%, thus showing good repeatability of the calibration curve. The respective coefficients of determination was r2 = 0.9996. The analytical procedure was verified from the linearity of the assayed method as expressed by the correlation coefficient obtained, which was evaluated by analyzing each sample spiked with a known amount of the analyte at low, medium and high concentrations, which showed that the response was proportional to the concentrations of the samples. The respective coefficients of determination were r2 = 0.9992 for thiamethoxam in soil and water and r2 = 0.9990 for thiamethoxam in leaves and rhizomes.
3.2.2. Detection and Quantification Limits
The detection limit was 0.36 µg∙mL–1 for thiamethoxam based on the signal-to-noise ratio (S\N) for 3 (Table 1).
The quantitation limit was defined as the lowest concentration level that provided a peak area with a signalto-noise ratio higher than 10, a precision (% CV) within ±5% and accuracy (% recovery) between 98% and 102%. The quantification limits by LC was 1.20 µg∙L–1 for thiamethoxam (Table 1). The results suggest that substances in plants at these concentrations can be estimated accurately.
3.2.3. Extraction Recovery
The efficiency of each analytical procedure was evaluated by calculating the recovery values.
Recovery results were between 81.16% and 99.93%, showing that the procedure employed to extract the thiamethoxam was efficient with a relative standard deviation lower than ±5% (Table 2), thus ensuring good recovery of this substance in soil, water, rhizomes and leaves.
3.2.4. Stability
The identification of thiamethoxam and of the samples (extracts of soil, leaves, water and rhizomes in studied) was performed by comparing their retention times with authentic standards through adding standard solutions to the samples analyzed by LC and by comparing its UVVis spectrum [6]. There were no alterations detected in the working solutions of the thiamethoxam prepared after
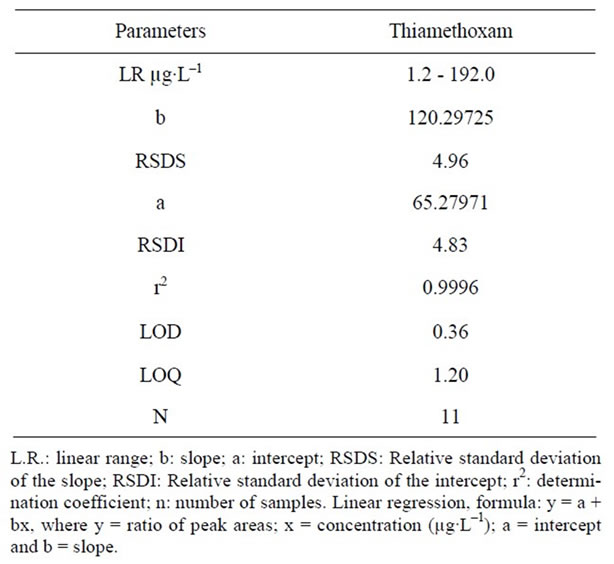
Table 1. Regression data of the analytical calibration curve for quantitative determination of the thiamethoxam by LC.
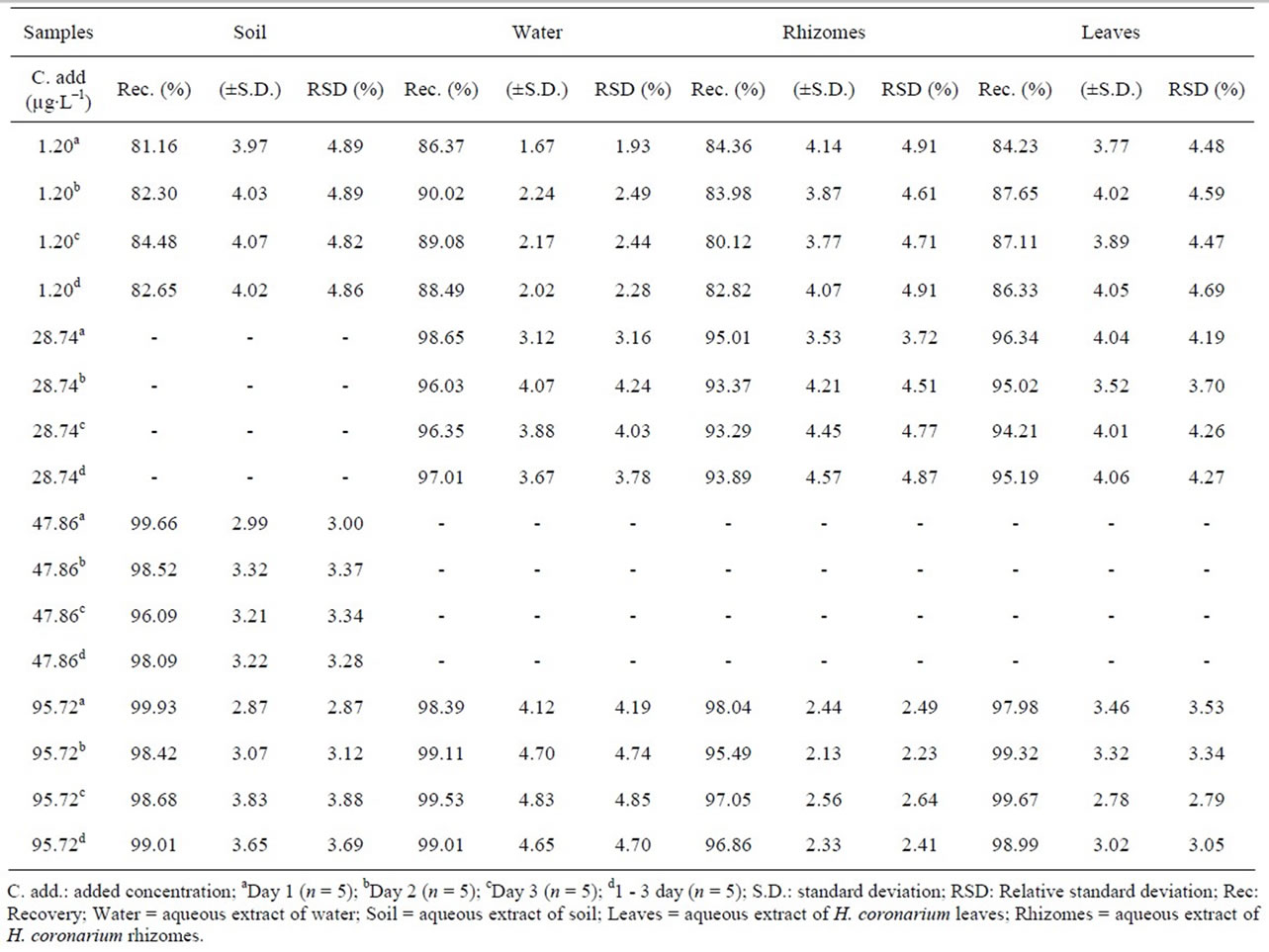
Table 2. Recovery of the thiamethoxam in aqueous extracts in samples (n = 5 for each sample).
24 hours at 22˚C, two months at 4˚C and six months of storage at –20˚C. Therefore, this validated method for the determination of the thiamethoxam can be considered adequate to indicate the stability of the solutions.
3.2.5. Accuracy and Precision
As for the precision of the assay, inter-day coefficients of variation were lower than ±5%. The international acceptance criteria (RSD for precision better than 15% and recovery values) were therefore met [25]. The inter-day variability of the tested method was determined at low, medium and high concentrations. The results are presented in Table 3. These data indicate that the method was reproducible on three different days.
3.3. Contents in Samples of the Thiamethoxam
The results from this method led to a variation coefficient lower than 5% when using the same sample. In the experiment in a greenhouse employing different samples (Table 4) to measure the contents of thiamethoxam led to variation coefficient lower than 8%, indicating that the plants did not present considerable variations.
Thiamethoxam was quantified from the extracts of leaves of H. coronarium, water and soil that were used in the experiment, but was not identified in extracts of rhizomes of H. coronarium in the experiment in the greenhouse after 21 days (Table 4).
4. Conclusions
The present study makes a contribution to reducing the waste load generated by the use of organic solvents during the stages of preparation of extracts of the analysis from thiamethoxam. This approach requires little organic solvent, which is easily discarded after the preparation steps. To our knowledge, the literature does not report the studies of aqueous extraction of the thiamethoxam in samples. The experiments demonstrated the presence of thiamethoxam in leaves, but not in rhizomes of H. coronarium after the period of twenty one days in the greenhouse. Finally, it is expected that the present approach can contribute to the study of other plant species, water and soil by providing a rapid and important strategy for
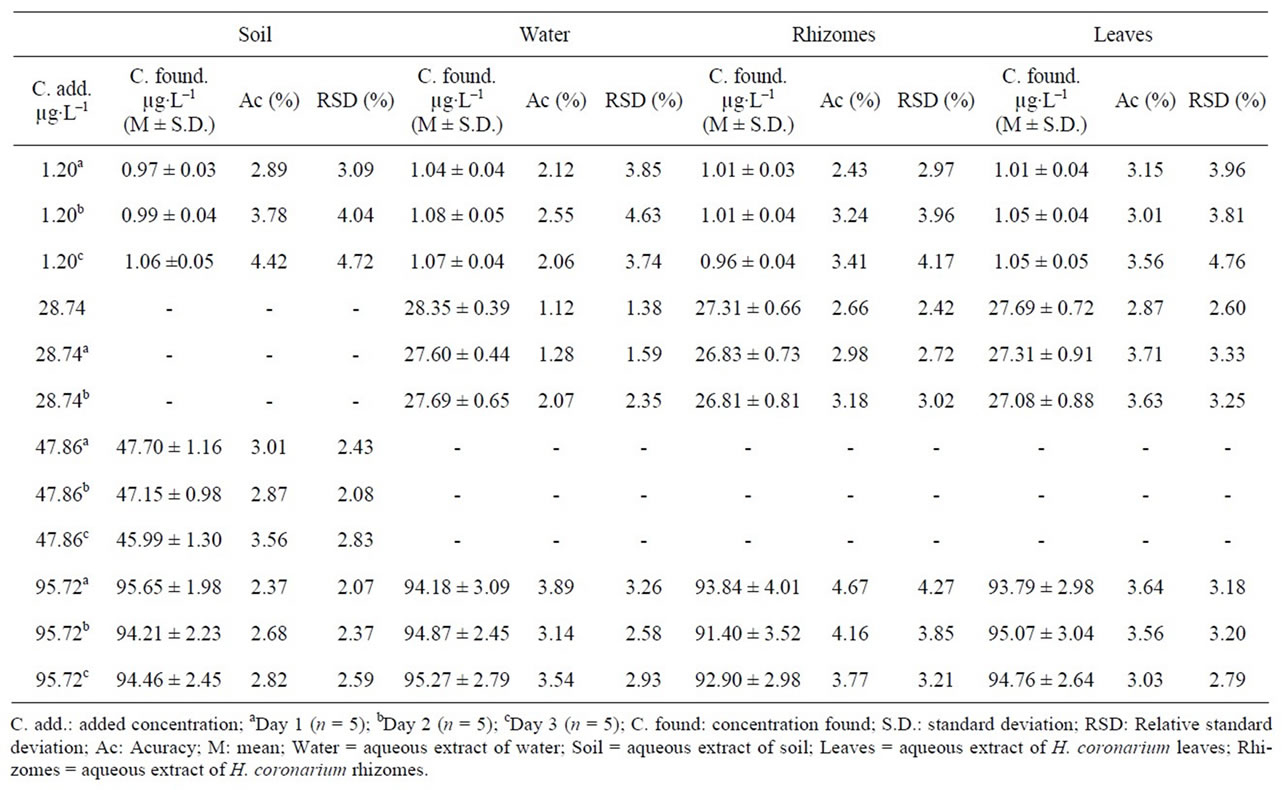
Table 3. Inter-day accuracy and precision of the LC method for determining the thiamethoxam in samples (n = 5 for each sample).

Table 4. Contents in μg∙Kg–1 in samples of the greenhouse (mean ± S.D.) of the thiamethoxam employing the LC method.
the determination of thiamethoxam.
5. Acknowledgements
The authors thank the Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
REFERENCES
- P. Maienfisch, M. Angst, F. Brandl, W. Fischer, D. Hofer, H. Kayser, W. Kobel, A. Rindlisbacher, R. Senn, A. Steinemann and H. Widmer, “Chemistry and Biology of Thiamethoxam: A Second Generation Neonicotinoid,” Pest Management Science, Vol. 57, No. 10, 2001, pp. 906-913. doi:10.1002/ps.365
- S. B. Soloway, A. C. Henry, W. D. Kollmeyer, W. M. Padgett, J. E. Powell, S. A. Roman, C. H. Tieman, R. A. Corey and C. A. Home, “Advances in Pesticides Sciences,” H. Geissbuhler, G. T. Brooks and P. C. Kearney, Eds., Pergamon Press, New York, 1979, p. 206.
- I. Denholm, G. Devine, S. Foster, K. Gorman and R. Nauen, “Incidence and Management of Insecticide Resistance to Neonicotinoids,” Proceeding of Brighton Crop Protection Conference—Pests and Diseases, Brighton, 2002, p. 161.
- M. Rancan, S. Rossi and A. G. Sabatini, “Determination of Thiamethoxam Residues in Honeybees by High Performance Liquid Chromatography with an Electrochemical Detector and Post-Column Photochemical Reactor,” Journal of Chromatography, Vol. 1123, No. 1, 2006, pp. 60-65. doi:10.1016/j.chroma.2006.05.006
- A. Peña; J. A. Rodríguez-Liébana and M. D. Mingorance, “Persistence of Two Neonicotinoid Insecticides in Wastewater, and in Aqueous Solutions of Surfactants and Dissolved Organic Matter,” Chemosphere, Vol. 84, No. 4, 2011, pp. 464-470. doi:10.1016/j.chemosphere.2011.03.039
- S. Seccia, P. Fidenteb, D. Montesano and P. Morrica, “Determination of Neonicotinoid Insecticides Residues in Bovine Milk Samples by Solid-Phase Extraction Clean- Up and Liquid Chromatography with Diode-Array Detection,” Journal of Chromatography, Vol. 1214, No. 1-2, 2008, pp. 115-120. doi:10.1016/j.chroma.2008.10.088
- M. M. Galera, G. A. Frenich, J. L. M. Vidal and P. P. Velázquez, “Resolution of Imidacloprid Pesticide and Its Metabolite 6-Chloronicotinic Acid Using Cross-Sections of Spectrochromatograms Obtained by High-Performance Liquid Chromatography with Diode-Array Detection,” Journal of Chromatography A, Vol. 799, No. 1-2, 1998, pp. 149-154. doi:10.1016/S0021-9673(97)01086-8
- S. Baskaran, R. S. Kookana and R. Naidu, “Determination of the Insecticide Imidacloprid in Water and Soil Using High-Performance Liquid Chromatography,” Journal of Chromatography A, Vol. 787, No. 1-2, 1997, pp. 271-275. doi:10.1016/S0021-9673(97)00652-3
- J. M. Bonmatin, I. Moineau, R. Charvet, C. Fleche, M. E. Colin and E. R. Bengsch, “A LC/APCI-MS/MS Method for Analysis of Imidacloprid in Soils, in Plants, and in Pollens,” Analytical Chemistry, Vol. 75, No. 9, 2003, pp. 2027-2033. doi:10.1021/ac020600b
- M. M. Macke, “Quantitation of Imidacloprid in Liquid and Solid Formulations by Reversed-Phase Liquid Chromatography: Collaborative Study,” Journal of AOAC International, Vol. 81, No. 2, 1998, pp. 344-348.
- S. B. Singh, G. D. Foster and S. U. Khan, “Microwave-Assisted Extraction for the Simultaneous Determination of Thiamethoxam, Imidacloprid, and Carbendazim Residues in Fresh and Cooked Vegetable Samples,” Journal of Agricultural and Food Chemistry, Vol. 52, No. 1, 2004, pp. 105-109. doi:10.1021/jf030358p
- J. L. Vilchez, R. El-Khattabi, J. Fernández, A. González-Casado and A. Navalón, “Determination of Imidacloprid in Water and Soil Samples by Gas Chromatography-Mass Spectrometry,” Journal of Chromatography A, Vol. 746, No. 2, 1996, pp. 289-294. doi:10.1016/0021-9673(96)00402-5
- L. M. MacDonald and T. R. Meyer, “Determination of Imidacloprid and Triadimefon in White Pine by Gas Chromatography/Mass Spectrometry,” Journal of Agricultural and Food Chemistry, Vol. 46, No. 8, 1998, pp. 3133- 3138. doi:10.1021/jf9800357
- R. G. Lodevico and Q. X. Li, “Determination of Total Imidacloprid Residues in Coffee by Gas Chromatography-Mass Spectrometry,” Analytical Letters, Vol. 35, No. 2, pp. 315-326. doi:10.1081/AL-120002532
- D. Hou, “Flora of Taiwan,” 2nd Edition, Epoch Publishing Co. Ltd., Taipei, 1978.
- T. Morikawa, H. Matsuda, Y. Sakamoto, K. Ueda, M. Yoshikawa, “New Farnesane-Type Sesquiterpenes, Hedychiols A and B 8,9-Diacetate, and Inhibitors of Degranulation in RBL-2H3 Cells from the Rhizome of Hedychium coronarium,” Chemical and Pharmaceutical Bulletin, Vol. 50, No. 8, 2002, pp. 1045-1049. doi:10.1248/cpb.50.1045
- S. Joshi, C. Chanotiya, G. Agarwal, O. Prakash, A. K. Pant and C. S. Mathela, “Terpenoid Compositions, and Antioxidant And Antimicrobial Properties of the Rhizome Essential Oils of Different Hedychium species,” Chemistry and Biodiversity, Vol.5, No. 2, 2008, pp. 299- 309. doi:10.1002/cbdv.200890027
- H. Itokawa, H. Morita, I. Katou, K.Takeya, A.-J. Cavalheiro, R.-C. B. Oliveira, M. Ishige and M. Motidome, “Cytotoxic Diterpenes from the Rhizomes of Hedychium coronarium,” Planta Medica, Vol. 54, No. 4, 1988, pp. 311-315. doi:10.1055/s-2006-962442
- H. Matsuda, T. Morikawa, T. Sakamoto, I. Toguchida and M. Yoshikawa, “Labdane-Type Diterpenes with Inhibitory Effects on Increase in Vascular Permeability and Nitric Oxide Production from Hedychium coronarium,” Bioorganic and Medicinal Chemistry, Vol. 10, No. 8, 2002, pp. 2527-2534. doi:10.1016/S0968-0896(02)00121-9
- N. Chimnoi, S. Pisutjaroenpong, L. Ngiwsara, D. Dechtrirut, D. Chokchaichamnankit, N. Khunnawutmanotham, C. Mahidol and S. Techasakul, “Labdane Diterpenes from the Rhizomes of Hedychium coronarium,” Natural Product Research, Vol. 22, No. 14, 2008, pp. 1249-1256. doi:10.1080/14786410701726434
- H. Itokawa, H. Morita, K. Takeya and M. Motidome, “Diterpenes from Rhizomes of Hedychium coronarium,” Chemical and Pharmaceutical Bulletin, Vol. 36, No. 7, 1988, pp. 2682-2684. doi:10.1248/cpb.36.2682
- J.-C. Ho, “Antimicrobial, Mosquito Marvicidal and Antioxidant Properties of the Leaf and Rhizome of Hedychium coronarium,” Journal of the Chinese Chemical Society, Vol. 58, No. 4, 2011, pp. 563-567.
- R. A. Almeida, D. P. S. Pitaluga and R. P. A. Reis, “Treatment of Wastewater by Root Zone Proceeded by Septic Tank,” Journal Biosciences, Vol. 16, No. 1, 2010, pp. 73-81.
- W. Vilegas, C. A. L. Cardoso and A. E. Pires, “Natural Products Chemistry, New Drugs and Modern Pharmacognosy,” Univali, Itajaí, 2009.
- V. P. Shah, K. K. Midha, S. Dighe, I. McGilvery, J. P. Skelly, A. Yakobi, T. Layloff, C. T. Viswanathan, C. E. Cook, R. D. McDowall, K. A. Pittman and S. Spector, “Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies,” Pharmaceutical Research, Vol. 9, No. 4, 1992, pp. 588-592. doi:10.1023/A:1015829422034
NOTES
*Corresponding author.