Open Journal of Rheumatology and Autoimmune Diseases
Vol.4 No.3(2014), Article ID:49106,14 pages
DOI:10.4236/ojra.2014.43025
NF-κB Controls Resistance of Human Salivary Gland (HSG) Cells to Apoptosis in an in Vitro Model of Sjogren’s Syndrome
Yan Wang1, Syed A. Jamal2,3, Luis F. Torres-Romero4, Agostino Molteni4, Alexander Shnyra5, Carole McArthur1*
1Department of Oral and Craniofacial Science, School of Dentistry, Kansas City, USA
2Department of Molecular Biosciences, Lawrence, USA
3Department of Chemistry, Rockhurst University, Kansas Ciy, USA
4Department of Molecular Biosciences, Lawrence, USA
5Kansas City University of Medicine and Biosciences, Kansas City, USA
Email: frankmdy@gmail.com, Syed.Jamal@rockhurst.edu, Luis.TorresRomero@tmcmed.org, moltenia@umkc.edu, ashnyra@kcumb.edu,*mcarthurc@umkc.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 June 2014; revised 17 July 2014; accepted 17 August 2014
ABSTRACT
Aim: To elucidate the anti-apoptotic properties of nuclear factor kappa light-chain-enhancer of activated B cells (NF-κB) and feedback regulation of NF-κB by nuclear factor of kappa light-chain-enhancer of activated B-cells inhibitor alpha (IκBα). Methods: We developed an in vitro model of Sjogren’s syndrome by transfecting human salivary gland (HSG) and acinar cells (NS-SV-AC) with a plasmid-encoding IκBαM (pCMV-IκBαM), a degradation-resistant IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha)-mutant, and examined TNF-induced apoptosis and anti-apoptotic properties of NF-κB. Apoptosis and induction of pro-apoptotic and anti-apoptotic genes were investigated by cDNA arrays, RT-PCR, electrophoretic mobility shift assays, and western blot. Results: In the presence of NF-κB inhibitors, TNF-induced apoptosis was markedly increased in both salivary gland and acinar cells. Increased caspase-3 activity was present in both HSG and NS-SV-AC cells. IκBαM-transfected salivary gland cells were more sensitive to TNF-induced apoptosis than IκBαM-transfected acinar cells. Transcription of pro-apoptotic genes was confirmed in both HSG and NS-SV-AC cells that were transfected with IκBαM. Results from caspase-3 activity assay confirmed previous experiments showing an apoptotic role for NF-κB. Conclusion: Data from gene expression arrays suggest that different mechanisms may operate during TNF-induced apoptosis in salivary gland ductal and acinar cells.
Keywords:Acinar, Ductal, Cytokines, Salivary Glands, Sjogren’s Syndrome, Tumor Necrosis Factor-Alpha

1. Introduction
Sjogren’s Syndrome (SS) is an autoimmune disease that involves inflammation of salivary glands, lacrimal glands, and other tissues of the body [1] . Apoptosis of salivary gland tissue is a hallmark of the disease. There are two forms of SS: primary SS occurs in the absence of another autoimmune disorder; secondary SS occurs in conjunction with another autoimmune disorder such as lupus or rheumatoid arthritis. According to the latest figures published by the Sjogren’s syndrome Foundation, 4 million people worldwide have primary SS. Symptoms of SS are seen primarily in women between the ages of 45 to 55: symptoms include dry mouth, dry eyes, itchiness and burning sensation in the eyes, dysphagia, and the presence of auto-antibodies against the salivary glands as well as anti-bodies against nuclear antigens [2] . Although the pathogenesis of SS is poorly understood, both genetic and infectious factors have been implicated [3] . Both cell-mediated and humoral responses accompanied by pro-inflammatory cytokines, such as TNF-alpha, IL-1, IL-6, IL-8, IL 17, IL-23, and the recently discovered IL-34, transcription factors such as NF-κB and STAT3, caspases and other proteins that mediate programmed cell death have been implicated in the pathogenesis of SS [4] [5] . Symptoms result from tissue destruction but the mechanism of destruction, generally agreed to be induced by cytokines, is poorly understood.
Numerous studies suggest that cytokine-mediated apoptosis of epithelial cells within the salivary gland is involved [6] . Furthermore, salivary gland cells from patients with SS exhibit greater expression of Fas receptor (CD95), a death domain-containing member of the TNF receptor superfamily, which mediates apoptosis triggered by Fas ligand (FasL)-expressing lymphocytes [7] . Apoptosis in salivary tissue can be induced in vitro by Tumor Necrosis Factor (TNF): TNF, previously known as TNF-α, is a pleiotrophic cytokine produced by immune cells in response to infection, environmental challenges and/or other danger signals [8] -[10] . As an early pro-inflammatory cytokine, TNF stimulates production of various cytokines, chemokines, coagulation factors, and other inflammatory mediators, and up-regulates the expression of adhesion molecules [11] . More importantly, TNF binds to TNF receptor 1 (TNFR1) and triggers a caspase-dependent apoptosis in target cells that can be prevented by a concurrent activation of NF-κB [12] . Tumor necrosis factor is increased in the peripheral blood and in the parenchyma of salivary glands in patients with SS, providing support for the hypothesis that cytokines mediate apoptosis [12] .
Understanding the regulation of apoptosis involving the salivary glands is important in the development of new therapies for SS. Nuclear Transcription Factor Kappa B (NF-κB) is a transcription factor that regulates the expression of inflammatory genes in response to a wide range of stimuli such as stress, cytokines, and bacterial or viral antigens. NF-κB plays a key role in regulating the immune response to infection [12] . However, aberrant induction or dysregulation of NF-κB has been linked to cancer, chronic inflammatory and autoimmune diseases, septic shock, viral infection and improper immune development [13] -[16] . NF-κB belongs to the category of “rapid-acting” primary transcription factors because it preexists in cytoplasm in a latent form that can be promptly activated without de novo protein synthesis [12] -[14] . The activity of NF-κB is regulated by an inhibitor of nuclear transcription factor NF-κB, known as IκBα [14] . Activation of the classical NF-κB signaling pathway by TNF results in rapid phosphorylation and dissociation of IκBα from the complex with NF-κB, which culminates in IκBα degradation by a cytosolic proteasome [14] [15] . This allows nuclear translocation of p50/ p65 heterodimers and transcriptional induction of various genes including anti-apoptotic genes [14] -[16] . In most patients with SS, there is a defect in the proteasomal low-molecular weight protein subunit (LMP2), a protein that is mandatory for the activation and subsequent translocation of NF-κB, followed by the transcription of survival genes in activated T cells [17] . These observations led to the formulation of our hypothesis that inhibition of NF-κB signaling would lead to enhanced rates of apoptosis in immortalized human salivary gland ductal (HSG) and acinar cells (NS-SV-AC).
We tested the above hypothesis by developing an in vitro model of SS comprised of Human Salivary Gland (HSG) ductal cells and human acinar cells (NS-SV-AC). TNF-dependent apoptosis and anti-apoptotic properties of NF-κB were examined in HSG and NS-SV-AC salivary cells both in the absence and presence of NF-κB inhibitors. In addition, TNF-induced apoptosis was studied in HSG and NS-SV-AC cells transfected with a pCMV-IκBαM plasmid-encoding IκBαM, a degradation-resistant IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha)-mutant. Apoptosis and transcriptional induction of genes encoding pro-and anti-apoptotic proteins were quantified by using a caspase-3 activity assay, cDNA arrays, and semiquantitative RT-PCR, respectively. The findings of our study support an important role for NF-κB signaling in the apoptosis of salivary gland cells in SS. Evidence in this model suggests that inhibition of NF-κB signaling sensitizes ductal cells to a greater degree of TNF-induced apoptosis as compared to the induction of apoptosis in acinar cells. Findings suggest targets for SS and other autoimmune diseases.
2. Materials and Methods
2.1. Approval
This study was approved by the Ethics Committee of the University of Missouri-Kansas City.
2.2. Reagents
Cell-permeable proteosome inhibitors PSI (N-carboben-zoxy-L-isoleucyl-L-glutamyl (O-tert-Butyl)-L-alanylL-leucinal) and MG132 were purchased from Calbiochem (San Diego, CA) and used as nonspecific NF-κB inhibitors. CAPE (Caffeic Acid Phenethyl Ester, an inhibitor of P65 nuclear binding), NF-κB SN50 (AAVALLPAVLLALLAPVQRKRQKLMP, a cell-permeable peptide inhibitor of NF-κB nuclear translocation) and Capsaicin (8-Methyl-N-Vanilly-6-Nonenamide, an NF-κB inhibitor) were purchased from Calbiochem (San Diego, CA). Cycloheximide was purchased from Sigma (St. Louis, MO). Recombinant human TNF was purchased from R&D system Inc. (Minneapolis, MN). The HSG cell line was a gift from Dr. Hinda Klienman at the National Institute of Dental and Craniofacial Research. HSG cells were expanded and sub-cultured at passage 26. NS-SV-AC cells were a gift from Professor M Sato at Tokushima University, Japan. Industrial grade carbon dioxide was supplied by Airgas (Kansas City). Buffers were made from reagent grade chemicals (Sigma Chemical Company, St Louis, MO).
2.3. Plasmids and Transfection
HSG (human salivary gland ductal) and human salivary gland acinar (NS-SV-AC) cells were transfected with IκBαM vector (Figure 1(a)) using a Lipofectin® Reagent and the manufacturer’s protocol from Invitrogen (Carlsbad, CA). The cDNA plasmid encodes a super-repressor IκBαM, a mutant form of IκBα with a serine-to-alanine mutation at residues 32 and 36. This mutation prevents phosphorylation and subsequent degradation of IκBα in proteosomes—an outcome that leads to the retention of NF-κB in a latent form outside of the nucleus. Clones with stable expression of IκBαM were selected using media containing 10% FBS and 500 μg/ml of Geneticin (G418) and were confirmed by Western blot.
2.4. Cells
Two well-characterized cell lines, HSG (salivary gland ductal) and NS-SV-AC (salivary gland acinar), were used. Cells were grown in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% fetal calf serum (FBS) and penicillin (100 μ/ml) and streptomycin (100 μg/ml). Cell cultures were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37˚C. HSG and NS-SV-AC cells were transfected as follows. To induce transfection, lipofection, reagent Pfx-4 (Invitrogen, CA) was combined with the cells and plasmid, mixed and incubated at 37˚C for 8 hours. Clones with IκBαM were selected by media containing 10% FBS and 500 μg/ml of geneticin (G418). Positions within IκBαM clones were confirmed by Western blot. HSG and NS-SVAC cells were transfected with a pCMV-IκBαM plasmid encoding IκBαM, a mutant degradation-resistant form of IκBα, and then subcloned into IκBαM(+) cell lines.
2.5. Transfection of IκBαM into HSG and NS-SV-AC Cells in Vitro and Analysis of IκBαM Protein Expression
The objective was to observe the effects of inhibition of NF-κB on apoptosis. The insertion of the 110 kd IκBαM was confirmed in each cell line following transfection using western blot prior to analyzing the effect of the repressor on TNF-induced apoptosis.
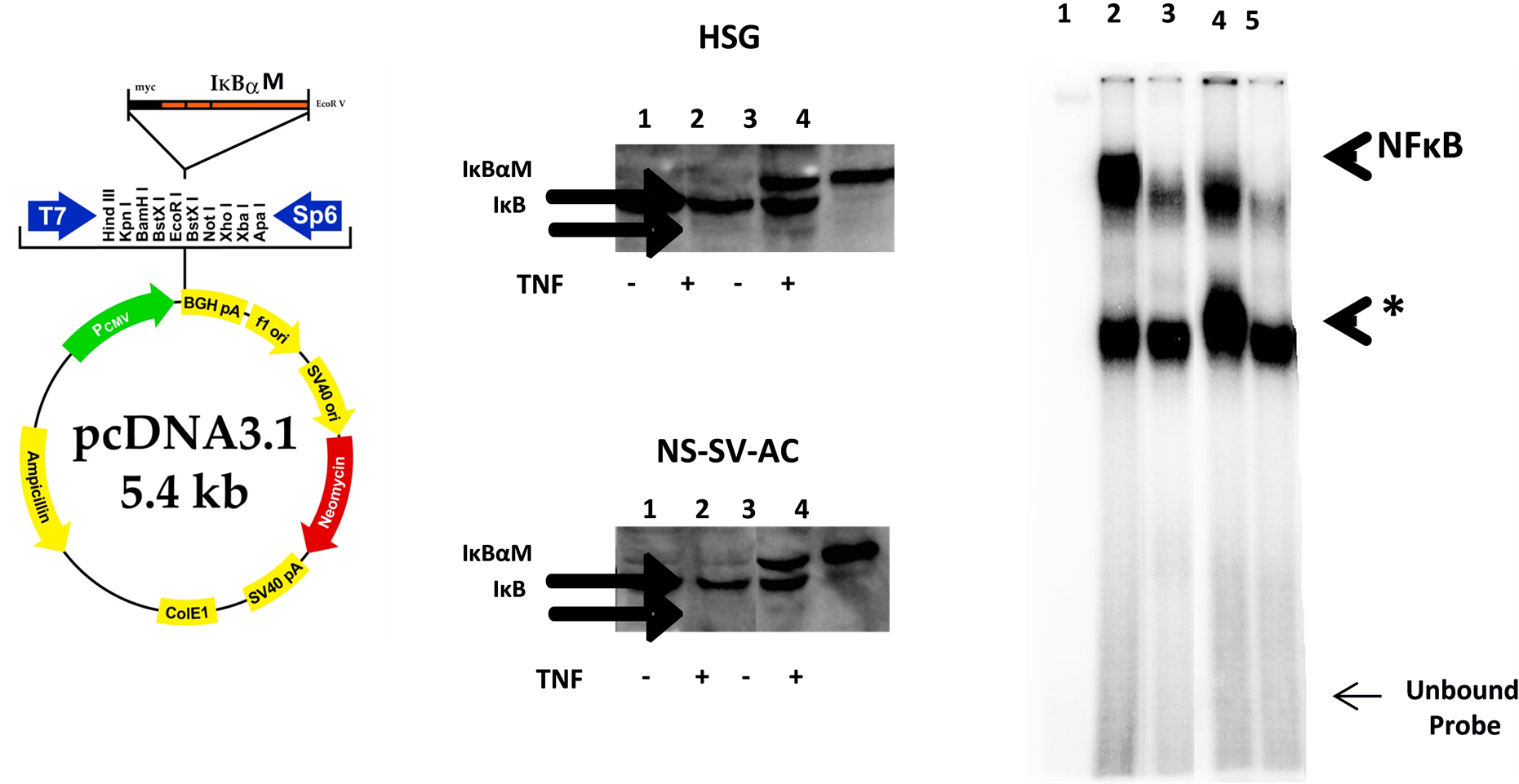 (a) (b) (c)
(a) (b) (c)
Figure 1. Analysis of expression and activity of IκBαM protein in HSG and NS-SV-AC cells stably transfected with an IκBαM plasmid. (a) The diagram shows the IκBαM plasmid used for cell transfection; (b) The IκB and IκBαM proteins in salivary gland cells were analyzed by Western blotting. Fifty micrograms of the whole cell extracts were subjected to SDSPAGE, transferred into a Hybond-P membrane, and then probed with anti-I B antibody as described. Lane 1 and 2 show the I B protein levels in resting and TNF-challenged I BαM(−) salivary gland cells, respectively. Lane 3 and 4 show the expression of both IκB and IκBαM proteins in resting and TNF-stimulated IκBαM(+) cells; (c) Electrophoretic mobility shift assay of NF-κB-DNA binding in nuclear extracts prepared from IκBαM(−) and IκBαM(+) cells. Lane 1 shows probe alone (negative control). Lanes 2 and 4 show the NF-κB binding activity in TNF-stimulated IκBαM(−) and HSG and NS-SV-AC cells, respectively. Lanes 3 and 5 show markedly reduced NF-κB activation in TNF-stimulated IκBαM(+) HSG and NS-SV-AV cells, respectively. Specific (NF-κB) and nonspecific (*) complexes are indicated.
A well-established Electrophoretic Mobility Shift Assay (EMSA) was used to confirm that the molecular weight changed with NF-κB activity following transfection.
2.6. Caspase-3 Activity Assay
Since the apoptotic pathway is activated by TNF ultimately activating pro-caspase-3 to caspase-3, an assay for caspase-3 activity is utilized to determine apoptotic activity. Cell apoptosis was measured using a commercial caspase-3 activity assay according to the manufacturer’s instructions (Oncogene Research Products, San Diego, CA). After cells were incubated into a 96-well plate with extraction buffer at 4˚C for 30 min, assay buffer was added to all the cells. Caspase-3 fluorescent substrate conjugate was added, incubated 1 hour at 37˚C, and the plate read using a fluorescent plate reader (ELX800, Bio-Tek Instrument, Inc. VT) capable of measuring excitation at 400 mm and emission at 505 mm. The data were expressed as the relative fluorescent unit (RFU) after subtraction of the relative signal produced by the appropriate buffer controls.
2.7. Nuclear and Cytosolic Extract Preparation
Cells were seeded into 150-mm plastic flasks and treated with TNF-α after 24 hours. Nuclear and cytosolic extracts were obtained as described by the manufacturer of NE-PER Nuclear and Cytoplasmic Extraction Reagents I & II (CER I-CER II, Pierce, Rockford, IL). In brief, cells were washed twice with ice-cold PBS before suspension in 400 μl of ice-cold CER I buffer for 10 min. CER II buffer was added and the lysates vortexed before centrifugation using a microcentrifuge. The supernatants of this centrifugation were designated as cytosolic extract. Each nuclear pellet was resuspended in 100 μl of Nuclear Extraction Reagent (NER) buffer and placed on ice for 40 min. Protein concentration was determined by the Bradford method (Bio-Rad Lab, Inc, Hercules, CA).
2.8. Western Immunoblotting
Sample cytosolic extracts were mixed with 50 μl Gel loading buffer (pH 6.8). Then samples were incubated at 100˚C for 5 min then were separated by electrophoresis on 10% SDS-PAGE gels with running buffer (25 mM Tris, 19.2 mM glycerin, 0.1% SDS, pH 8.3). After 1 hour SDS-PAGE running, samples were transferred to a Hybond-P membrane (Amersham Pharmacia Biotech Inc., Piscataway, NJ) with Tris/glycine buffer (20% methanol). After protein samples was transferred to the membrane, membranes were blocked with 5% (w/v) skimmed milk powder in TBS (20 mM Tris, 500 mM NaCl, pH 7.5), then were probed with a primary monoclonal anti-IκB antibody (Chemicon International, Inc., Temecula, CA) (1:1000) diluted in TTBS (0.05% Tween). Following washing with TTBS, bound antibodies were detected using a peroxidase-conjugated anti-mouse IgG (Pierce, Rockford, IL). After further extensive washing with TTBS, peroxidase reactivity was detected using chemiluminescence (Pierce, Rockford, IL). Densitometric analysis of blots was performed with a GS-250 molecular imaging system (Bio-Rad, Hercules, CA).
2.9. RNA Preparation, Reverse Transcription and Microarray Analysis
Total RNA was extracted by RNAqueous-Midi total RNA Isolation system (Ambion, CA) as described by the manufacturer. Total RNA (0.5 ug) was treated with P solution (deoxyribonuclease I) and preincubated at 37˚C for 10 min followed by reverse transcription for 30 min at 37˚C using the Ampolabeling-LPR kit, (SuperArray, MA).
cDNA array mRNA expression was evaluated using selectively targeted gene microarrays focusing on genes known to be involved in human apoptosis. This specific Human Apoptosis GEArray S series (SuperArray Inc., Bethesda, MD) contains 67 genes known to be involved in the mechanism of apoptosis. Detailed information about the cDNA array including the description of gene probes, experiment protocol, and data analysis software can be obtained at the supplier’s website (www.superarray.com).
2.10. Transcription
0.5 ug of total RNA was reverse-transcribed into cDNA with Ampolabeling-LPR kit in presence of P32 dCTP (Perkin Elmer, Boston, MA). Labeled cDNA samples were then hybridized overnight to human extracellular matrix and adhesion molecule gene-specific probes that were spotted onto the commercially available GEArray membranes. After the membranes were washed, they were scanned to produce raw data that was analyzed by OptiQuant Acquisition Analysis Program. The signals from the expression of each gene on the array were normalized to the signal derived from an internal GAPDH standard on the same membrane and expressed as a percentage expression of GAPDH, using the following formula:
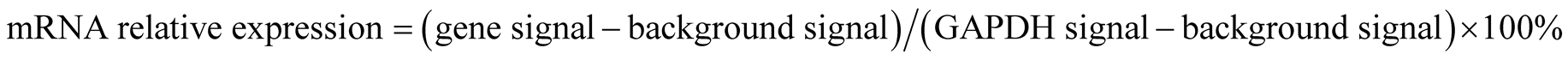
2.11. Labeling of Oligonucleotides and Electrophoretic Mobility Shift Assay (EMSA)
The probe for NF-κB consists of NF-κB-specific double stranded oligonucleotide having a sequence 5’-AGTTGAGGGGACTTTCCCAGGC-3’ containing the κB binding site. Oligonucleotides were end-labeled with (γ-32 P) ATP using polynucleotide kinase (Ambion, Austin, TX ) and unincorporated (γ-32 P) ATP was removed by Sephadex G25-packed spin columns (Ambion, Austin, TX). EMSA was carried out as follows: 1.0 μg of nuclear extract was mixed with the labeled probe in a 20 μl volume of buffer (10 mM Hepes, pH 7.9, 50 mM KCL, 0.2 mM EDTA, 2.5 mM DTT, 10% glycerol, and 0.05% Nonidet P-40). The specificity of the complex in response to dexamethasone treatment was analyzed by incubation with an excess of unlabeled competitor oligonucleotide (100-fold molar excess of label probe). Samples were run on 7.5% non-denaturing polyacrylamide gel dried at 80˚C for 2 hours and exposed in a GS-250 molecular imaging system (Bio-Rad, Hercules, CA). Images were analyzed using Molecular Analyst computer program (Bio-Rad, Hercules, CA).
2.12. Statistical Analysis
We assessed differences between samples using a T-test. A p-value less than 0.05 was taken as statistically significant.
3. Results
We have observed increased susceptibility of human salivary gland cells to TNF-induced apoptosis in the presence of NF-κB inhibitors and in the presence of degradation resistant IκB mutant (Figure 2 and Figure 3). To complement the results of the microarray analysis, the EMSA was used to study NF-κB activity in HSG and
 (a)
(a)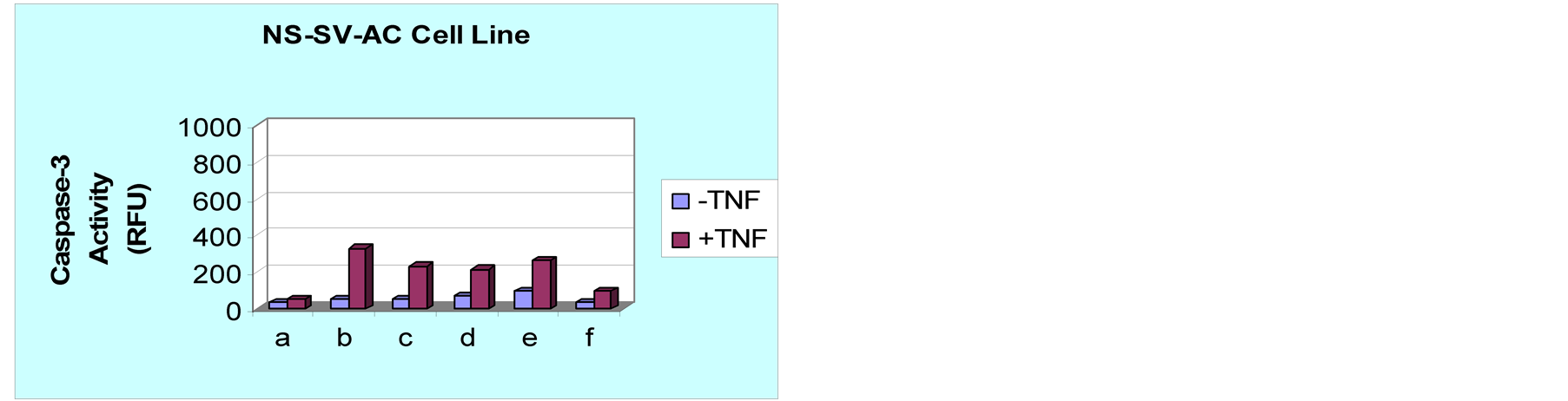 (b)
(b)
Figure 2. NF-κB inhibitors enhance TNF-α-induced apoptosis. HSG and NS-SV-AC cells were treated for 12 hr with either (a) No inhibitor, (b) PSI (N-carboben-zoxy-L-isoleucyl-L-glutamyl (O-tert-Butyl)-L-alanyl-L-leucinal) (1µg/ml) (c) CAPE (Caffeic Acid Phenethyl Ester) (10 µg/ml) (d) MG132 (1 μM) (e) Capsaicin, (8-Methyl-N-Vanillyl-6- Nonenamide) (5 µg/ml) and (f) Cell-Permeable inhibitor peptide (20 μM, 12 hours). Pretreated cells were then incubated in the absence or presence of TNF for 4 h and apoptosis induced was evaluated by measuring caspase-3 activity in cell lysates. The activity is expressed in relative fluorescence units (RFU). (a) HSG cells were more vulnerable to apoptosis in the presence of all NF-κB inhibitors except cell-permeable peptide inhibitor (p < 0.05). (b) Acinar cells showed lower differences in apoptotic rates as reflected by caspase-3 induction. statistically significant differences were observed in the presence of all inhibitors of NF-κB (p < 0.05).
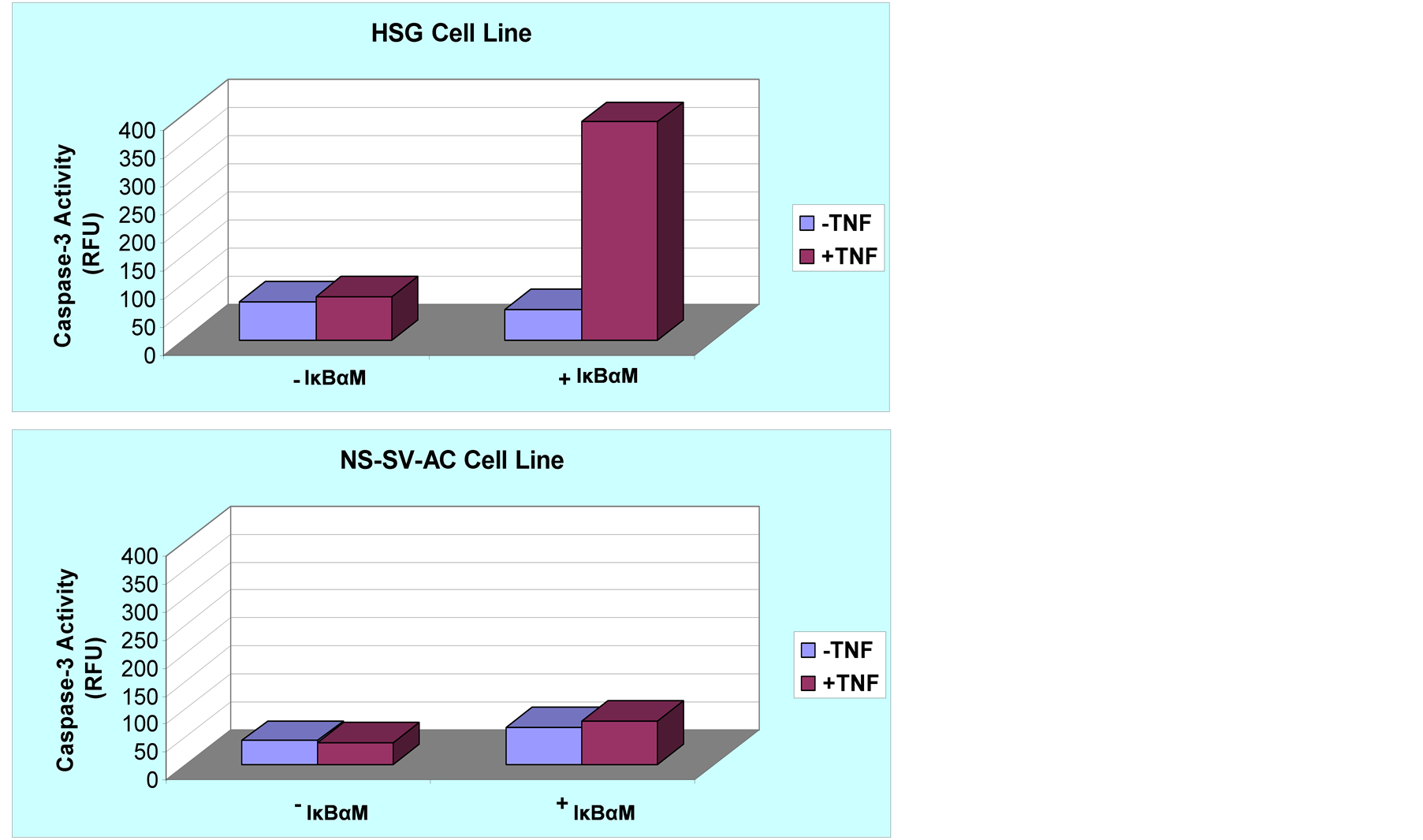
Figure 3. Selective augmentation of TNF-induced apoptosis in IκBαM(+) HSG cells. Apoptosis in IκBαM(−) cells and IκBαM(+) cells was quantified by measuring caspase-3 activity in cell lysates. The caspase-3 activity is expressed in relative fluorescence units (RFU) after subtraction of the background controls produced by reactions where no cell lysates were added. Top panel shows that in IκBαM-transfected human salivary gland cells, apoptosis was significantly greater in the presence of TNF-α (p << 0.05). Lower panel shows that acinar cells were more resistant to TNF-α-induced apoptosis even when transfected with IκBαM.
NS-SV-AC cells (Figure 1(c)) which had been stimulated with TNF. These results showed a decrease in NF-κB activity as predicted from the gene microarray data. NF-κB suppression differentially regulates apoptosis-related genes in ductal and acinar cells. In both IκBαM-HSG and NS-SV-AC cells, TNF induced a transient decrease in IκB protein within 20 - 30 min of stimulation (data not shown) which was restored upon a 24-hr treatment due to NF-κB dependent transactivation of IκB gene and de novo synthesis of IκB protein (Figure 1(c), lane 2). In TNF-treated IκBαM+ cells, however, depleted IκB protein was not restored (Figure 1(c), lane 4) suggesting IκBαM inhibited nuclear translocation of NF-κB which prevented transactivation of IκB gene and synthesis of IκB protein. These findings were further corroborated by the results of EMSA. Figure 1(c) shows that TNF induced a band shift in wild type (IκBαM−) ductal and acinar cells which indicates a classical activation and nuclear translocation of NF-κB (Figure 1(c), lanes 2 and 4). In contrast, activation and nuclear translocation of NF-κB was prevented in HSG and NS-SV-AC cells expressing a mutant IκBαM+ protein (Figure 1(c), lanes 3 and 5).
Oκur cDNA array experiments were designed to elucidate regulatory mechanisms underlying the dichotomy in resistance of HSG and NS-SV-AC cells to TNF-α-induced apoptosis upon inhibition of NF-κB signaling pathway. We expected acinar cells to be more sensitive to apoptosis since they are the component markedly destroyed in vivo leaving a few ducts remaining amidst fibrotic scar tissue. Caspase-8 is considered the canonical initiator of death receptor mediated apoptosis, an apoptotic pathway studied in these experiments [17] . Upon TNF (TNFSF1A) binding to a death receptor (TNFRSF) expressed at the surface of target cell, a complex between cytoplasmic domain of TNFRSF and Fas-associated protein with death domain (FADD) is formed that recruits and activates caspase-8 (Figure 4). Targeted profiling of proand anti-apoptotic genes in HSG and NSSV-AC cells was performed by using a pathway-specific cDNA gene array (GE Array Q series, Human apoptosis gene array). The cDNA array allowed simultaneous measurements of mRNAs of 97 genes related to apoptosis. In IκBαM+ HSG cells challenged with TNF, the expression of 47 genes was up-regulated by more than twofold as compared to mRNA levels in wild type HSG cells stimulated with TNF. The ratio of up-regulated (n = 35) to down-regulated (n = 12) genes was approximate. Similarly, 48 apoptosis-relevant genes changed their transcriptional expression (a twofold cut off) upon TNF challenge in apoptosis-resistant IκBαM+ NS-SV-AC cells. However, the number of down-regulated genes (n = 23) was higher and comparable to that of up-regulated genes (n = 25) (Table 1).
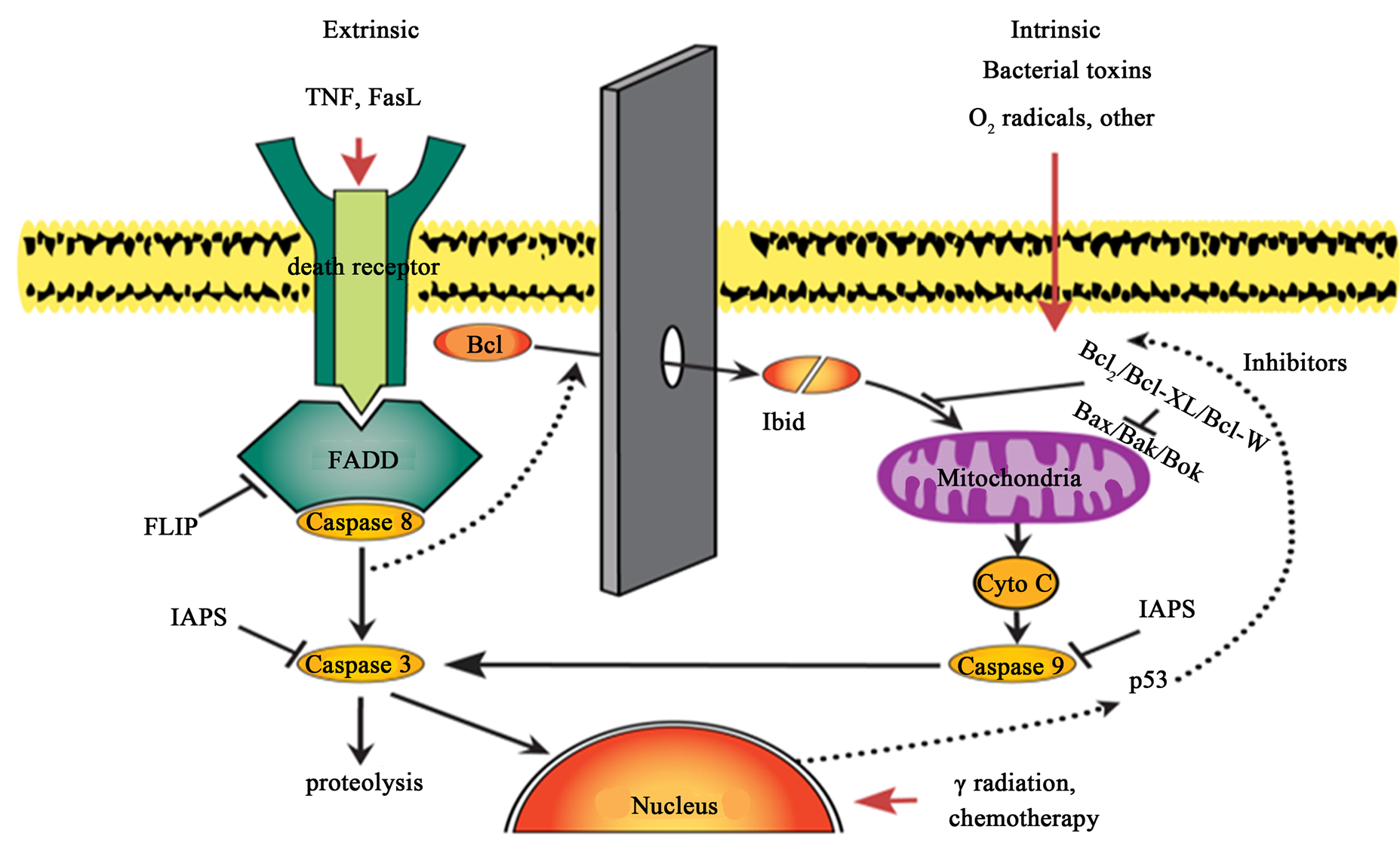
Figure 4. A simplified diagram of the relationship between the intrinsic and extrinsic apoptotic pathways.

Table 1. Gene array analysis of HSG and NS-SV-AC cell lines with or without Inserted IκBαM.
4. Discussion
Primary SS is a chronic autoimmune disease of the exocrine glands manifested by an excessive activation of T and B cells, hypergammaglobulinemia and destruction of salivary tissue [17] [18] -[20] . Identification of apoptotic mechanisms controlling the loss of salivary glands function in SS is a key step in the development of effective immunotherapies for treatment of the disease. The emerging evidence has strongly implicated the involvement of TNFSF and TNFRSF members in pathogenic mechanisms of various autoimmune diseases [18] . In this report, we provide experimental evidence that inhibition of NF-κB signaling pathway sensitizes ductal, but not acinar, cells to apoptosis induced by TNF, an inflammatory cytokine long suspected to be a key pathogenic factor in SS (Figure 2 and Figure 3).
These unexpected findings were corroborated by the results of cDNA array experiments which showed a differential display of apoptosis-related genes modulated in the cells by TNF. As expected, TNF up-regulated expression of CASP8 gene in both IκBαM+ HSG and IκBαM+ NS-SV-AC cells. However, other initiator caspases such as capsase-2 and caspase-10 were differentially expressed in ductal and acinar cells undergoing TNF-induced apoptosis. Although caspase-2 is required to initiate stress-induced apoptosis in vitro, studies on caspase-2 in murine modeld provide evidence that the role of caspase-2 in cell death is redundant and can be compensated by other caspases [18] -[20] . As to differential patterns of CASP10 mRNAs in the cells, emerging evidence suggests a strong homology between caspase-8 and -10 and, therefore, both enzymes may share similar substrate specificity and biological function [21] . The cDNA array data showed no significant differences in transcriptional expression of executioner caspases-3 and -7 in IκBαM+ HSG and IκBαM+ NS-SV-AC cells (Figure 3 lower panel) and a slightly up-regulated (2-fold) expression of caspase-6 in HSG cells (Table 1).
Caspase-8 cleaves the executioner pro-caspase-3 that activates the downstream caspases including effector caspase-6 [21] . However, it is unlikely that an increased activity of CASP6 gene in IκBαM+ HSG (ductal cells) is a key factor in selective susceptibility of these cells to TNF-induced apoptosis. This assumption is supported by experimental evidence that caspase-6 is only responsible for the apoptotic cleavage of lamin A in nuclei of the cells which express this protein [22] . In contrast, transcription of other caspase genes, such as CASP1, CASP4, and CASP5, is altered to various degrees in TNF-challenged IκBαM+ HSG and IκBαM+ NS-SV-AC cells (Table 1). Although these caspases are known to regulate an inflammatory arm of apoptosis, the observed strong up-regulation of CASP1 and CASP4 genes in IκBαM+ HSG cells would be expected to reflect, but not to cause, the undergoing apoptosis. Collectively, our cDNA array data indicate that the dichotomy in the susceptibility of ductal and acinar cells to TNF-induced apoptosis upon a suppression of NF-κB may well be controlled by other regulatory mechanisms beyond the expression levels of caspases.
The NF-κB pathway is an important mechanism in regulation of apoptosis induced by various members of the TNF superfamily (TNFSF) including TNF (TNFSF1A) [23] . Specifically, NF-κB signaling can activate transcription of various inhibitors of apoptosis (IAPs) such as cIAP1, cIAP2, XIAP, and survivin [22] -[24] . IAPs directly bind and inactivate executioner caspase-3 and caspase-7 [23] . Our cDNA array experiments revealed a striking difference between IκBαM+ HSG and IκBαM+ NS-SV-AC cells in their ability to transactivate BIRC1 gene coding for a baculoviral IAP repeat-containing 1 protein (BIRC1) (Table 1). BIRC1 is known to inhibit autocatalytic processing of procaspase-9 and subsequent cleavage of procaspase-3 thereby preventing apoptosis at the initiation stage of apoptosome formation [22] [23] . In our experiments, another anti-apoptotic caspase regulator CASP8 and FADD-like apoptosis regulator (CFLAR), was found to be up-regulated in IκBαM+ NSSV-AC cells; up-regulation of CFLAR may also contribute to the relatively unexpected cell resistance to apoptosis that was observed in the acinar cells (Table 1). CFLAR happens to be a key interface between survival and death pathways in mammalian cells: CFLAR is up-regulated by the NF-κB pathway and it prevents activation of caspase-8 by blocking recruitment and processing of caspase-8 at the death-inducing signaling complex (DISC) leading to cell survival in response to TNF [22] . In contrast, IκBαM+ HSG cells challenged with TNF exhibited a strong transactivation of CASP8AP2 gene coding for caspase-8 associated protein 2. CASP8AP2, similar to FLASH—a mouse apoptotic protein, controls the extent of cell death by promoting caspase-8 activation in response to cell stimulation via death receptors [24] . We speculate that transactivation of BIRC1 and CFLAR genes in IκBαM+ NS-SV-AC cells may, at least in part, account for a relative resistance of these cells to TNF-induced apoptosis whereas transcriptional suppression of these anti-apoptotic genes and concurrent induction of proapoptotic CASP8AP2 gene in IκBαM+ HSG cells may render these cells susceptible to apoptosis.
In the inflamed tissues, epithelial cells may serve as non-professional antigen presenting cells (APCs) presenting self-antigens and/or providing co-stimulatory signals which may result in loss of tolerance and development of autoimmunity [25] . Interestingly, our cDNA array data showed that transcriptional expression of TNFSF7 (CD-70) is strongly up-regulated (>13-fold) in TNF-treated IκBαM+ HSG ductal, but not in IκBαM+ NS-SV-AC acinar (<2-fold), cells (Table 1). CD70 expressed on dendritic cells plays a key role in inducing effective CD8+ and CD4+ T cell responses by providing a co-stimulatory signal for activation of naive T lymphocytes [26] . This could be related to the lymphocytic infiltrates seen in vivo. Furthermore, it has been shown that antibody-mediated blockage of CD70-CD27 signaling pathway halts the development of experimental autoimmune colitis and collagen-induced arthritis in mice [26] -[29] . It is tempting to speculate that up-regulated expression of CD70 in epithelial cells of the salivary glands could lead to the lymphocytic infiltrate and destruction of cells observed in vivo. Under such a scenario, the CD70-CD27 axis could be a potential target for immune intervention in patients with SS.
There were some limitations in our study that could account for the fact that we noticed greater TNF alphainduced apoptosis as reflected by increased caspase-3 expression in human ductal cells compared to human acinar cells transfected with degradation-resistant IκB. First, the apparent resistance of human acinar cells to TNFalpha-induced apoptosis, which is contrary to what is seen in vivo, suggests that redundant mechanisms occur or that this model using human cells lines from different individuals may not be reflective of the in vivo situation. Second, since our cells were not cultured on matrigel or on laminin-coated plates, it is highly likely that the balance between the expression of pro-apoptotic and anti-apoptotic genes may have been skewed in these cells [30] . Third, under in vivo conditions laminin-integrin signaling, balance between matrix metalloproteases (MMPs) and tissue inhibitors of matrix metalloproteases (TIMPS), and postganglionic acetylcholine play a role in acinar and ductal cell survival [30] . Lastly, transformation of cell lines and gender of donors may also have played a role.
The results of our apoptosis experiments on human salivary gland ductal and human acinar cells suggest that the NF-κB signaling pathways play a prominent role in the survival mechanisms in salivary cells in spite of the following conflicting data. In acinar cells transfected with IκBαM, pro-apoptotic genes were down-regulated. Down-regulation of pro-apoptotic genes in the absence of translocation of NF-κB to the nucleus would delimit the anti-apoptotic role of NF-κB and suggest the presence of other mechanisms that would protect against apoptosis in acinar cells. An opposing trend was observed in human salivary gland cells transfected with IκBαM, where transcription of some pro-apoptotic genes, including BNIP 3, CASP1, TNFSF10, BFAR, TNFSF14, CASP2, TNFSF5, TNFRSF5, TNFSF5, had increased (Table 1).
5. Conclusions
Apoptosis in ductal and acinar cells of the salivary glands is differentially controlled through an incompletely defined mechanism involving the NF-κB signaling pathway. We speculate that HSG ductal cells represent type I cells which are able to activate high amounts of initiator caspase-8 at the DISC, thus facilitating the downstream activation of an executioner caspase-3 [31] . In contrast, acinar epithelial cells may belong to type II cells which activate low amounts of caspase-8 and thus become resistant in this model to the extrinsic apoptotic pathway initiated by ligand-induced activation of the death receptors at the plasma membrane [31] . As type II cells, however, apoptosis in acinar cells may be controlled by the intrinsic cell death pathway that activates caspase-3 in a caspase-9-dependent manner involving the apoptosome. Our data also suggest that differential expression of regulatory proteins, such as BIRC1, CFLAR, and CASP8AP2, which physically bind to the appropriate caspase and regulate its activity, may contribute to the differences in TNF-induced apoptosis in ductal and acinar cells via suppression of NF-κB signaling. In addition, these data highlight the importance of the CD70-CD27 axis in breaking tolerance, and suggest that CD70 may be a target for immunotherapy.
All these experiments had been highly reproducible and carried out many times. This model has been examined elsewhere by our laboratory and we have shown that cleavage of autoantigens SSA (Ro), SSB (La), alpha fodrin and caspase-3 occurs in response to TNF stimulation [32] . These observations are consistent with in vivo findings and suggest a potential mechanism for autoimmunity since the cleaved products are expressed upon the cell surface where they could stimulate the production of auto-antibodies associated with Sjogren’s syndrome. Considered collectively, our in vitro data suggest that delineation of the inflammatory cascade that accompanies the onset of SS, especially the role of Th17 cytokines and the CD27-CD70 axis, is an important area for potential immunomodulation.
Acknowledgements
This work was supported by the Arthritis Foundation.
Conflicts of Interest
We report no conflicts of interest.
References
- Sankar, V., Noll, J.L. and Brennan, M.T. (2014) Diagnosis of Sjogren’s Syndrome. American-European and the American College of Rheumatology Classification Criteria. Oral and Maxillofacial Clinics of North America, 26, 13-22.
- Fox, P.C., Brennan, M. and Di Sun, P. (1999) Cytokine Expression in Human Labial Minor Salivary Gland Epithelial Cells in Health and Disease. Archives of Oral Biology, 44, S49-S52.http://dx.doi.org/10.1016/S0003-9969(99)90018-3
- Burbelo, P.D., Ambatipudi, K. and Alevizos, I. (2014) Genome-Wide Association Studies in Sjogren’s Syndrome: What Do the Genes Tell Us about Disease Pathogenesis. Autoimmun Review, 13,756-761. .http://dx.doi.org/10.1016/j.autrev.2014.02.002
- Ciccia, F., Guggino, G., Alessandro, R., Carubbi, F., Giardina, A., Cipriani, P., Ferrante, A., Canizzaro, A., Giacomelli, R. and Triolo, G. (2014) Rituximab Modulates IL-17 Expression in the Salivary Glands of Patients with Primary Sjogren’s Syndrome. Rheumatology, 53, 532-539.
- Bostrem, E.A. and Lundburg, P. (2013) The Newly Discovered Cytokine IL-34 Is Expressed in Gingival Fibroblasts, Shows Enhanced Expression by Pro-Inflammatory Cytokines, and Stimulates Osteoclast Differentiation. PLoS One, 8, e81665. http://dx.doi.org/10.1371/journal.pone.0081665
- Jin, J. and Yu, Q. (2013) T-Cell Associated Cytokines in the Pathogenesis of Sjogren’s Syndrome. Journal of Clinical Cellular Immunology, S, 11742.
- Singh, N. and Cohen, P.L. (2012) The T Cell in Sjogren’s Syndrome: Force Majeure, Not Spectateur. Journal of Autoimmunity, 39, 229-233.
- Maruyama, T., Saito, I., Hayashi, Y., Kompfner, E., Fox, R.I., Burton, D.R., et al. (2004) Molecular Analysis of the Human Autoantibody Response to α-Fodrin in Sjogren’s Syndrome Reveals Novel Apoptosis-Induced Specificity. The American Journal of Pathology, 165, 53-61. http://dx.doi.org/10.1016/S0002-9440(10)63274-9
- Tracey, D., Klareskog, L., Sasso, E.H., et al. (2008) Tumor Necrosis Factor antagonist mechanisms of action: A comprehensive review. Pharmacology & Therapeutics, 117, 244-279. http://dx.doi.org/10.1016/j.pharmthera.2007.10.001
- Beutler, B. and Cerami, A. (1989) The Biology of Cachectin/TNF-Primary Mediator of the Host Response. Annual Review of Immunology, 7, 625-655. http://dx.doi.org/10.1146/annurev.immunol.7.1.625
- Wang, J., Chun, H.J., Wong, W., Spencer, D.M. and Lenardo, M.J. (2001) Caspase-10 Is an Initiator Caspase in Death Receptor Signaling. Proceedings of the National Academy of Sciences of the United States of America, 98, 13884-13888. http://dx.doi.org/10.1073/pnas.241358198
- Mannel, D.N. and Echtenacher, B. (2000) TNF in the Inflammatory Response. Chemical Immunology, 74, 141-161.http://dx.doi.org/10.1159/000058757
- Karin, M. and Lin, A. (2002) NF-kappaB at the Crossroads of Life and Death. Nature Immunology, 3, 221-227.http://dx.doi.org/10.1038/ni0302-221
- Müller, F.J., Synder, E.Y. and Loring, J.F. (2006) Gene Therapy: Can Neural Stem Cells Deliver? Nature Reviews Neuroscience, 7, 75-84. http://dx.doi.org/10.1038/nrn1829
- Peng, B., Ling, J., Lee, A.J., Wang, Z., Chang, Z., Jin, W., Kang, Y., Zhang, R., Shim, D., Wang, H., Fleming, J., Zheng, H., Sun, S. and Chiao, P. (2010) Defective Feedback Regulation of NF-κB Underlies Sjogren’s Syndrome in Mice with Mutated κB Enhancers of the IκBα Promoter. Proceedings of the National Academy of Sciences of the United States of America, 107, 15193-15198. http://dx.doi.org/10.1073/pnas.1005533107
- Hoesel, B. and Schmid, J.A. (2013) The Complexity of NF-κB Signaling in Inflammation and Cancer. Molecular Cancer, 12, 86. http://dx.doi.org/10.1186/1476-4598-12-86
- Vallabhapurapu, S. and Karin, M. (2009) Regulation and Function of NF-κB Transcription Factors in the Immune System. Annual Review of Immunology, 27, 293-733. http://dx.doi.org/10.1146/annurev.immunol.021908.132641
- Kumar, S. (2007) Caspase Function in Programmed Cell Death. Cell Death and Differentiation, 14, 32-43.http://dx.doi.org/10.1038/sj.cdd.4402060
- Lassus, P., Opitz-Araya, X. and Lazebnik, Y. (2002) Requirement for Caspase-2 in Stress-Induced Apoptosis before Mitochondrial Permeabilization. Science, 297, 1352-1354. http://dx.doi.org/10.1126/science.1074721
- Konimani, K., Nakabayshi, J., Nagai, T., Tsujimura, Y., Chiba, K., Kimura, H., Miyawaki, A., Sawasaki, T., Yokota, H., Manabe, N. and Sakamaki, K. (2012) The Molecular Mechanisms of Caspase-8 Activation: Quantitative Experimental Validation of a Mathematical Model. Biochimica et Biophysica Acta-Molecular Cell Research, 182, 1825-1840.
- Ruchaud, S., Korfali, N., Villa, P., Kottke, T.J., Dingwall, C., Kaufmann, S.H. and Earnshaw, W.C. (2002) Caspase-6 Gene Disruption Reveals a Requirement for Lamin A Cleavage in Apoptotic Chromatin Condensation. The EMBO Journal, 21, 1967-1977. http://dx.doi.org/10.1093/emboj/21.8.1967
- Micheau, O. and Tschopp, J. (2003) Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell, 114, 181-190. http://dx.doi.org/10.1016/S0092-8674(03)00521-X
- Berthelet, J. and Dubrez, I. (2013) Regulation of Apoptosis by Inhibitors of Apoptosis. Cells, 2, 163-187.
- Imai, Y., Kimura, T., Murakami, A., Yajima, N., Sakamaki, K. and Yonehara, S. (1999) The CED-4-Homologous Protein FLASH Is Involved in Fas-Mediated Activation of Caspase-8 during Apoptosis. Nature, 398, 777-785.http://dx.doi.org/10.1038/19709
- Faustman, D. and Davis, M. (2010) TNF Receptor 2 Pathway: Drug Target for Autoimmune Diseases. Nature Reviews Drug Discovery, 9, 482-493. http://dx.doi.org/10.1038/nrd3030
- O’Reilly, L.A., Ekert, P., Harvey, N., Marsden, V., Cullen, L., Vaux, D.L., Hacker, G., Magnusson, C., Pakusch, M., Cecconi, F., Kuida, K., Strasser, A., Huang, D.C. and Kumar, S. (2002) Caspase-2 Is Not Required for Thymocyte or Neuronal Apoptosis Even though Cleavage of Caspase-2 Is Dependent on both Apaf-1 and Caspase-9. Cell Death and Differentiation, 9, 832-841. http://dx.doi.org/10.1038/sj.cdd.4401033
- Oflazoglu, E., Boursalian, T.E., Zeng, W., Edwards, A.C., Duniho, S., McEarchern, J.A., Law, C.L., Gerber, H.P. and Grewal, I.S. (2009) Blocking of CD27-CD70 Pathway by Anti-CD70 Antibody Ameliorates Joint Disease in Murine Collagen-Induced Arthritis. The Journal of Immunology, 183, 3770-3777. http://dx.doi.org/10.4049/jimmunol.0901637
- Hashimoto-Okada, M., Kitawaki, T., Kadowaki, N., Iwata, S., Morimoto, C., Hori, T. and Uchiyama, T. (2009) The CD70-CD27 Interaction during the Stimulation with Dendritic Cells Promotes Naive CD4+ T Cells to Develop into T Cells Producing a Broad Array of Immunostimulatory Cytokines in Humans. International Immunology, 21, 891-904.http://dx.doi.org/10.1093/intimm/dxp056
- Yu, S.E., Park, S.H. and Jang, Y.K. (2010) Epigenetic Silencing of TNSF7(CD70) by DNA Methylation during Progression to Breast Cancer. Molecules and Cells, 29, 217-221. http://dx.doi.org/10.1007/s10059-010-0052-9
- Laine, M., Vertanin, I., Porola, P., Rotar, Z., Rozman, B., Poduval, P. and Konttinen, Y.P. (2008) Acinar Epithelial Cells Laminin-Receptors in Labial Salivary Glands in Sjogren’s Syndrome. Clinical and Experimental Rheumatology, 26, 807-813.
- Scaffidi, C., Fulda, S., Srinivasan, A., Friesen, C., Li, F., Tomaselli, K.J., Debatin, K.M., Krammer, P.H. and Peter, M.E. (1998) Two CD95 (APO-1/Fas) Signaling Pathways. The EMBO Journal, 17, 1675-1687.http://dx.doi.org/10.1093/emboj/17.6.1675
- Wang, Y., Virji, A.S., Howard, P., Sayani, Y., Zhang, J., Achu, P. and McArthur, C.P. (2006) Detection of Cleaved α-Fodrin Autoantigen in Sjogren’s Syndrome: Apoptosis and Co-Localisation of Cleavedα-Fodrin with Activated Caspase-3 and Cleaved Poly (ADP-Ribose) Polymerase (PARP) in Labial Salivary Glands. Archives of Oral Biology, 51, 558-566. http://dx.doi.org/10.1016/j.archoralbio.2005.11.008
NOTES

*Corresponding author.